

Abstract
Oxygen (O2) is essential for the viability and function of most metazoan organisms and thus is closely monitored at both the organismal and the cellular levels. However, alveoli often encounter decreased O2 levels (hypoxia), leading to activation of physiological or pathophysiological responses in the pulmonary arteries. Such changes are achieved by activation of transcription factors. The hypoxia‐inducible factors (HIFs) are the most prominent hypoxia‐regulated transcription factors in this regard. HIFs bind to hypoxia‐response elements (HREs) in the promoter region of target genes, whose expression and translation allows the organism, amongst other factors, to cope with decreased environmental O2 partial pressure (pO2). However, prolonged HIF activation can contribute to major structural alterations, especially in the lung, resulting in the development of pulmonary hypertension (PH). PH is characterized by a rise in pulmonary arterial pressure associated with pulmonary arterial remodelling, concomitant with a reduced intravascular lumen area. Patients with PH develop right heart hypertrophy and eventually die from right heart failure. Thus, understanding the molecular mechanisms of HIF regulation in PH is critical for the identification of novel therapeutic strategies. This review addresses the relationship of hypoxia and the HIF system with pulmonary arterial dysfunction in PH. We particularly focus on the cellular and molecular mechanisms underlying the HIF‐driven pathophysiological processes.
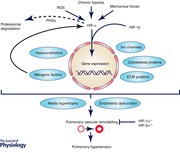
Biological role of oxygen in the lung
Ambient air contains 21% O2 at sea level, which corresponds to approximately 160 Torr. The O2 concentration drops to ∼14% O2 (100 Torr) in the pulmonary alveoli (Semenza, 2001). Finally, the partial pressure of O2 (pO2) in arterial and venous blood is ∼100 Torr (∼14% O2) and ∼40 Torr (6% O2), respectively (Prabhakar & Semenza, 2012). Moderate hypoxia (10% O2; simulated altitude of 5000 m) corresponds to 75–82 Torr (Suzuki et al. 1999). Following hypoxic exposure alveolar O2 concentration drops to ∼40 Torr, and thus the pO2 in arterial blood is ∼40 Torr (Bland et al. 1977).
Cells rely on O2 for energy production via oxidative phosphorylation in the mitochondrial respiratory chain. However, precise regulation of cellular pO2 is necessary to minimize the production of reactive oxygen species (ROS), which are associated with cellular damage (Semenza, 2000 a). Under acute hypoxic conditions (ranging from seconds to minutes) homeostatic mechanisms in the cardiovascular and respiratory systems (Semenza, 2004) are altered, primarily causing changes in cellular redox state, signalling, protein configuration and/or phosphorylation (Semenza, 2000 b). These alterations also lead to hypoxic pulmonary vasoconstriction (HPV), an essential mechanism used to adapt pulmonary blood flow to the local alveolar ventilation situation. During regional alveolar hypoxia, HPV shifts blood flow from less ventilated and thus hypoxic areas of the lung to better oxygenated areas, optimizing and maintaining gas exchange. However, under generalized hypoxia, HPV results in constriction of precapillary pulmonary arteries, increasing pulmonary vascular resistance and thus contributing to PH. In addition, if hypoxia is chronic (ranging from hours to days), gene expression is changed (Semenza, 2004), leading to pulmonary arterial remodelling and PH. This remodelling process is mainly characterized by hypertrophy of the artery media caused by proliferation of the vascular smooth muscle cells starting around day 4 of hypoxia (Paddenberg et al. 2007) and is established after 3 weeks of hypoxic exposure (Semenza, 2001; Taraseviciene‐Stewart et al. 2001). Prominent mediators of pulmonary vascular remodelling are the hypoxia‐inducible factors (HIFs).
The HIF system
The HIFs are heterodimeric transcription factors, consisting of an α and a β subunit. The β subunit is identical to the aryl hydrocarbon nuclear translocator (ARNT). Both subunits are continuously transcribed and translated and belong to the PAS family of basic helix–loop–helix (bHLH) transcription factors (Wang et al. 1995). Changes in pO2 show no influence on either HIF‐α or HIF‐β mRNA expression (Huang et al. 1996). Moreover, at the protein level, HIF‐β is constitutively expressed and not affected by pO2. In contrast, the HIF‐α protein is the regulatory subunit and remarkably unstable in normoxic (21% O2 at sea level) cells (Wang et al. 1995). The half‐life is about 5 min (Huang et al. 1996) under normoxic conditions. However, under hypoxia the abundance of HIF‐α protein is increased.
Under normoxic conditions HIF‐α is hydroxylated by prolyl hydroxylase domain proteins (PHDs) at conserved prolines 402 and 564 (Epstein et al. 2001; Ivan et al. 2001) in the oxygen‐dependent degradation (ODD) domain (Huang et al. 1998). Removal of the entire ODD domain renders HIF‐α stable even in normoxic cells, resulting in heterodimerization, DNA binding and transactivation, independent of hypoxia (Huang et al. 1998). PHDs utilize molecular O2, Fe(II) and 2‐oxoglutarate as co‐substrate for hydroxylation (Epstein et al. 2001). HIF‐α hydroxylation is recognized by the von Hippel‐Lindau tumour suppressor protein (pVHL), a component of the multi‐subunit E3 ubiquitin ligase complex (Ivan et al. 2001), which marks the HIF‐α complex for proteasomal degradation (Huang et al. 1998). Under hypoxic conditions, however, HIF‐α hydroxylation by PHD is inhibited (Epstein et al. 2001). HIF‐α accumulates and translocates to the cell nucleus where it dimerizes with the HIF‐β subunit, forming with transcriptional co‐activators (CREB‐binding protein (CBP) and p300) a functional transcription factor (Martin et al. 2010). Binding of the transcription factor via its bHLH domain to the HREs initiates expression of hypoxia‐specific genes (Fig. 1). To date, several thousand human HIF target genes have been identified (Semenza, 2014). Amongst others they ensure sufficient O2 delivery (angiogenesis), metabolic adaptations (genes involved in glycolytic pathways) and erythropoiesis (O2 transport), cell proliferation and vascular remodelling (Semenza, 2000 b, 2011).
Figure 1. Schematic diagram displaying the regulatory mechanisms of HIF activation in pulmonary hypertension .
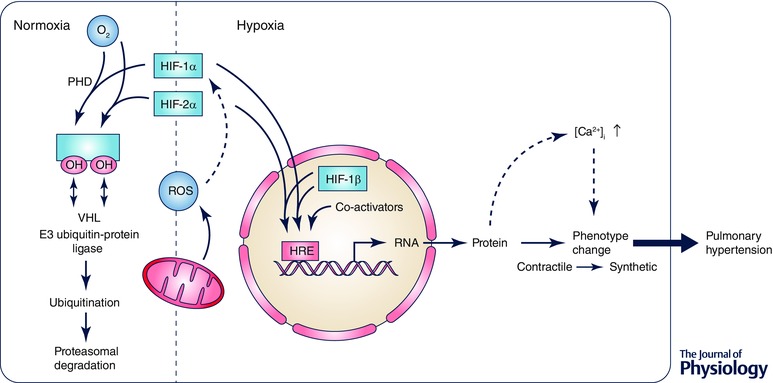
Under hypoxic conditions the HIF‐1α and HIF‐2α subunits are stabilized, among others by reactive oxygen species (ROS). They translocate from the cytoplasm to the nucleus where they dimerize with the β‐subunit forming, together with co‐activators, an active transcription factor. HIF binding to the hypoxia response elements (HREs) allows the expression of HIF target genes, contributing to the pathogenesis of pulmonary hypertension. During normoxia, HIF hydroxylation carried out by prolyl‐hydroxylase (PHD) targets the α‐subunit for proteasomal degradation through the von Hippel–Lindau (VHL) protein, a member of the E3 ubiquitin proteasome ligase family. However, HIFs might also be stabilized by ROS under normoxic conditions leading to dimerization translocation and expression of target genes.
Thus far, three ubiquitously expressed HIF‐α subunits are known with variable tissue expression and abundance, but similar O2‐dependent regulation, at least for short hypoxic exposure (Heidbreder et al. 2003; Clerici & Planes, 2009). HIF‐1α is present in all nucleated cell types, HIF‐2α expression is restricted to the vascular endothelium and type II pneumocytes (Wiesener et al. 2003), whereas HIF‐3α can be found in the cortex, hippocampus, lung, heart, liver and kidney (Heidbreder et al. 2003). HIF‐3α expression is induced by HIF‐1 in hypoxic cells, suggesting that HIF‐3α may act as a negative‐feedback factor attenuating HIF‐1 activity during prolonged hypoxia to prevent overshoot phenomena (Makino et al. 2007; Augstein et al. 2011). In addition, over‐expression of HIF‐3α decreases the hypoxia‐induced increase in the HIF‐1α target gene VEGF (Augstein et al. 2011).
HIF‐1α and HIF‐2α share high sequence homology, whereas the HIF‐3α sequence is quite different. HIF‐3α, produces multiple splice variants that contain extra DNA binding elements and protein–protein interaction motifs (Maynard et al. 2005). Nevertheless, all HIF‐α subunits are subjected to the same PHD‐dependent degradation machinery.
The HIF‐1 subunit has been most extensively studied. It was identified in 1991 as a protein that bound, under hypoxic conditions, to the HRE of the erythropoietin (EPO) gene (Semenza et al. 1991), which is critically involved in red blood cell production.
HIF‐induced pulmonary arterial smooth muscle cell dysfunction in pulmonary hypertension
PH is a life threatening disease with multifactorial causes. It is well established that prolonged exposure to hypoxia due to chronic lung diseases or residence at high altitude leads to PH development (Arias‐Stella & Saldana, 1963; Stenmark et al. 2006); however, short exposure to hypoxia induces a tremendous change in gene expression (Kwapiszewska et al. 2005). PH is characterized by a pronounced pulmonary arterial remodelling process, leading in the case of chronic hypoxia to muscularization of previously non‐muscularized arteries and an increase in the degree of muscularization of already muscularized pulmonary arteries. In this regard, pulmonary arterial smooth muscle cell (PASMC) hyper‐proliferation in the media layer of the pulmonary artery is suggested to be the key event in vascular remodelling and the main determinant of elevated pulmonary vascular resistance in hypoxia‐induced PH (Pak et al. 2007).
Recently, considerable work has been done to investigate the molecular and cellular responses to alveolar hypoxia. The direct impact of the HIF system on PH pathogenesis has been confirmed in vivo, using haplo‐insufficient HIF‐1α and HIF‐2α knockout mice. Homozygous HIF‐1α and HIF‐1β mice are not viable due to cardiac and vascular defects (Iyer et al. 1998). Loss of HIF‐2α causes fetal death in 50% of embryos (Compernolle et al. 2002; Brusselmans et al. 2003). However, mice with partial HIF‐1α or HIF‐2α deficiency are viable and develop normally. When exposed to chronic hypoxia (10% O2) the outcome of these mice is improved due to a smaller increase in pulmonary arterial pressure, right ventricular hypertrophy and pulmonary vascular remodelling, as compared to respective controls (Yu et al. 1999; Brusselmans et al. 2003), suggesting a role of the HIF system in the impaired development of PH (Yu et al. 1999). Recent data in PASMCs also indicate that HIF‐1α regulates the expression of an ion channel subunit that mitigates vasoconstriction (Ahn et al. 2012). Finally, the role of HIF‐1α in modulating pulmonary vascular tone and pulmonary vascular remodelling was addressed in more detail using smooth muscle cell specific inducible HIF‐1α knockout mice. This study showed that selective deletion of HIF‐1α leads to attenuated pulmonary vascular remodelling and pulmonary hypertension in chronic hypoxic mice. However, right ventricular hypertrophy was unchanged despite attenuated pulmonary pressures (Ball et al. 2014), suggesting that right heart hypertrophy in the absence of PH may be caused by a direct cardiac HIF induction, independent of elevated pulmonary artery pressure. However, others observed that a complete loss of HIF‐1α in smooth muscle cells raised right ventricular systolic pressure in normoxic and hypoxic mice, compared to respective wild‐type littermates. The number of muscularized arteries in murine lungs was not changed. Moreover, myosin light chain (MLC) phosphorylation, which determines vascular tone, was higher in PASMCs isolated from smooth muscle specific HIF‐1α knockout mice compared to control, during both normoxia and after acute hypoxia. Thus, results from this group indicate a primary role for smooth muscle HIF‐1α in modulating vascular tone specifically, and not vascular remodelling (Kim et al. 2013). By using HIF‐2α overexpressing mice, caused by a single missense mutation in HIF‐2α, a clear association of HIF‐2α with the development of PH in mice was shown (Tan et al. 2013).
In addition, there are studies investigating the indirect effect of HIF on PH pathogenesis. In this regard, mice with mutation of the von Hippel–Lindau (VHL) tumour suppressor protein, a Chuvash polycythaemia disease model, possess elevated HIF‐2α levels and increased pulmonary arterial pressure and pulmonary vessel muscularization (Hickey et al. 2010). The role of HIF in PH is also strengthened by (1) elevated HIF‐2α protein expression in Fawn‐hooded rats, (2) enhanced HIF‐1α levels in plexiform lesions of PH patients, and (3) increased HIF‐1α expression in the pulmonary vasculature of chronic hypoxic mice (Tuder et al. 2001; Bonnet et al. 2006; Mizuno et al. 2011). However, in rat pulmonary arteries HIF‐1α protein levels decline with increasing time of hypoxia (Jiang et al. 2007), indicating that there might be species‐dependent differences in HIF stability in chronic hypoxia or that other factors than hypoxia contribute to HIF stabilization.
Recent in vitro findings from our group indicate enhanced PASMC proliferation following hypoxic exposure or HIF‐1α over‐expression, compared to control (Malczyk et al. 2013; Veith et al. 2014). Moreover, our data indicate that the contractile PASMC phenotype is replaced by a synthetic phenotype during hypoxia (Veith et al. 2014).
Thus, while a global decrease of HIF‐1α may mitigate against the development of pulmonary hypertension, the specific role in PASMCs needs further investigation, and HIF‐1α stabilization in cell types other than PASMCs should also be considered as potential contributors to hypoxia‐induced remodelling/muscularization, e.g. via HIF‐1α mediated induction of growth factors.
HIF‐regulated ion channel expression in pulmonary hypertension
Both pulmonary vascular remodelling and active PASMC contraction can cause a decrease in lumen diameter, and an increase in pulmonary vascular resistance and pulmonary arterial pressure, resulting in PH. Several studies indicate the involvement of the HIF system in altered ion channel and ion transporter expression leading to contraction of the pulmonary vasculature.
Hypoxia and HIF‐dependent regulation is proposed for classical transient receptor potential (TRPC) 1 and TRPC6 proteins in murine and rat PASMCs (Wang et al. 2006; Malczyk et al. 2013). TRPCs are non‐selective cation channels, permeable to Ca2+ ions. An increase in cytosolic free Ca2+ concentration is suggested as a major trigger for PASMC proliferation in PH. In this regard, TRPC1 silencing by specific siRNA transfection or TRPC1 knockout impaired hypoxia‐induced PASMC proliferation in vitro. In contrast, TRPC6 has no effect on PASMC hyperplasia and pulmonary vascular remodelling, but is necessary for acute hypoxic responses (pulmonary vasoconstriction) (Weissmann et al. 2006; Malczyk et al. 2013).
Previous studies indicate that alterations in PASMC pH homeostasis are crucial for the development of hypoxia‐induced pulmonary hypertension. In line with this hypothesis, hypoxia and HIF‐1α‐dependent regulation of the Na+/H+ exchanger isoform 1 (NHE1) was observed (Shimoda et al. 2006), leading to an alkaline shift in pH. Both activation of the Na+/H+ exchanger and an alkaline shift in pH are suggested to be required for PASMC proliferation in response to growth factors (Quinn et al. 1996).
In addition to TRPC and NHE, the family of voltage‐gated K+ (Kv) channels Kv1.5 and Kv2.1 have altered expression following hypoxic exposure. Chronic hypoxia reduces K+ currents in PASMCs (Wang et al. 1997), leading to membrane depolarization, activation of voltage‐dependent Ca2+ channels (VDCCs) and Ca2+ influx. However, in PASMCs isolated from heterozygous HIF‐1α mice, the reduction in voltage‐gated K+ currents following chronic hypoxia was absent (Shimoda et al. 2001), indicating a role of HIF‐1α activation in regulating both pulmonary vascular remodelling and vascular tone.
The HIF system in endothelial dysfunction underlying PH
Plexiform lesions are a hallmark of severe PH in humans. They are characterized by a dysregulated proliferation of pulmonary arterial endothelial cells and/or decreased cell death (Fig. 2), leading to vascular occlusions. HIF‐1α and HIF‐1β are highly expressed in endothelial cells of plexiform lesions in patients with severe PH (Tuder et al. 2001). Thus, they might play a critical role in the expression of mitogenic factors and vasoconstrictors and their receptors such as vascular endothelial growth factor (VEGF), placental growth factor (PGF), platelet‐derived growth factor (PDGF), angiopoietin‐1 and ‐2 (ANGPT1 and ‐2) and serotonin (5‐HT) (Kelly et al. 2003; Esteve et al. 2007; Haugen et al. 2012). Similar results were recently obtained with cultured endothelial cells isolated from patients with idiopathic pulmonary arterial hypertension. These cells have greater proliferation rate and decreased apoptosis, higher levels of phosphorylated STAT3 and increased expression of its downstream pro‐survival target, Mcl‐1 (Masri et al. 2007).
Figure 2. Schematic diagram displaying the molecular mechanisms of vascular alterations in pulmonary hypertension .
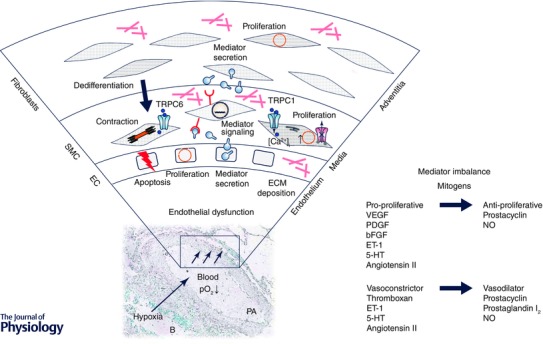
A reduction in environmental O2 partial pressure (pO2↓ = hypoxia) leads to less oxygenated blood. Hypoxia is sensed within the pulmonary artery leading to endothelial dysfunction, mediator secretion, changes in gene expression, deposition of extracellular matrix (ECM) proteins, membrane depolarization due to reduced outward K+ currents and activation of inward Ca2+ currents and increase in intracellular Ca2+ concentration ([Ca2+]i). Elevated [Ca2+]i positively affects contraction of smooth muscle cells (SMC) and as well as cell proliferation. The imbalance between pro‐ and anti‐proliferative mitogens and vasodilators and vasoconstrictors contributes to pulmonary vascular remodelling and pulmonary hypertension. Abbreviations: EC, endothelial cells; B, bronchus; PA, pulmonary artery; VEGF, vascular endothelial growth factor; PDGF, platelet‐derived growth factor; bFGF, basic fibroblast growth factor; ET‐1, endothelin 1; 5‐HT, 5‐hydroxytryptamine; NO, nitric oxide.
In contrast to endothelial cells, hypoxic smooth muscle cells possess elevated VEGF levels, but decreased ANGPT2 expression and no changes in PDGF and ANGPT1 expression (Kelly et al. 2003), indicating that each cell type shows a different expression pattern of HIF target genes.
There is controversy as to whether angiogenesis increases or decreases in PH. Some groups report elevated hypoxia‐induced angiogenesis in the pulmonary circulation (Howell et al. 2003), while others do not (Hislop & Reid, 1976; Rabinovitch et al. 1979; Partovian et al. 2000). However, it is well established that chronic hypoxia induces expression of VEGF, a potent inducer of angiogenesis, in a HIF‐1α and/or HIF‐2α dependent manner (Tuder et al. 1995; Clerici & Planes, 2009).
In addition to the HIF‐dependent regulation of mitogens, growth factors (PDGF, epidermal growth factor (EGF), fibroblast growth factor 2 (FGF‐2) and thrombin), cytokines and vasoactive substances (endothelin‐1) themselves can induce HIF‐1 target gene expression via the mitogen‐activated protein kinase and/or phosphatidylinositol 3‐kinase signal transduction pathways (Laughner et al. 2001; Fukuda et al. 2003; Schultz et al. 2006; Whitman et al. 2008), thus utilizing a positive feedback loop to amplify the HIF activity.
Crosstalk of the HIF system and reactive oxygen species
Currently, there is intense debate about whether there is an increase or a decrease in ROS in hypoxia‐induced PH (Ward, 2006; Weir & Archer, 2006). We and others have evidence that reduced O2 levels can lead to increased ROS levels in acute and chronic hypoxia (Chandel et al. 1998; Liu et al. 2006; Mittal et al. 2007; Nozik‐Grayck & Stenmark, 2007; Sanders & Hoidal, 2007; Ismail et al. 2009; Nisbet et al. 2009; Waypa & Schumacker, 2010; Weissmann et al. 2012).
Accordingly, complex III in the mitochondrial electron transport chain, plasma membrane bound NADPH oxidases or cytochromes (Klimova & Chandel, 2008; Nisbet et al. 2009; Weissmann et al. 2012; Veit et al. 2013) are suggested as ROS sources. Superoxide radicals can be reduced to hydrogen peroxide by superoxide dismutase (SOD). Hydrogen peroxide is much more stable than superoxide and can cross cell membranes, and may thus act as a potent signalling molecule. Recently, much literature has emerged concerning the possible role of ROS in HIF induction and PH development (Beckman & Koppenol, 1996; Liu et al. 2006; Mittal et al. 2007; Clerici & Planes, 2009).
There are several theories as to how ROS can lead to HIF‐α stabilization. One theory suggests that hydrogen peroxide can act as a diffusible second messenger, activating redox‐sensitive transcription factors, including HIF‐1α and the activation and expression of voltage‐gated Kv channels (Tuder et al. 2007). Another theory is that under hypoxia, mitochondria scavenge available O2 and PHDs are then deprived of their co‐substrate and cannot hydroxylate HIF‐α, marking it for proteasomal degradation (Hagen et al. 2003). A third possibility is that ROS interfere with the regulation of PHDs. In this regard, hydrogen peroxide might inhibit PHDs by oxidizing non‐haem‐bound Fe2+ to Fe3+ (Hagen, 2012). However, other findings postulate low sensitivity of PHDs to hydrogen peroxide (Masson et al. 2012).
There is also evidence, that inhibitors of mitochondrial ROS or electron transport chain reduce HIF‐1α and HIF‐2α protein accumulation (Chandel et al. 1998, 2000; Mansfield et al. 2005), suggesting that the HIF‐α subunit requires a functional electron transport chain for stabilization under hypoxic conditions but not under anoxic conditions (Schroedl et al. 2002).
Recently, HIF‐1 regulation by non‐hypoxia generated ROS has gained considerable interest. In this regard, CoCl2 treatment of normoxic Hep3B cells generates ROS, leading to HIF‐1 DNA binding and expression of HIF target genes (Chandel et al. 1998). Moreover, angiotensin‐II treatment of vascular smooth muscle cells mediates hydrogen peroxide generation and HIF‐1 stabilization under normoxic conditions (Page et al. 2002, 2008).
The HIF system and cytoskeletal and extracellular matrix proteins
A growing body of evidence suggests the importance of cytoskeletal proteins in dysregulated PASMC function in PH. In this regard, we identified the cytoskeletal scaffold proteins four‐and‐a‐half LIM domains 1 (FHL‐1) and paxillin as novel hypoxia and HIF‐1α target genes in primary human PASMCs. Both are up‐regulated following hypoxic exposure and negatively affected by HIF‐1α silencing (Kwapiszewska et al. 2008; Veith et al. 2012). However, HIF‐2α knockdown affects FHL‐1, but not paxillin, mRNA expression (Kwapiszewska et al. 2008; and unpublished observations). Along these lines, we observed enhanced paxillin levels following HIF‐1α over‐expression (Veith et al. 2014). In addition, phosphorylation of the cytoskeletal proteins cofilin and paxillin is hypoxia and HIF‐1α dependent (Veith et al. 2013, 2014). All three of them are predominantly localized in the media of small intrapulmonary vessels, major sites of vascular remodelling in PH (Jeffery & Wanstall, 2001), suggesting them as effectors of HIF‐induced PASMC dysfunction.
Cellular copper (Cu) plays an important role in extracellular matrix remodelling. Elevated Cu levels in vascular smooth muscle cells have been demonstrated to be associated with hypoxia‐induced PH. In this context, it was shown that the Cu‐uptake transporter 1 (CTR1) and the Cu‐efflux pump (ATP7A) were both up‐regulated in lung tissue and pulmonary arteries of mice with hypoxia‐induced PH. The hypoxia‐mediated up‐regulation of CTR1 is mediated in a HIF‐1α‐dependent manner as HIF‐1α silencing by siRNA transfection attenuated the hypoxia‐mediated CTR1 up‐regulation. Hypoxia also significantly increased expression and activity of lysyl oxidase (LOX), a Cu‐dependent enzyme that crosslinks collagen and elastin in the extracellular matrix and is involved in PASMC proliferation. Crosslinking of extracellular matrix proteins leads to increased pulmonary arterial stiffness which, in combination with the increased PASMC proliferation and migration, further contributes to the development and progression of PH (Zimnicka et al. 2014).
Non‐hypoxia‐driven HIF activation
In addition to hypoxia‐induced HIF activation, research is focusing on HIF‐1 regulation by non‐hypoxic stimuli as, for example, the endothelium is not only exposed to decreased pO2, but also to mechanical forces due to vessel constriction. In this regard, elevated HIF‐1α and HIF‐2α levels were detected in stretch‐induced but not shear‐stress‐induced angiogenesis in rat skeletal muscle (Milkiewicz et al. 2007). Next to mechanical forces, stimulation with mitogens such as angiotensin II, which is upregulated in PH, increases transcription and functional HIF‐1α protein levels (Page et al. 2002) as well as increases VEGF expression in vascular smooth muscle cells (Page et al. 2002). In addition, it is well established that the non‐hypoxia driven ROS generation may also interfere with HIF signalling (Page et al. 2002, 2008). Moreover, it was shown that dysfunction of the two Krebs cycle enzymes α‐ketoglutarate dehydrogenase and succinate dehydrogenase leads to accumulation of succinate and fumarate which inactivate PHDs and thus stabilize HIF‐1α (Isaacs et al. 2005; Selak et al. 2005).
Therapeutic intervention targeting the HIF–PHD axis
Recently, numerous pharmacological studies have emerged targeting the HIF–PHD axis. In this regard cardiac glycosides, such as digoxin, ouabain or proscillaridin A, inhibit HIF protein synthesis and HIF target gene expression (Zhang et al. 2008; Yoshida et al. 2010). In vivo, digoxin prevents and reverses the development of chronic hypoxia‐induced PH in mice (Abud et al. 2012). Acriflavine, which is the most potent inhibitor of HIF subunit dimerization (Lee et al. 2009), leads to reduced hypoxia‐induced PH in rats (Abud et al. 2012). Finally, the HIF–PHD axis can be directly targeted by iron supplementation, since PHD activity is iron sensitive (Smith et al. 2008). Interestingly, infusion of iron reduced, whereas iron chelation increased, elevated pulmonary arterial pressure in response to hypoxia (Smith et al. 2008, 2009).
Summary and conclusions
In summary, hypoxia‐activated HIF signalling pathways in part drive the pathogenesis of PH, as they regulate the expression of several genes critical for pulmonary vascular remodelling and disease progression. Understanding the molecular mechanisms of the HIF signalling axis can help to develop new therapeutic approaches for PH treatment.
Additional information
Competing interests
None declared.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (DI 514/8‐1 and Excellence Cluster Cardio‐Pulmonary System and SFB‐TR 84).
Biography
Christine Veith studied biology at Justus‐Liebig‐University Giessen, Germany and joined the department of Internal Medicine there in 2008. After finishing her dissertation in 2012 she became a postdoc. Her research is mainly focused on the dysregulation of cytoskeletal proteins in the pathogenesis of pulmonary hypertension, with special interest in hypoxia/HIF‐regulated pathways contributing to pulmonary vascular remodelling in pulmonary hypertension. Norbert Weissmann studied biology at Justus‐Liebig‐University, Giessen. After his graduation as PhD, he worked as a postdoc researcher and subsequently as research assistant and University Lecturer at the Department of Internal Medicine at Justus‐Liebig‐University. There he became Extraordinary Professor for Pathophysiology and Experimental Pneumology in 2007, and full Professor for Molecular Mechanisms of Emphysema, Hypoxia and Lung Aging in 2008 at the Excellence Cluster Cardio‐Pulmonary System (ECCPS). His research centred on pulmonary oxygen sensing and signalling, including pulmonary hypertension. Moreover, he is interested in mechanisms of chronic obstructive lung disease and accelerated ageing of the lung. Ralf P. Brandes studied medicine at Hannover Medical School, Hannover, Germany and Emory University, Atlanta, GA, USA. After a clinical period in the Department of Cardiology, Hannover Medical School, he joined the Institute of Cardiovascular Physiology at Goethe‐University, Frankfurt, Germany. There he became assistant professor in 2002, associate professor in 2006 and full professor and chairman in 2008. His research interests are particularly vascular redox regulation but also other aspects of endothelial cell biology like epigenetic control and lipid signalling. Ralph T. Schermuly studied biology in Giessen. After his dissertation in 1996 at Justus‐Liebig‐University Giessen, he joined the Department of Internal Medicine there in 2003. He did his habilitation in pathophysiology and experimental pneumology in 2005. Since 2011 he holds a professorship for Pulmonary Pharmacotherapy at Justus‐Liebig‐University. Examples of his ‘from bench to bedside’ research include preclinical investigations of inhaled prostanoids, phosphodiesterase inhibitors, stimulators of soluble guanylate cyclase, receptor tyrosine kinases, or transbronchial surfactant application, providing the basis for worldwide approval of inhaled iloprost, oral sildenafil and riociguat for treatment of pulmonary arterial hypertension.

References
- Abud EM, Maylor J, Undem C, Punjabi A, Zaiman AL, Myers AC, Sylvester JT, Semenza GL & Shimoda LA (2012). Digoxin inhibits development of hypoxic pulmonary hypertension in mice. Proc Natl Acad Sci U S A 109, 1239–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn YT, Kim YM, Adams E, Lyu SC, Alvira CM & Cornfield DN (2012). Hypoxia‐inducible factor‐1α regulates KCNMB1 expression in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 302, L352–L359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias‐Stella J & Saldana M (1963). The terminal portion of the pulmonary arterial tree in people native to high altitudes. Circulation 28, 915–925. [DOI] [PubMed] [Google Scholar]
- Augstein A, Poitz DM, Braun‐Dullaeus RC, Strasser RH & Schmeisser A (2011). Cell‐specific and hypoxia‐dependent regulation of human HIF‐3α: inhibition of the expression of HIF target genes in vascular cells. Cell Mol Life Sci 68, 2627–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball MK, Waypa GB, Mungai PT, Nielsen JM, Czech L, Dudley VJ, Beussink L, Dettman RW, Berkelhamer SK, Steinhorn RH, Shah SJ & Schumacker PT (2014). Regulation of hypoxia‐induced pulmonary hypertension by vascular smooth muscle hypoxia‐inducible factor‐1α. Am J Respir Crit Care Med 189, 314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman JS & Koppenol WH (1996). Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol Cell Physiol 271, C1424–C1437. [DOI] [PubMed] [Google Scholar]
- Bland RD, Demling RH, Selinger SL & Staub NC (1977). Effects of alveolar hypoxia on lung fluid and protein transport in unanesthetized sheep. Circ Res 40, 269–274. [DOI] [PubMed] [Google Scholar]
- Bonnet S, Michelakis ED, Porter CJ, Andrade‐Navarro MA, Thebaud B, Bonnet S, Haromy A, Harry G, Moudgil R, McMurtry MS, Weir EK & Archer SL (2006). An abnormal mitochondrial‐hypoxia inducible factor‐1α‐Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 113, 2630–2641. [DOI] [PubMed] [Google Scholar]
- Brusselmans K, Compernolle V, Tjwa M, Wiesener MS, Maxwell PH, Collen D & Carmeliet P (2003). Heterozygous deficiency of hypoxia‐inducible factor‐2α protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J Clin Invest 111, 1519–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC & Schumacker PT (1998). Mitochondrial reactive oxygen species trigger hypoxia‐induced transcription. Proc Natl Acad Sci USA 95, 11715–11720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM & Schumacker PT (2000). Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia‐inducible factor‐1α during hypoxia: a mechanism of O2 sensing. J Biol Chem 275, 25130–25138. [DOI] [PubMed] [Google Scholar]
- Clerici C & Planes C (2009). Gene regulation in the adaptive process to hypoxia in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol 296, L267–L274. [DOI] [PubMed] [Google Scholar]
- Compernolle V, Brusselmans K, Acker T, Hoet P, Tjwa M, Beck H, Plaisance S, Dor Y, Keshet E, Lupu F, Nemery B, Dewerchin M, Van Veldhoven P, Plate K, Moons L, Collen D & Carmeliet P (2002). Loss of HIF‐2α and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med 8, 702–710. [DOI] [PubMed] [Google Scholar]
- Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ & Ratcliffe PJ (2001). C. elegans EGL‐9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107, 43–54. [DOI] [PubMed] [Google Scholar]
- Esteve JM, Launay JM, Kellermann O & Maroteaux L (2007). Functions of serotonin in hypoxic pulmonary vascular remodeling. Cell Biochem Biophys 47, 33–44. [DOI] [PubMed] [Google Scholar]
- Fukuda R, Kelly B & Semenza GL (2003). Vascular endothelial growth factor gene expression in colon cancer cells exposed to prostaglandin E2 is mediated by hypoxia‐inducible factor 1. Cancer Res 63, 2330–2334. [PubMed] [Google Scholar]
- Hagen T (2012). Oxygen versus reactive oxygen in the regulation of HIF‐1α: the balance tips. Biochem Res Int 2012, 436981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen T, Taylor CT, Lam F & Moncada S (2003). Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science 302, 1975–1978. [DOI] [PubMed] [Google Scholar]
- Haugen M, Dammen R, Svejda B, Gustafsson BI, Pfragner R, Modlin I & Kidd M (2012). Differential signal pathway activation and 5‐HT function: the role of gut enterochromaffin cells as oxygen sensors. Am J Physiol Gastrointest Liver Physiol 303, G1164–G1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidbreder M, Frohlich F, Johren O, Dendorfer A, Qadri F & Dominiak P (2003). Hypoxia rapidly activates HIF‐3α mRNA expression. FASEB J 17, 1541–1543. [DOI] [PubMed] [Google Scholar]
- Hickey MM, Richardson T, Wang T, Mosqueira M, Arguiri E, Yu H, Yu QC, Solomides CC, Morrisey EE, Khurana TS, Christofidou‐Solomidou M & Simon MC (2010). The von Hippel‐Lindau Chuvash mutation promotes pulmonary hypertension and fibrosis in mice. J Clin Invest 120, 827–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hislop A & Reid L (1976). New findings in pulmonary arteries of rats with hypoxia‐induced pulmonary hypertension. Br J Exp Pathol 57, 542–554. [PMC free article] [PubMed] [Google Scholar]
- Howell K, Preston RJ & McLoughlin P (2003). Chronic hypoxia causes angiogenesis in addition to remodelling in the adult rat pulmonary circulation. J Physiol 547, 133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LE, Arany Z, Livingston DM & Bunn HF (1996). Activation of hypoxia‐inducible transcription factor depends primarily upon redox‐sensitive stabilization of its α subunit. J Biol Chem 271, 32253–32259. [DOI] [PubMed] [Google Scholar]
- Huang LE, Gu J, Schau M & Bunn HF (1998). Regulation of hypoxia‐inducible factor 1α is mediated by an O2‐dependent degradation domain via the ubiquitin‐proteasome pathway. Proc Natl Acad Sci U S A 95, 7987–7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs JS, Jung YJ, Mole DR, Lee S, Torres‐Cabala C, Chung YL, Merino M, Trepel J, Zbar B, Toro J, Ratcliffe PJ, Linehan WM & Neckers L (2005). HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 8, 143–153. [DOI] [PubMed] [Google Scholar]
- Ismail S, Sturrock A, Wu P, Cahill B, Norman K, Huecksteadt T, Sanders K, Kennedy T & Hoidal J (2009). NOX4 mediates hypoxia‐induced proliferation of human pulmonary artery smooth muscle cells: the role of autocrine production of transforming growth factor‐β1 and insulin‐like growth factor binding protein‐3. Am J Physiol Lung Cell Mol Physiol 296, L489–L499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS & Kaelin WG Jr (2001). HIFα targeted for VHL‐mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292, 464–468. [DOI] [PubMed] [Google Scholar]
- Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY & Semenza GL (1998). Cellular and developmental control of O2 homeostasis by hypoxia‐inducible factor 1α. Genes Dev 12, 149–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery TK & Wanstall JC (2001). Pulmonary vascular remodeling: a target for therapeutic intervention in pulmonary hypertension. Pharmacol Ther 92, 1–20. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Dai A, Li Q & Hu R (2007). Hypoxia induces transforming growth factor‐β1 gene expression in the pulmonary artery of rats via hypoxia‐inducible factor‐1α. Acta Biochim Biophys Sin (Shanghai) 39, 73–80. [DOI] [PubMed] [Google Scholar]
- Kelly BD, Hackett SF, Hirota K, Oshima Y, Cai Z, Berg‐Dixon S, Rowan A, Yan Z, Campochiaro PA & Semenza GL (2003). Cell type‐specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia‐inducible factor 1. Circ Res 93, 1074–1081. [DOI] [PubMed] [Google Scholar]
- Kim YM, Barnes EA, Alvira CM, Ying L, Reddy S & Cornfield DN (2013). Hypoxia‐inducible factor‐1α in pulmonary artery smooth muscle cells lowers vascular tone by decreasing myosin light chain phosphorylation. Circ Res 112, 1230–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimova T & Chandel NS (2008). Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ 15, 660–666. [DOI] [PubMed] [Google Scholar]
- Kwapiszewska G, Wilhelm J, Wolff S, Laumanns I, Koenig IR, Ziegler A, Seeger W, Bohle RM, Weissmann N & Fink L (2005). Expression profiling of laser‐microdissected intrapulmonary arteries in hypoxia‐induced pulmonary hypertension. Respir Res 6, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwapiszewska G, Wygrecka M, Marsh LM, Schmitt S, Trosser R, Wilhelm J, Helmus K, Eul B, Zakrzewicz A, Ghofrani HA, Schermuly RT, Bohle RM, Grimminger F, Seeger W, Eickelberg O, Fink L & Weissmann N (2008). Fhl‐1, a new key protein in pulmonary hypertension. Circulation 118, 1183–1194. [DOI] [PubMed] [Google Scholar]
- Laughner E, Taghavi P, Chiles K, Mahon PC & Semenza GL (2001). HER2 (neu) signaling increases the rate of hypoxia‐inducible factor 1α (HIF‐1α) synthesis: novel mechanism for HIF‐1‐mediated vascular endothelial growth factor expression. Mol Cell Biol 21, 3995–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Zhang H, Qian DZ, Rey S, Liu JO & Semenza GL (2009). Acriflavine inhibits HIF‐1 dimerization, tumor growth, and vascularization. Proc Natl Acad Sci U S A 106, 17910–17915. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Liu JQ, Zelko IN, Erbynn EM, Sham JS & Folz RJ (2006). Hypoxic pulmonary hypertension: role of superoxide and NADPH oxidase (gp91phox). Am J Physiol Lung Cell Mol Physiol 290, L2–L10. [DOI] [PubMed] [Google Scholar]
- Makino Y, Uenishi R, Okamoto K, Isoe T, Hosono O, Tanaka H, Kanopka A, Poellinger L, Haneda M & Morimoto C (2007). Transcriptional up‐regulation of inhibitory PAS domain protein gene expression by hypoxia‐inducible factor 1 (HIF‐1): a negative feedback regulatory circuit in HIF‐1‐mediated signaling in hypoxic cells. J Biol Chem 282, 14073–14082. [DOI] [PubMed] [Google Scholar]
- Malczyk M, Veith C, Fuchs B, Hofmann K, Storch U, Schermuly RT, Witzenrath M, Ahlbrecht K, Fecher‐Trost C, Flockerzi V, Ghofrani HA, Grimminger F, Seeger W, Gudermann T, Dietrich A & Weissmann N (2013). Classical transient receptor potential channel 1 in hypoxia‐induced pulmonary hypertension. Am J Respir Crit Care Med 188, 1451–1459. [DOI] [PubMed] [Google Scholar]
- Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT & Simon MC (2005). Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF‐α activation. Cell Metab 1, 393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DS, Khosravi M, Grocott MP & Mythen MG (2010). Concepts in hypoxia reborn. Crit Care 14, 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masri FA, Xu W, Comhair SA, Asosingh K, Koo M, Vasanji A, Drazba J, Anand‐Apte B & Erzurum SC (2007). Hyperproliferative apoptosis‐resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 293, L548–L554. [DOI] [PubMed] [Google Scholar]
- Masson N, Singleton RS, Sekirnik R, Trudgian DC, Ambrose LJ, Miranda MX, Tian YM, Kessler BM, Schofield CJ & Ratcliffe PJ (2012). The FIH hydroxylase is a cellular peroxide sensor that modulates HIF transcriptional activity. EMBO Rep 13, 251–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard MA, Evans AJ, Hosomi T, Hara S, Jewett MA & Ohh M (2005). Human HIF‐3α4 is a dominant‐negative regulator of HIF‐1 and is down‐regulated in renal cell carcinoma. FASEB J 19, 1396–1406. [DOI] [PubMed] [Google Scholar]
- Milkiewicz M, Doyle JL, Fudalewski T, Ispanovic E, Aghasi M & Haas TL (2007). HIF‐1α and HIF‐2α play a central role in stretch‐induced but not shear‐stress‐induced angiogenesis in rat skeletal muscle. J Physiol 583, 753–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal M, Roth M, Konig P, Hofmann S, Dony E, Goyal P, Selbitz AC, Schermuly RT, Ghofrani HA, Kwapiszewska G, Kummer W, Klepetko W, Hoda MA, Fink L, Hanze J, Seeger W, Grimminger F, Schmidt HH & Weissmann N (2007). Hypoxia‐dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ Res 101, 258–267. [DOI] [PubMed] [Google Scholar]
- Mizuno S, Bogaard HJ, Kraskauskas D, Alhussaini A, Gomez‐Arroyo J, Voelkel NF & Ishizaki T (2011). p53 Gene deficiency promotes hypoxia‐induced pulmonary hypertension and vascular remodeling in mice. Am J Physiol Lung Cell Mol Physiol 300, L753–L761. [DOI] [PubMed] [Google Scholar]
- Nisbet RE, Graves AS, Kleinhenz DJ, Rupnow HL, Reed AL, Fan TH, Mitchell PO, Sutliff RL & Hart CM (2009). The role of NADPH oxidase in chronic intermittent hypoxia‐induced pulmonary hypertension in mice. Am J Respir Cell Mol Biol 40, 601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozik‐Grayck E & Stenmark KR (2007). Role of reactive oxygen species in chronic hypoxia‐induced pulmonary hypertension and vascular remodeling. Adv Exp Med Biol 618, 101–112. [DOI] [PubMed] [Google Scholar]
- Paddenberg R, Stieger P, von Lilien AL, Faulhammer P, Goldenberg A, Tillmanns HH, Kummer W & Braun‐Dullaeus RC (2007). Rapamycin attenuates hypoxia‐induced pulmonary vascular remodeling and right ventricular hypertrophy in mice. Resp Res 8, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page EL, Chan DA, Giaccia AJ, Levine M & Richard DE (2008). Hypoxia‐inducible factor‐1α stabilization in nonhypoxic conditions: role of oxidation and intracellular ascorbate depletion. Mol Biol Cell 19, 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page EL, Robitaille GA, Pouyssegur J & Richard DE (2002). Induction of hypoxia‐inducible factor‐1α by transcriptional and translational mechanisms. J Biol Chem 277, 48403–48409. [DOI] [PubMed] [Google Scholar]
- Pak O, Aldashev A, Welsh D & Peacock A (2007). The effects of hypoxia on the cells of the pulmonary vasculature. Eur Respir J 30, 364–372. [DOI] [PubMed] [Google Scholar]
- Partovian C, Adnot S, Raffestin B, Louzier V, Levame M, Mavier IM, Lemarchand P & Eddahibi S (2000). Adenovirus‐mediated lung vascular endothelial growth factor overexpression protects against hypoxic pulmonary hypertension in rats. Am J Respir Cell Mol Biol 23, 762–771. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR & Semenza GL (2012). Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia‐inducible factors 1 and 2. Physiol Rev 92, 967–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn DA, Dahlberg CG, Bonventre JP, Scheid CR, Honeyman T, Joseph PM, Thompson BT & Hales CA (1996). The role of Na+/H+ exchange and growth factors in pulmonary artery smooth muscle cell proliferation. Am J Respir Cell Mol Biol 14, 139–145. [DOI] [PubMed] [Google Scholar]
- Rabinovitch M, Gamble W, Nadas AS, Miettinen OS & Reid L (1979). Rat pulmonary circulation after chronic hypoxia: hemodynamic and structural features. Am J Physiol Heart Circ Physiol 236, H818–H827. [DOI] [PubMed] [Google Scholar]
- Sanders KA & Hoidal JR (2007). The NOX on pulmonary hypertension. Circ Res 101, 224–226. [DOI] [PubMed] [Google Scholar]
- Schroedl C, McClintock DS, Budinger GR & Chandel NS (2002). Hypoxic but not anoxic stabilization of HIF‐1α requires mitochondrial reactive oxygen species. Am J Physiol Lung Cell Mol Physiol 283, L922–L931. [DOI] [PubMed] [Google Scholar]
- Schultz K, Fanburg BL & Beasley D (2006). Hypoxia and hypoxia‐inducible factor‐1α promote growth factor‐induced proliferation of human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 290, H2528–H2534. [DOI] [PubMed] [Google Scholar]
- Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB & Gottlieb E (2005). Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF‐α prolyl hydroxylase. Cancer Cell 7, 77–85. [DOI] [PubMed] [Google Scholar]
- Semenza GL (2000. a). Expression of hypoxia‐inducible factor 1: mechanisms and consequences. Biochem Pharmacol 59, 47–53. [DOI] [PubMed] [Google Scholar]
- Semenza GL (2000. b). HIF‐1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol 88, 1474–1480. [DOI] [PubMed] [Google Scholar]
- Semenza GL (2001). Hypoxia‐inducible factor 1: oxygen homeostasis and disease pathophysiology. Trends Mol Med 7, 345–350. [DOI] [PubMed] [Google Scholar]
- Semenza GL (2004). O2‐regulated gene expression: transcriptional control of cardiorespiratory physiology by HIF‐1. J Appl Physiol 96, 1173–1177; discussion 1170–1172. [DOI] [PubMed] [Google Scholar]
- Semenza GL (2011). Oxygen sensing, homeostasis, and disease. N Engl J Med 365, 537–547. [DOI] [PubMed] [Google Scholar]
- Semenza GL (2014). Oxygen sensing, hypoxia‐inducible factors, and disease pathophysiology. Annu Rev Pathol 9, 47–71. [DOI] [PubMed] [Google Scholar]
- Semenza GL, Nejfelt MK, Chi SM & Antonarakis SE (1991). Hypoxia‐inducible nuclear factors bind to an enhancer element located 3’ to the human erythropoietin gene. Proc Natl Acad Sci U S A 88, 5680–5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoda LA, Fallon M, Pisarcik S, Wang J & Semenza GL (2006). HIF‐1 regulates hypoxic induction of NHE1 expression and alkalinization of intracellular pH in pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol 291, L941–L949. [DOI] [PubMed] [Google Scholar]
- Shimoda LA, Manalo DJ, Sham JS, Semenza GL & Sylvester JT (2001). Partial HIF‐1α deficiency impairs pulmonary arterial myocyte electrophysiological responses to hypoxia. Am J Physiol Lung Cell Mol Physiol 281, L202–L208. [DOI] [PubMed] [Google Scholar]
- Smith TG, Balanos GM, Croft QP, Talbot NP, Dorrington KL, Ratcliffe PJ & Robbins PA (2008). The increase in pulmonary arterial pressure caused by hypoxia depends on iron status. J Physiol 586, 5999–6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TG, Talbot NP, Privat C, Rivera‐Ch M, Nickol AH, Ratcliffe PJ, Dorrington KL, Leon‐Velarde F & Robbins PA (2009). Effects of iron supplementation and depletion on hypoxic pulmonary hypertension: two randomized controlled trials. JAMA 302, 1444–1450. [DOI] [PubMed] [Google Scholar]
- Stenmark KR, Fagan KA & Frid MG (2006). Hypoxia‐induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res 99, 675–691. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Noda M, Sugita M, Ono S, Koike K & Fujimura S (1999). Impairment of transalveolar fluid transport and lung Na+‐K+‐ATPase function by hypoxia in rats. J Appl Physiol 87, 962–968. [DOI] [PubMed] [Google Scholar]
- Tan Q, Kerestes H, Percy MJ, Pietrofesa R, Chen L, Khurana TS, Christofidou‐Solomidou M, Lappin TR & Lee FS (2013). Erythrocytosis and pulmonary hypertension in a mouse model of human HIF2A gain of function mutation. J Biol Chem 288, 17134–17144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraseviciene‐Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF & Tuder RM (2001). Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death‐dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J 15, 427–438. [DOI] [PubMed] [Google Scholar]
- Tuder RM, Chacon M, Alger L, Wang J, Taraseviciene‐Stewart L, Kasahara Y, Cool CD, Bishop AE, Geraci M, Semenza GL, Yacoub M, Polak JM & Voelkel NF (2001). Expression of angiogenesis‐related molecules in plexiform lesions in severe pulmonary hypertension: evidence for a process of disordered angiogenesis. J Pathol 195, 367–374. [DOI] [PubMed] [Google Scholar]
- Tuder RM, Flook BE & Voelkel NF (1995). Increased gene expression for VEGF and the VEGF receptors KDR/Flk and Flt in lungs exposed to acute or to chronic hypoxia. Modulation of gene expression by nitric oxide. J Clin Invest 95, 1798–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuder RM, Yun JH, Bhunia A & Fijalkowska I (2007). Hypoxia and chronic lung disease. J Mol Med (Berl) 85, 1317–1324. [DOI] [PubMed] [Google Scholar]
- Veit F, Pak O, Egemnazarov B, Roth M, Kosanovic D, Seimetz M, Sommer N, Ghofrani HA, Seeger W, Grimminger F, Brandes RP, Schermuly RT & Weissmann N (2013). Function of NADPH oxidase 1 in pulmonary arterial smooth muscle cells after monocrotaline‐induced pulmonary vascular remodeling. Antioxid Redox Signal 19, 2213–2231. [DOI] [PubMed] [Google Scholar]
- Veith C, Marsh LM, Wygrecka M, Rutschmann K, Seeger W, Weissmann N & Kwapiszewska G (2012). Paxillin regulates pulmonary arterial smooth muscle cell function in pulmonary hypertension. Am J Pathol 181, 1621–1633. [DOI] [PubMed] [Google Scholar]
- Veith C, Schmitt S, Veit F, Dahal BK, Wilhelm J, Klepetko W, Marta G, Seeger W, Schermuly RT, Grimminger F, Ghofrani HA, Fink L, Weissmann N & Kwapiszewska G (2013). Cofilin, a hypoxia‐regulated protein in murine lungs identified by 2DE: role of the cytoskeletal protein cofilin in pulmonary hypertension. Proteomics 13, 75–88. [DOI] [PubMed] [Google Scholar]
- Veith C, Zakrzewicz D, Dahal BK, Balint Z, Murmann K, Wygrecka M, Seeger W, Schermuly RT, Weissmann N & Kwapiszewska G (2014). Hypoxia‐ or PDGF‐BB‐dependent paxillin tyrosine phosphorylation in pulmonary hypertension is reversed by HIF‐1α depletion or imatinib treatment. Thromb Haemost 112, 1288–1303. [DOI] [PubMed] [Google Scholar]
- Wang GL, Jiang BH, Rue EA & Semenza GL (1995). Hypoxia‐inducible factor 1 is a basic‐helix‐loop‐helix‐PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 92, 5510–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Juhaszova M, Rubin LJ & Yuan XJ (1997). Hypoxia inhibits gene expression of voltage‐gated K+ channel alpha subunits in pulmonary artery smooth muscle cells. J Clin Invest 100, 2347–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Weigand L, Lu W, Sylvester JT, Semenza GL & Shimoda LA (2006). Hypoxia inducible factor 1 mediates hypoxia‐induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res 98, 1528–1537. [DOI] [PubMed] [Google Scholar]
- Ward JP (2006). Point: Hypoxic pulmonary vasoconstriction is mediated by increased production of reactive oxygen species. J Appl Physiol 101, 993–995; discussion 999. [DOI] [PubMed] [Google Scholar]
- Waypa GB & Schumacker PT (2010). Hypoxia‐induced changes in pulmonary and systemic vascular resistance: where is the O2 sensor? Respir Physiol Neurobiol 174, 201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir EK & Archer SL (2006). Counterpoint: Hypoxic pulmonary vasoconstriction is not mediated by increased production of reactive oxygen species. J Appl Physiol 101, 995–998; discussion 998. [DOI] [PubMed] [Google Scholar]
- Weissmann N, Dietrich A, Fuchs B, Kalwa H, Ay M, Dumitrascu R, Olschewski A, Storch U, Mederos y Schnitzler M, Ghofrani HA, Schermuly RT, Pinkenburg O, Seeger W, Grimminger F & Gudermann T (2006). Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc Natl Acad Sci USA 103, 19093–19098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissmann N, Sydykov A, Kalwa H, Storch U, Fuchs B, Mederos y Schnitzler M, Brandes RP, Grimminger F, Meissner M, Freichel M, Offermanns S, Veit F, Pak O, Krause KH, Schermuly RT, Brewer AC, Schmidt HH, Seeger W, Shah AM, Gudermann T, Ghofrani HA & Dietrich A (2012). Activation of TRPC6 channels is essential for lung ischaemia‐reperfusion induced oedema in mice. Nat Commun 3, 649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitman EM, Pisarcik S, Luke T, Fallon M, Wang J, Sylvester JT, Semenza GL & Shimoda LA (2008). Endothelin‐1 mediates hypoxia‐induced inhibition of voltage‐gated K+ channel expression in pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol 294, L309–L318. [DOI] [PubMed] [Google Scholar]
- Wiesener MS, Jurgensen JS, Rosenberger C, Scholze CK, Horstrup JH, Warnecke C, Mandriota S, Bechmann I, Frei UA, Pugh CW, Ratcliffe PJ, Bachmann S, Maxwell PH & Eckardt KU (2003). Widespread hypoxia‐inducible expression of HIF‐2α in distinct cell populations of different organs. FASEB J 17, 271–273. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Zhang H, Iwase T, Shen J, Semenza GL & Campochiaro PA (2010). Digoxin inhibits retinal ischemia‐induced HIF‐1α expression and ocular neovascularization. FASEB J 24, 1759–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, Sham JS, Wiener CM, Sylvester JT & Semenza GL (1999). Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia‐inducible factor 1α. J Clin Invest 103, 691–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, Rey S, Hammers H, Chang D, Pili R, Dang CV, Liu JO & Semenza GL (2008). Digoxin and other cardiac glycosides inhibit HIF‐1α synthesis and block tumor growth. Proc Natl Acad Sci USA 105, 19579–19586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimnicka AM, Tang H, Guo Q, Kuhr FK, Oh MJ, Wan J, Chen J, Smith KA, Fraidenburg DR, Choudhury MS, Levitan I, Machado RF, Kaplan JH & Yuan JX (2014). Upregulated copper transporters in hypoxia‐induced pulmonary hypertension. PLoS One 9, e90544. [DOI] [PMC free article] [PubMed] [Google Scholar]