

Abstract
At high altitude, barometric pressure falls and with it inspired , potentially compromising O2 delivery to the tissues. With sufficient acclimatisation, the erythropoietic response increases red cell mass such that arterial O2 content () is restored; however arterial remains low, and the diffusion of O2 from capillary to mitochondrion is impaired. Mitochondrial respiration and aerobic capacity are thus limited, whilst reactive oxygen species (ROS) production increases. Restoration of with supplementary O2 does not fully restore aerobic capacity in acclimatised individuals, possibly indicating a peripheral impairment. With prolonged exposure to extreme high altitude (>5500 m), muscle mitochondrial volume density falls, with a particular loss of the subsarcolemmal population. It is not clear whether this represents acclimatisation or deterioration, but it does appear to be regulated, with levels of the mitochondrial biogenesis factor PGC‐1α falling, and shows similarities to adapted Tibetan highlanders. Qualitative changes in mitochondrial function also occur, and do so at more moderate high altitudes with shorter periods of exposure. Electron transport chain complexes are downregulated, possibly mitigating the increase in ROS production. Fatty acid oxidation capacity is decreased and there may be improvements in biochemical coupling at the mitochondrial inner membrane that enhance O2 efficiency. Creatine kinase expression falls, possibly impairing high‐energy phosphate transfer from the mitochondria to myofibrils. In climbers returning from the summit of Everest, cardiac energetic reserve (phosphocreatine/ATP) falls, but skeletal muscle energetics are well preserved, possibly supporting the notion that mitochondrial remodelling is a core feature of acclimatisation to extreme high altitude.
Abbreviations
- 31P‐MRS
31P magnetic resonance spectroscopy
- BNIP3
BCL2/adenovirus E1B 19 kDa protein‐interacting protein 3
- ETC
electron transport chain
- HIF
hypoxia‐inducible factor
- HOAD
3‐hydroxyacyl‐CoA dehydrogenase
- PGC
peroxisome proliferator‐activated receptor gamma co‐activator
partial pressure of O2
- PCr
phosphocreatine
- PPARα
peroxisome proliferator‐activated receptor alpha
- ROS
reactive oxygen species
- UCP3
uncoupling protein 3
- Vmax
maximum rate of mitochondrial oxygen consumption
maximum rate of whole‐body oxygen consumption
Introduction
Barometric pressure decreases with altitude and thus inspired falls, such that at high altitudes O2 delivery from the lungs to oxidative tissues such as heart and skeletal muscle is challenged. Within the body, the majority of oxygen consumption occurs at cytochrome c oxidase (complex IV) of the mitochondrial electron transport chain (ETC). The concept of symmorphosis postulates that biological systems are economical, with structural parameters matched to functional capacity (Weibel et al. 1991). Accordingly, for the respiratory system, mitochondrial volume in the muscles would be proportional to total aerobic capacity (Weibel et al. 1991). Thus, at high altitude, when aerobic capacity becomes limited, alterations in mitochondrial volume and function could intuitively form part of the process of human acclimatisation (Murray, 2014). Alternatively, the mitochondria might be viewed as potential targets of damage during the less well understood process of high altitude deterioration (Ward, 1954) as they are sites of reactive oxygen species (ROS) production.
Over the past 25 years, several studies have reported changes in mitochondrial volume density, subcellular distribution, oxidative enzyme activity and protein expression in the muscles of climbers returning from extreme high altitude (>5500 m) and these findings are reviewed here (Fig. 1). Whilst no direct measures of mitochondrial function have yet been made above 5500 m, 31P magnetic resonance spectroscopy (31P‐MRS) has been used by our research group to study heart and skeletal muscle energetics as an indirect measure of mitochondrial function in climbers at sea level, prior to and within a few weeks of their return from extreme altitude. In addition, a small number of informative studies of mitochondrial respiratory function have been made in permeabilised fibres of skeletal muscle biopsies at altitudes between 3400 and 4500 m using high‐resolution respirometry, and these findings have also been included here. Finally, whilst the focus of this review is on human mitochondrial function at extreme high altitude, reference is made to relevant mechanistic insights gleaned from high altitude adapted species, experimental animal models and cells cultured under hypoxic conditions. For a more comprehensive account of the metabolic changes that occur in the skeletal muscle of rodents and humans in response to real or simulated altitudes >3000 m, readers are referred to our recent review (Horscroft & Murray, 2014).
Figure 1.

Timeline of key events and studies pertaining to the study of mitochondrial function in heart and skeletal muscle at extreme high altitude and discussed in this review
What underlies the possible change in mitochondrial function at extreme high altitude?
Upon ascent to altitudes over 5500 m, barometric pressure, and thus inspired , falls to less than half of that at sea level, with the inspired on the summit of Mt Everest (8848 m) being less than one‐third of that at sea level (43.1 vs. 148.0 mmHg) (West et al. 1983). The successful ascent of Everest without supplementary oxygen by Reinhold Messner and Peter Habeler in 1978 (a feat repeated by Messner and others since) demonstrated the remarkable capacity of the human body to perform at such extreme altitude. It is clear that successful acclimatisation during a gradual ascent is paramount to any such attempt to reach extreme altitudes.
Features of acclimatisation that improve O2 delivery at altitude have been well documented. Upon arrival at altitude, resting heart rate increases and cardiac output is elevated for a given amount of physical work. The sea‐level relationship between cardiac output and workload is later restored as arterial O2 content () returns to sea level values, although notably the maximum achievable cardiac output falls at higher altitudes (Peacock, 1998). Ventilatory aspects of acclimatisation show a distinct time course, with an initial increase in ventilation (the hypoxic ventilatory response) followed by a brief reduction back towards control values (hypoxic ventilatory decline) before a sustained period of acclimatisation, during which ventilation continues to increase for up to about 2 weeks (West et al. 2007). Meanwhile, an initial increase in haematocrit can result from a fall in plasma volume due to dehydration, but in response to sustained hypoxia the juxtaglomerular apparatus of the kidneys increases production of erythropoietin (EPO), which stimulates bone marrow to increase red cell output. EPO levels can rise within hours of arrival at altitude, but the increase in red cell mass can take several weeks (Peacock, 1998; West et al. 2007). In the face of falling arterial , and therefore haemoglobin‐O2 saturation, the erythropoietic response allows to be maintained at, or even above, sea level values up to 7000 m, falling only at more extreme altitudes (Grocott et al. 2009).
Whilst the process of acclimatisation can take many weeks, a prolonged stay at extreme high altitude can result in high altitude deterioration. This process is rather less well understood than acclimatisation, but is characterised by weight loss (including muscle wasting), poor appetite, lethargy, slow recovery from fatigue and possibly low systemic blood pressure (Ward, 1954). Altitude exposure can decrease energy intake by as much as half (Guilland & Klepping, 1985; Rose et al. 1988), resulting in an energy deficit which underlies the loss of body mass (Westerterp & Kayser, 2006). The cachectic response to high altitude, whilst traditionally viewed as detrimental, may afford some protection through, for example, the release of metabolic substrates and signalling molecules (Murray & Montgomery, 2014; Levett et al. 2015), an increased ratio of capillaries to muscle fibre cross sectional area (Hoppeler et al. 1990 a) or simply by decreasing whole‐body O2 demand due to the loss of body mass. Changes in diet and energy balance are therefore confounding factors, which must be considered in studies of metabolism at extreme altitude.
Even with adequate acclimatisation, however, and a equivalent to that at sea level, exercise capacity and are impaired at altitude (Cerretelli, 1976; Levett et al. 2011, 2012). Moreover, the loss of at any given altitude is not discernibly different between unacclimatised and fully acclimatised subjects (Cerretelli, 1976). Thus, whilst successful acclimatisation can help to stave off some of the potentially adverse consequences of high altitude (most notably, acute mountain sickness) (West et al. 2007), exercise capacity is not improved. This somewhat counterintuitive finding illustrates the importance of capillary , as opposed to , in supporting O2 transport and utilisation. Convective O2 delivery is restored by acclimatisation, but because capillary remains low, the capacity for diffusion between the capillary and mitochondrion is limited – a concept proposed by Peter Wagner and co‐workers (Wagner, 1996, 2000), and supported by experimental evidence in hypoxic dogs (Hogan et al. 1988) and retrospective analysis of data collected from human subjects during Operation Everest II, in which subjects underwent progressive decompression to the equivalent of 8840 m in a chamber at sea level (Sutton et al. 1988; Wagner, 2010). Whilst muscle wasting at altitude might conceivably improve diffusion to the mitochondria by decreasing mean diffusion distance, in dogs with muscle wasting following leg immobilisation, O2 conductance was not improved (Hepple et al. 2000).
Therefore, at extreme high altitude capillary is decreased, even in fully acclimatised subjects. What then of the mitochondrial ? Measurements of the distribution of within working skeletal muscle fibres suggest a sharp immediate fall in at the interface between capillary and myocyte, but with the across the remaining width of the myocyte being reasonably uniform and low, due to the presence of myoglobin in aiding distribution (Gayeski & Honig, 1986 a,b). Within the mitochondrion itself, at cytochrome c oxidase (the terminal complex of the ETC and site of O2 consumption) is extremely low, probably less than 1 mmHg (0.13 kPa) even in the absence of environmental hypoxia (Chance, 1957; Chance et al. 1962). As such, mitochondrial has often been considered negligible and thus assumed to be zero in many models of O2 diffusion and consumption. This assumption cannot be correct, however, because oxidative phosphorylation is dependent upon O2, and if were zero then so too would be (Cano et al. 2013). A recently refined model of whole‐body O2 transport and utilisation has allowed for greater heterogeneity in across muscle regions, with mitochondrial increasing under conditions where mitochondrial respiratory capacity (V max) is relatively low compared with O2 delivery (Cano et al. 2015). Using data from Operation Everest II, the model predicts a progressive fall in mitochondrial of exercising muscle at altitude, from around 1 mmHg at sea level to a mean value of 0.119 mmHg on the summit of Everest (Cano et al. 2015). This is below measurements made in vitro of the P 50 of mitochondrial respiration i.e. the at which respiration would fall by half (Gnaiger et al. 1998) and as such respiration would be limited. In addition, another expected consequence of such low mitochondrial at extreme altitude would be a sharp increase in ROS production (Cano et al. 2014), the implications of which are discussed later in this review.
The fall in arterial (and thus capillary) at extreme altitude thus limits O2 diffusion, exaggerating the mismatch between the capacity of the tissue to consume available O2 (V max) and the capacity to deliver it – a situation already present to some degree in humans at sea level (Boushel et al. 2011) and seeming to contradict the concept of symmorphosis (Weibel et al. 1991). At altitude, can be improved by the use of O2 supplementation to elevate arterial (and thus capillary) , but curiously it is not completely restored to sea level values in acclimatised subjects, despite their elevated haematocrit which actually results in them having an enhanced compared with that at sea level (Cerretelli, 1976). This finding provided the earliest evidence for a peripheral impairment either in local O2 delivery, as Paolo Cerretelli suggested at the time, or in the oxidative capacity of the muscle – a subject that was to form the basis of much of his later research (Ferretti, 2003). In further support of this, does not return to pre‐expedition values for several weeks after acclimatised subjects return to sea level (Grassi et al. 1996). Intriguingly, recent measures of impaired blood flow through the sublingual microvasculature using sidestream darkfield imaging suggest that local O2 delivery might indeed be worsened in acclimatised subjects at altitude (Martin et al. 2009), perhaps due to increased blood viscosity secondary to a raised haematocrit or endothelial dysfunction. Note, however, that comparable measures have not yet been made in the skeletal muscle microcirculation and how generalisable this finding is remains of debate.
Morphological changes in mitochondria – volume density and subcellular distribution
Altitude thus poses a sustained challenge to oxidative phosphorylation. An interpretative hypothesis, based on observational data from the muscles from high altitude adapted animals, had previously suggested that an enhanced oxidative capacity was a necessary component of high altitude acclimatisation (Hochachka et al. 1983). This was supported by earlier evidence of high oxidative capacities and myoglobin concentrations in the sartorius muscle of some Andean highlanders compared with lowlanders (Reynafarje, 1962), although these measurements may have been confounded by differences in training status between subjects. Endurance training is a potent stimulant for mitochondrial biogenesis, and was found to be as effective in Andean natives as it is in lowland residents (Desplanches et al. 1996). Certainly in lowlanders acclimatising to high altitude this is not the case; oxidative capacities fall with prolonged exposure, and at extreme altitude the mitochondrial volume density of human skeletal muscle decreases. A loss of 20–30% of mitochondrial density has consistently been seen in climbers returning from the summit of Everest (Hoppeler et al. 1990; Levett et al. 2012), alongside the accumulation of lipofuscin, a substance believed to result from lipid peroxidation and thus indicating possible oxidative stress (Howald & Hoppeler, 2003). It is tempting to view this phenomenon as a feature of high altitude deterioration; interestingly, there was no apparent loss of mitochondria in the muscles of the subjects of Operation Everest II (MacDougall et al. 1991) whose rapid ascent to the equivalent altitude of the summit of Everest (over 40 days) and immediate return to normal barometric pressure (Houston et al. 1987) limited their exposure to extreme altitude in comparison with a traditional Everest expedition. Intriguingly, however, in muscle biopsies from Sherpas, a highly adapted subgroup of native Tibetan highlanders reputed to be strong performers at extreme altitude (Gilbert‐Kawai et al. 2014), lower mitochondria densities have also been reported (Kayser et al. 1991) and this also appears to be the case in lowland‐dwelling Tibetans who have never resided at altitude (Kayser et al. 1996). Therefore, the loss of mitochondrial density in acclimatising lowlanders may be an adaptive response that mimics the adaptations of high altitude natives, rather than simple deterioration.
Curiously, there appear to be differential effects of extreme altitude on two discrete subpopulations of mitochondria in human muscle. Within the cell, mitochondria do not generally exist as distinct, individual, rod‐shaped organelles, but instead form a dynamic reticular network, which undergoes continual fusion and fission (Picard et al. 2011). Within skeletal and cardiac muscle, this network forms spatially distinct pools, with a subpopulation of subsarcolemmal mitochondria lying alongside the cell membrane and interfibrillar mitochondria positioned in rows alongside the myofibrils (Ferreira et al. 2010; Hollander et al. 2014). In climbers returning from extreme altitude, there is a greater percentage loss of the less abundant subsarcolemmal mitochondria than the more populous, deep‐lying interfibrillar mitochondria (Hoppeler et al. 1990; Levett et al. 2012) (Fig. 2). Correspondingly, whilst the lower mitochondrial density in Tibetans compared with lowland Nepalese appears to be driven by lower densities of both subsarcolemmal and interfibrillar mitochondria, a greater component of the difference is accounted for by the lower subsarcolemmal population (Kayser et al. 1996). These findings are somewhat counterintuitive, as a selective loss of the subsarcolemmal population would result in a potentially counterproductive increase in mean diffusion distance for O2 between the capillary and remaining (largely interfibrillar) mitochondria. This may in part be mitigated by an increase in capillary density, due to muscle fibre wasting (Hoppeler et al. 1990 a; Ferretti, 2003; Murray & Montgomery, 2014), and possibly by an increased myoglobin content to facilitate intracellular O2 diffusion. Increased myoglobin levels have been reported in rodents exposed to environmental hypoxia (Clark et al. 1952; Vaughan & Pace, 1956) and dogs residing at high altitude (Hurtado et al. 1937). In humans, however, the picture is less clear. Elevated myoglobin levels were reported in some high altitude natives (Reynafarje, 1962), although again this observation may have been confounded by training status. Expression levels of myoglobin were not increased in climbers returning from the summit of Everest, however (Levett et al. 2015), and may only become upregulated when exercise training is imposed on top of the hypoxic stimulus (Hoppeler & Vogt, 2001; Desplanches et al. 2014). Moreover, in one study muscle levels of myoglobin fell by 35% in human subjects after 7–9 days at 4559 m, alongside the downregulation of other iron‐related proteins and loss of total muscle iron content – effects that were attributed to the need to mobilise intramuscular iron to support the erythropoietic response (Robach et al. 2007).
Figure 2. Subcellular distribution of mitochondria within the myocyte in A, unacclimatised lowlanders; B, acclimatised lowlanders or adapted Tibetan highlanders; and C, adapted bar‐headed geese .
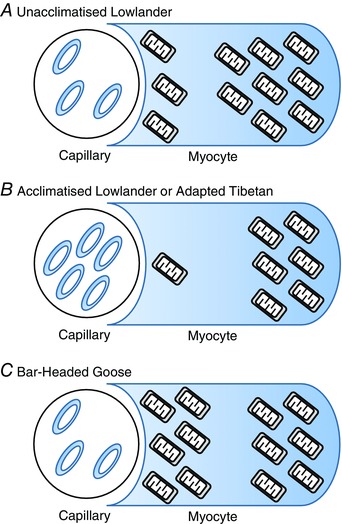
Acclimatisation in humans is associated with a greater loss of subsarcolemmal mitochondria, compared with the interfibrillar population. Meanwhile high altitude adapted Tibetans have lower mitochondrial densities than lowlanders with a greater proportion of the difference being accounted for by a relatively low subsarcolemmal population. Hence, high altitude acclimatisation appears to mimic the adapted‐highlander phenotype. In bar‐headed geese, however, there is a preponderance of subsarcolemmal mitochondria, which appears to minimise the diffusion distance for O2. Bar‐headed geese appear to have an enhanced creatine kinase shuttle, to aid the distribution of high‐energy phosphate metabolites throughout the myocyte, whereas in humans at altitude, creatine kinase is downregulated.
The subcellular distribution of mitochondria is not, however, only important for the diffusion of O2 but also for the exchange of the high energy phosphate products of respiration between the mitochondrial source and the myofibrils (Hoppeler & Billeter, 1991). The latter process is aided in highly oxidative tissues such as cardiac and skeletal muscle by the creatine kinase shuttle, which acts as a temporal and spatial buffer of cellular ATP by transferring phosphate from ATP to creatine at the mitochondrion, forming phosphocreatine (PCr) and ADP, and catalysing the reverse reaction at the myofibrils. These reactions maintain [ADP] at the mitochondrion, supporting respiration, and [ATP] at myosin, supporting contraction. The creatine kinase shuttle also allows a faster distribution of the high energy phosphate from the mitochondrion to myofibril as the smaller PCr molecule diffuses more rapidly than ATP. At altitude, however, creatine kinase is downregulated (Vigano et al. 2008; Levett et al. 2015), potentially impairing high energy phosphate transfer. Muscle fibre wasting may mitigate this to some extent, by decreasing average diffusion distances, but it is possible that with a compromised capacity for PCr synthesis the preferred maintenance of mitochondria in intermyofibrillar regions circumvents some of the resulting limitations of high‐energy phosphate delivery to myosin.
In contrast with humans, in the muscles of certain species of bird adapted for flight at extreme high altitudes, such as the bar‐headed goose, a greater overall aerobic capacity is accompanied by a preponderance of subsarcolemmal mitochondria (Fig. 2), an adaptation presumed to be beneficial in minimising O2 diffusion distance (Scott, 2011). Training status may be a confounding factor here too though, because the low air density at these altitudes increases the work required to generate the lift and thrust to maintain flight (Bishop et al. 2015). Note, however, that mitochondrial ATP production in these birds is more strongly regulated by the creatine kinase shuttle than in species dwelling at low altitude (Scott et al. 2009). Perhaps this suggests that the geese have a more effective high energy phosphate delivery mechanism than humans, obviating the need for a more substantial population of interfibrillar mitochondria and allowing redistribution of mitochondria to the cell periphery. Moreover, these birds exhibit other adaptations, including high muscle capillarity and an enhanced hypoxic ventilatory response, which would partly counteract the fall in arterial at these altitudes (Scott, 2011).
The physiological significance of the apparent selective loss of subsarcolemmal mitochondria in humans at extreme altitude remains unclear, whilst the mechanism regulating it is undetermined. Indeed little is firmly known about the functional importance of these two subpopulations, although in some rat muscles (but not all) they have different respiratory capacities (Palmer et al. 1977; Philippi & Sillau, 1994) and differences in the proteome of the two subpopulations have also been noted (Ferreira et al. 2010). In humans at sea level, it is notable that the subsarcolemmal population is associated with much greater changes in volume density in response to endurance training than the interfibrillar mitochondria (Desplanches et al. 1993). It is possible that the loss of mitochondrial density, and in particular the subsarcolemmal population, with prolonged exposure to extreme high altitude may therefore simply represent a detraining response accompanying the lethargy associated with high altitude deterioration, rather than true acclimatisation. Moreover, the cachectic response to high altitude and changes in diet may also affect mitochondrial density and distribution. In cells, however, and increasingly in animal models and human subjects, there is evidence of regulated transcriptional modification of metabolism and mitochondrial density associated with hypoxic exposure, and this evidence is reviewed in the next section.
Regulation of mitochondrial density in prolonged hypoxia
The cellular response to hypoxia is predominantly governed by the hypoxia‐inducible factor (HIF) family of transcription factors. HIF‐1α and 2α subunits are constitutively expressed, but degraded following O2‐dependent hydroxylation by prolyl‐hydroxylase enzymes (PHD1–3) (Semenza, 2007). At low tissue , the HIF α‐subunits are thus stable and dimerise with the nuclear HIF‐1β subunit, activating the transcription of genes that contain hypoxia‐response elements in their promoter regions, including those encoding EPO and vascular endothelial growth factor (VEGF) (Semenza, 2007). There appears to be a timing‐dependent component to this response, termed the HIF switch, whereby HIF‐1 stabilisation underpins an initial response to hypoxia (<24 h) whilst the more sustained response is supported by HIF‐2 (Koh & Powis, 2012). Systemically, HIF‐2 activation underlies the erythropoietic response, whilst in cultured cells HIF‐1‐dependent measures to decrease O2 demand include the downregulation of mitochondrial oxidative phosphorylation via inhibition of pyruvate dehydrogenase (Kim et al. 2006). An HIF‐dependent upregulation of the pro‐apoptotic protein BNIP3 (BCL2/adenovirus E1B 19 kDa protein‐interacting protein 3) also underlies a process of mitochondrial autophagy, and consequent loss of mitochondrial density, in some cells (Zhang et al. 2008). An increase in BNIP3 appears to occur in the hypoxic rat heart (Regula et al. 2002), but was not seen in human muscle at extreme altitude, although markers of chaperone‐mediated autophagy were present (Levett et al. 2015) (Fig. 3).
Figure 3. Possible mechanism underlying the regulation of mitochondrial volume density in response to cellular hypoxia at extreme high altitude .
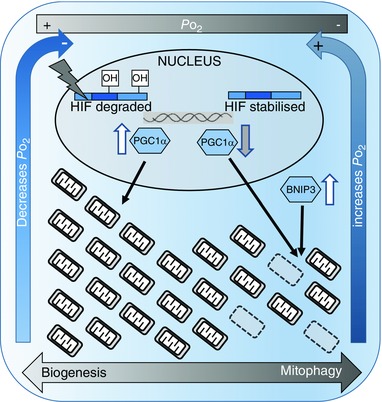
Under normoxic conditions (relatively high ), the hypoxia‐inducible factors (HIFs) are hydroxylated and degraded, whilst under hypoxic conditions (low ) they are stabilised, with increased mitochondrial ROS production enhancing HIF stability. HIF binds to hypoxia response elements on target genes activating transcription. Hypoxia can therefore increase mitophagy factors (e.g. BNIP3) and lead to the downregulation of factors that stimulate mitochondrial biogenesis (e.g. PGC‐1α). Thus, in sustained hypoxia, mitochondrial density falls, whereas in normoxic conditions mitochondrial biogenesis can be supported. An expanded population of mitochondria will consume O2 at a greater rate, decreasing cellular and leading to HIF stabilisation, whereas a loss of mitochondria will conversely restore leading to HIF degradation. Thus, the cell's capacity for oxygen consumption (i.e. mitochondrial density) is dynamically coupled to tissue , and oxygen demand therefore matched to supply.
In skeletal muscle, there is a possible interaction between the transcriptional activity of HIF and that of the mitochondrial biogenesis factor, peroxisome proliferator‐activated receptor γ co‐activator 1α (PGC‐1α). In cultured myocytes, PGC‐1α overexpression increased mitochondrial biogenesis and thus cellular O2 consumption, causing a fall in tissue , which in turn stabilised HIF‐1α (O'Hagan et al. 2009) (Fig. 3). Conversely, in renal carcinoma cells with constitutively stabilised HIF‐1, mitochondrial biogenesis was suppressed secondary to repression of C‐MYC activity and decreased expression of PGC‐1β (Zhang et al. 2007). Thus, mitochondrial density is dynamically regulated in response to O2 availability, with mitochondrial biogenesis supported when O2 is available (and HIF‐1 therefore not stabilised) but further mitochondrial biogenesis is inhibited if cellular O2 consumption increases to the extent that falls below a critical point and HIF‐1 becomes stabilised. Levels of PGC‐1α were decreased in climbers returning from the summit of Everest and biopsied at 5300 m, suggesting a suppression of mitochondrial biogenesis had occurred (Levett et al. 2012). HIF‐1α levels were not elevated in these subjects, although in line with the HIF switch this would not perhaps have been expected after 66 days of exposure. Unfortunately, levels of HIF‐2α, which mediates the more sustained response to hypoxia, were not measured in these subjects. In the same study, subjects biopsied within a few days of arrival at 5300 m following ascent did show enhanced HIF‐1α, but no loss of mitochondrial density or PGC‐1α at this stage (Levett et al. 2012).
Interestingly, the altitude‐dependent loss of mitochondria and remodelling of mitochondrial function do appear to be dependent on length of exposure or altitude reached (Horscroft & Murray, 2014), perhaps reflecting a time‐dependent regulation of the transcriptional response in line with the HIF switch. Notably, in two studies from Carsten Lundby's group, which used high‐resolution respirometry to measure respiratory function in skeletal muscle biopsies, relatively short‐term exposure (9–11 days) to 4559 m had no discernible effect on mitochondrial respiratory capacity (Jacobs et al. 2013), whilst 28 days of exposure to a lower altitude (3454 m) did result in a diminished respiratory capacity, albeit in the absence of a change in mitochondrial content (Jacobs et al. 2012). The findings from these studies are discussed in more detail in the next section.
Mitochondria themselves play a further, critical role in hypoxia signalling, forming the nexus of a feedback loop that maintains cellular redox homeostasis. Under hypoxic conditions, electron leak, particularly from complexes I and III of the highly reduced mitochondrial ETC, leads to increased production of ROS, such as the superoxide radical (O2 .−), potentially damaging proteins, lipids and DNA and thus impairing cellular function (Guzy & Schumacker, 2006; Tsutsui, 2006). ROS production is predicted to rise with altitude, increasing rapidly at altitudes above 7000 m (Cano et al. 2014). Oxidative stress enhances HIF stabilisation in cells (Chandel et al. 2000; Guzy et al. 2005; Guzy & Schumacker, 2006), thereby activating responses that in turn curb ROS generation, albeit at the possible cost of impaired ATP synthesis (Murray, 2009). An HIF‐regulated process of mitophagy or repression of mitochondrial biogenesis, in matching tissue O2 demand to a diminished supply, is thought to limit ROS production, thereby protecting the remaining mitochondria and preserving their respiratory function. Paradoxically, there is also evidence that ROS are necessary mediators for training‐induced mitochondrial biogenesis (Gomez‐Cabrera et al. 2008). These apparently opposing effects may be determined by the duration of the ROS stimulus, with an acute increase in ROS during exercise eliciting mitochondrial biogenesis, but sustained ROS production with chronic exposure to extreme altitude repressing biogenesis and activating mitophagy.
Qualitative changes in mitochondrial function – oxidative enzymes and respiratory coupling
Mitochondrial respiratory function itself exhibits plasticity under prolonged/severe hypoxia, beyond a modulation of mitochondrial content, with changes in substrate metabolism, ETC activity and respiratory coupling occurring in response to extreme high altitude, but also at more moderate high altitude over shorter periods of exposure when there may not yet be a loss of mitochondrial density (Horscroft & Murray, 2014). Regarding extreme altitude, the activity and/or expression of several tricarboxylic acid cycle enzymes have been found to be downregulated following real or simulated ascent above 5500 m, including citrate synthase (Green et al. 1989; Howald et al. 1990; Levett et al. 2012), isocitrate dehydrogenase (Levett et al. 2015), α‐ketoglutarate dehydrogenase (Levett et al. 2015) and succinate dehydrogenase (Green et al. 1989; Levett et al. 2015), which also comprises complex II of the ETC. Of the other ETC complexes, decreased protein levels or activity of subunits of complex I (Levett et al. 2012), complex III (Levett et al. 2015) and complex IV (Howald et al. 1990; Levett et al. 2012) have all been reported following exposure to extreme altitude. Furthermore, in high‐resolution respirometry studies of permeabilised muscle fibres, respiration capacity for both complex I and complex II substrates was decreased following 28 days at 3454 m with no loss in citrate synthase activity (Jacobs et al. 2012), but no such changes were seen with short‐term exposure to 4559 m (Jacobs et al. 2013). Downregulation of ETC complexes under hypoxic conditions may mitigate the potential rise in ROS production via electron leak, by restricting electron entry into the chain. In hypoxic human fibroblasts, upregulation of an HIF‐regulated microRNA, miR‐210, suppressed the expression of ETC complex assembly proteins, decreasing the levels and respiratory capacity of complexes I and IV (Colleoni et al. 2013). This effect was also seen in human placentas at 3100 m (Colleoni et al. 2013) and may explain the downregulation of these two complexes in the muscle of climbers returning from the summit of Everest (Levett et al. 2012) (Fig. 4).
Figure 4. Summary of intramitochondrial changes that occur during acclimatisation to extreme high altitude .
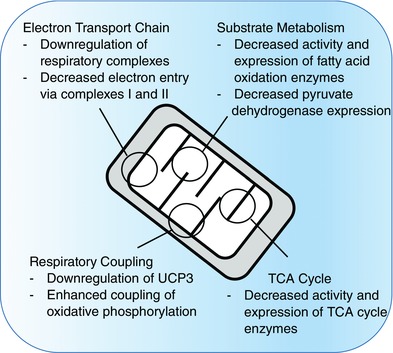
Studies at extreme high altitude have shown changes in enzyme activities and protein levels that suggest downregulation of electron transport chain (ETC) complexes, tricarboxylic acid (TCA) cycle enzymes and substrate oxidation enzymes, alongside decreased levels of uncoupling protein 3 (UCP3). High resolution respirometry of human muscle fibres at more moderate high altitude has shown that sustained hypoxia is associated with decreased electron entry into the ETC and improved mitochondrial efficiency.
A further possible adaptive response might be one of improved mitochondrial O2 efficiency, resulting from a substrate switch away from fatty acid oxidation towards more O2‐efficient glucose oxidation, and a possible improvement in mitochondrial coupling. The capacity for fatty acid oxidation tends to fall at high altitude (Horscroft & Murray, 2014), with decreased activity of 3‐hydroxyacyl‐CoA dehydrogenase (HOAD) (Levett et al. 2012) and protein levels of several other β‐oxidation enzymes (Levett et al. 2015) reported in climbers following ascent of Everest (Fig. 4). HOAD activities were also lower in Tibetans compared with lowland Nepalese (Kayser et al. 1996). Several of these enzymes are transcriptional targets for a fatty acid‐activated transcription factor, peroxisome proliferator‐activated receptor α (PPARα), which, at least in some tissues, is downregulated in hypoxia in an HIF‐dependent manner (Narravula & Colgan, 2001). Notably, however, protein levels of pyruvate dehydrogenase were also lower in these subjects following ascent of Everest, perhaps arguing against a switch towards glucose oxidation and instead supporting a possible increased role for glycolytic ATP production and lactate production instead, bypassing the mitochondria.
With the downregulation of oxidative enzymes and loss of mitochondrial density, it is conceivable that anaerobic glycolysis might make a greater contribution to muscle ATP demands at extreme altitude, particularly during exercise. The genes for many glycolytic enzymes contain hypoxia‐response elements in their promoter regions (Semenza et al. 1994), and in hypoxic cells, the inhibition of pyruvate dehydrogenase (Kim et al. 2006) supports a Pasteur effect, in which glycolytically derived lactate is expelled from the cell. In humans, however, the evidence to support increased glycolysis at altitude is limited (Horscroft & Murray, 2014). Indeed, in muscle biopsies from humans returning from extreme altitude, the levels of several glycolytic enzymes were decreased (Levett et al. 2015) as was hexokinase activity (Levett et al. 2012). These observations might reflect the so‐called ‘lactate paradox’. In this phenomenon, acute exposure to high altitude is accompanied by greater blood lactate levels ([Lab]) at a given submaximal workload than at sea level, although following acclimatisation over a period of weeks, the same exercise challenge results in a lower [Lab], more comparable with that at sea level (West, 1986). Thus, acclimatisation may decrease the initial dependence on glycolysis to meet cellular ATP demand, perhaps through multiple adjustments that optimise O2 delivery and utilisation or through better coupling of pyruvate production and oxidative phosphorylation. Some studies, however, have suggested that the ‘lactate paradox’ is a more transient feature of acclimatisation and not applicable to those spending longer durations at extreme altitude (Lundby et al. 2000; van Hall et al. 2001, 2009).
The notion of improved respiratory coupling, resulting in enhanced O2 efficiency at extreme altitude, is also appealing, although mitochondrial uncoupling is likely to afford some antioxidant protection and may thus be beneficial. Levels of uncoupling protein 3 (UCP3), another PPARα target, decreased in the muscle of climbers following an ascent of Everest (Levett et al. 2012), and whilst the possible effects of UCP3 on mitochondrial efficiency do remain controversial, it is notable that in high‐resolution respirometry studies LEAK state respiration (a functional measure of uncoupling) decreased in subjects after a month at 3454 m (Jacobs et al. 2012) (Fig. 4). UCP3 levels correlated negatively with some measures of cycling efficiency in athletes at sea level (Mogensen et al. 2006), but the question of whether improved mitochondrial coupling at altitude translates into enhanced exercise efficiency remains controversial, with some studies showing improvements (Gore et al. 2007; Latshang et al. 2013) and others showing no change in economy (Lundby et al. 2007). The regulation of mitochondrial efficiency, and indeed exercise efficiency, may occur independently of changes in gene transcription but with improvements in phosphorylation efficiency measured in rat liver mitochondria (Gnaiger et al. 2000) and human muscle mitochondria (Schiffer et al. 2014) when respiration was measured under acute hypoxic conditions. In contrast to these mitochondrial changes, however, in the same study of human subjects gross efficiency actually decreased with exercise in acute hypoxia (Schiffer et al. 2014), perhaps suggesting an influence from factors other than mitochondrial efficiency, such as different muscle fibre recruitment or the additional energetic cost of ventilation under hypoxic conditions.
Energetic and functional consequences of mitochondrial remodelling at extreme high altitude
The energetic and functional consequences of altitude‐induced mitochondrial remodelling are difficult to establish firmly. Using 31P‐MRS, we found altered skeletal muscle energetics in climbers within 48 h of returning to sea level from altitudes >7950 m, following a total of 10–17 days of descent. Changes included an increase in the free energy available from ATP hydrolysis, and lower resting ADP concentrations (Edwards et al. 2010). There was no change in resting PCr levels, nor PCr recovery half‐time (taken to represent mitochondrial function) following a plantar flexion exercise, but this re‐synthesis of PCr appeared to be more tightly regulated, with no overshoot in PCr production following exercise (Edwards et al. 2010). Overall, however, it is remarkable how well preserved energy metabolism was in these subjects throughout an exercise challenge, in the face of significant skeletal muscle atrophy (Edwards et al. 2010), loss of mitochondrial density (Levett et al. 2012) and downregulation of creatine kinase (Levett et al. 2015). Whilst enhanced glycolytic ATP production may contribute, muscle pH following exercise was no lower following altitude exposure (Edwards et al. 2010). Our findings may instead indicate that the loss of mitochondrial density in these subjects was within the excess capacity known to exist in human muscle at sea level (Boushel et al. 2011). Interestingly, many of the changes seen following exposure to extreme altitude were recapitulated in subjects ascending no higher than 5300 m and spending less than 3 weeks at altitude (Edwards et al. 2010), an ascent profile that one would not expect to have resulted in a loss of mitochondria. Such maintenance of energetic function would support a notion of acclimatisation, rather than detrimental deterioration.
In parallel experiments on some of the same climbers returning from the summit of Everest, a loss of cardiac energetic reserve (PCr/ATP) was also seen, in association with impaired left ventricular filling during diastole (Holloway et al. 2011). Whilst it would be tempting to view this as deterioration, a similar loss of PCr/ATP has also been seen in trekkers returning from a relatively short sojourn to 5300 m (Holloway et al. 2011, 2014). Meanwhile, the Sherpa heart was found to have a lower PCr/ATP than that of lowlanders, perhaps indicating that this lower energetic reserve results from a beneficial adaptation (Hochachka et al. 1996). For obvious practical reasons, it has not been possible to establish whether a loss of cardiac mitochondrial density or qualitative changes in cardiac mitochondrial function occur in humans at extreme altitude, but studies in rats have shown that 14 days of hypoxic exposure (at 11 or 13% O2) decreases cardiac ATP levels, fatty acid oxidation capacity and ETC complex I activity, with no loss in cardiac mitochondrial content over this time (Heather et al. 2012; Ashmore et al. 2014).
Conclusions
Prolonged exposure to extreme high altitude results in a loss of mitochondrial volume density in skeletal muscle, with a modulation in function of the remaining mitochondria characterised by a loss of oxidative capacity, particularly for fatty acid substrates, and a possible improvement in biochemical coupling at the inner mitochondrial membrane. Such qualitative changes in mitochondrial function are also seen at more moderate high altitudes, in the absence of changes in mitochondrial volume density. It is not clear whether such changes should be considered a feature of acclimatisation or deterioration, and indeed both aspects may be present, but the changes do appear to be regulated and similarities between the metabolic phenotype of the acclimatised lowlander and adapted Tibetan highlander (e.g. decreased mitochondrial density particularly from subsarcolemmal mitochondria, decreased fatty acid oxidation) add support to this notion. Whilst impaired cardiac energetics have been reported in climbers returning from extreme high altitude, it is notable that skeletal muscle energetics are largely preserved during an exercise challenge, despite significant muscle atrophy, further supporting the concept of mitochondrial remodelling as a core feature of acclimatisation to extreme high altitude.
Additional information
Competing Interests
The authors declare that they have no competing interests.
Author Contributions
Both authors conceived and planned the review. J.A.H. carried out a review of the literature. A.J.M. wrote the manuscript and generated the figures. Both authors reviewed the manuscript and edited it for content.
Funding
A.J.M. thanks the Research Councils UK for supporting his Academic Fellowship, and the British Heart Foundation, BBSRC, Action Medical Research, Isaac Newton Trust and Oroboros Instruments for supporting research in his laboratory. J.A.H. thanks the BBSRC for funding his PhD Studentship.
Acknowledgements
The authors wish to thank their colleagues and collaborators, especially members of the Caudwell Xtreme Everest Oxygen Research Consortium, and the subjects and investigators who took part in Caudwell Xtreme Everest (2007) and Xtreme Everest 2 (2013). Particular thanks are given to Professor Mike Grocott, Professor Hugh Montgomery and Professor Kieran Clarke for their long‐standing support of our research.
Biography
Andrew Murray is a University Lecturer in Physiology at the Department of Physiology, Development & Neuroscience, University of Cambridge. Prior to this he studied for his first degree in Biochemistry, and a DPhil in Physiology, at the University of Oxford, working with Professor Kieran Clarke and supported by the British Heart Foundation. Dr Murray is a co‐Principal Investigator of the Xtreme Everest Oxygen Research Consortium, and carried out studies of mitochondrial function at altitude during Caudwell Xtreme Everest 2007 and Xtreme Everest 2 in 2013. James Horscroft studied Natural Sciences at the University of Cambridge, and is currently a PhD candidate in Dr Murray's group, supported by a BBSRC‐CASE studentship. His thesis concerns the regulation of mitochondrial function, and particularly fatty acid oxidation, in hypoxia. James was an investigator on Xtreme Everest 2, and was part of a team that carried out high‐resolution respirometry at Everest Base Camp.

References
- Ashmore T, Fernandez BO, Branco‐Price C, West JA, Cowburn AS, Heather LC, Griffin JL, Johnson RS, Feelisch M & Murray AJ (2014). Dietary nitrate increases arginine availability and protects mitochondrial complex I and energetics in the hypoxic rat heart. J Physiol 592, 4715–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop CM, Spivey RJ, Hawkes LA, Batbayar N, Chua B, Frappell PB, Milsom WK, Natsagdorj T, Newman SH, Scott GR, Takekawa JY, Wikelski M & Butler PJ (2015). The roller coaster flight strategy of bar‐headed geese conserves energy during Himalayan migrations. Science 347, 250–254. [DOI] [PubMed] [Google Scholar]
- Boushel R, Gnaiger E, Calbet JA, Gonzalez‐Alonso J, Wright‐Paradis C, Sondergaard H, Ara I, Helge JW & Saltin B (2011). Muscle mitochondrial capacity exceeds maximal oxygen delivery in humans. Mitochondrion 11, 303–307. [DOI] [PubMed] [Google Scholar]
- Cano I, Mickael M, Gomez‐Cabrero D, Tegner J, Roca J & Wagner PD (2013). Importance of mitochondrial PO2 in maximal O2 transport and utilization: a theoretical analysis. Respir Physiol Neurobiol 189, 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano I, Roca J & Wagner PD (2015). Effects of lung ventilation‐perfusion and muscle metabolism‐perfusion heterogeneities on maximal O2 transport and utilization. J Physiol 593, 1841–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano I, Selivanov V, Gomez‐Cabrero D, Tegner J, Roca J, Wagner PD & Cascante M (2014). Oxygen pathway modeling estimates high reactive oxygen species production above the highest permanent human habitation. PLoS One 9, e111068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerretelli P (1976). Limiting factors to oxygen transport on Mount Everest. J Appl Physiol 40, 658–667. [DOI] [PubMed] [Google Scholar]
- Chance B (1957). Cellular oxygen requirements. Fed Proc 16, 671–680. [PubMed] [Google Scholar]
- Chance B, Cohen P, Jobsis F & Schoener B (1962). Intracellular oxidation‐reduction states in vivo . Science 137, 499–508. [DOI] [PubMed] [Google Scholar]
- Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM & Schumacker PT (2000). Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia‐inducible factor‐1α during hypoxia: a mechanism of O2 sensing. J Biol Chem 275, 25130–25138. [DOI] [PubMed] [Google Scholar]
- Clark RT, Criscuolo D & Coulson CK (1952). Effects of 20000 ft simulated altitude on myoglobin content of animals with and without exercise. Fed Proc 11, 25‐25. [Google Scholar]
- Colleoni F, Padmanabhan N, Yung HW, Watson ED, Cetin I, Tissot van Patot MC, Burton GJ & Murray AJ (2013). Suppression of mitochondrial electron transport chain function in the hypoxic human placenta: a role for miRNA‐210 and protein synthesis inhibition. PLoS One 8, e55194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplanches D, Amami M, Dupre‐Aucouturier S, Valdivieso P, Schmutz S, Mueller M, Hoppeler H, Kreis R & Fluck M (2014). Hypoxia refines plasticity of mitochondrial respiration to repeated muscle work. Eur J Appl Physiol 114, 405–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplanches D, Hoppeler H, Linossier MT, Denis C, Claassen H, Dormois D, Lacour JR & Geyssant A (1993). Effects of training in normoxia and normobaric hypoxia on human muscle ultrastructure. Pflugers Arch 425, 263–267. [DOI] [PubMed] [Google Scholar]
- Desplanches D, Hoppeler H, Tuscher L, Mayet MH, Spielvogel H, Ferretti G, Kayser B, Leuenberger M, Grunenfelder A & Favier R (1996). Muscle tissue adaptations of high‐altitude natives to training in chronic hypoxia or acute normoxia. J Appl Physiol (1985) 81, 1946–1951. [DOI] [PubMed] [Google Scholar]
- Edwards LM, Murray AJ, Tyler DJ, Kemp GJ, Holloway CJ, Robbins PA, Neubauer S, Levett D, Montgomery HE, Grocott MP, Clarke K & Caudwell Xtreme Everest Research Group (2010). The effect of high‐altitude on human skeletal muscle energetics: P‐MRS results from the Caudwell Xtreme Everest expedition. PLoS One 5, e10681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira R, Vitorino R, Alves RM, Appell HJ, Powers SK, Duarte JA & Amado F (2010). Subsarcolemmal and intermyofibrillar mitochondria proteome differences disclose functional specializations in skeletal muscle. Proteomics 10, 3142–3154. [DOI] [PubMed] [Google Scholar]
- Ferretti G (2003). Limiting factors to oxygen transport on Mount Everest 30 years after: a critique of Paolo Cerretelli's contribution to the study of altitude physiology. Eur J Appl Physiol 90, 344–350. [DOI] [PubMed] [Google Scholar]
- Gayeski TE & Honig CR (1986. a). O2 gradients from sarcolemma to cell interior in red muscle at maximal Am J Physiol 251, H789–799. [DOI] [PubMed] [Google Scholar]
- Gayeski TE & Honig CR (1986. b). Shallow intracellular O2 gradients and absence of perimitochondrial O2 "wells" in heavily working red muscle. Adv Exp Med Biol 200, 487–494. [DOI] [PubMed] [Google Scholar]
- Gilbert‐Kawai ET, Milledge JS, Grocott MP & Martin DS (2014). King of the mountains: Tibetan and Sherpa physiological adaptations for life at high altitude. Physiology 29, 388–402. [DOI] [PubMed] [Google Scholar]
- Gnaiger E, Lassnig B, Kuznetsov A, Rieger G & Margreiter R (1998). Mitochondrial oxygen affinity, respiratory flux control and excess capacity of cytochrome c oxidase. J Exp Biol 201, 1129–1139. [DOI] [PubMed] [Google Scholar]
- Gnaiger E, Mendez G & Hand SC (2000). High phosphorylation efficiency and depression of uncoupled respiration in mitochondria under hypoxia. Proc Natl Acad Sci USA 97, 11080–11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Cabrera MC, Domenech E, Romagnoli M, Arduini A, Borras C, Pallardo FV, Sastre J & Vina J (2008). Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training‐induced adaptations in endurance performance. Am J Clin Nutr 87, 142–149. [DOI] [PubMed] [Google Scholar]
- Gore CJ, Clark SA & Saunders PU (2007). Nonhematological mechanisms of improved sea‐level performance after hypoxic exposure. Med Sci Sports Exerc 39, 1600–1609. [DOI] [PubMed] [Google Scholar]
- Grassi B, Marzorati M, Kayser B, Bordini M, Colombini A, Conti M, Marconi C & Cerretelli P (1996). Peak blood lactate and blood lactate vs. workload during acclimatization to 5050 m and in deacclimatization. J Appl Physiol (1985) 80, 685–692. [DOI] [PubMed] [Google Scholar]
- Green HJ, Sutton JR, Cymerman A, Young PM & Houston CS (1989). Operation Everest II: adaptations in human skeletal muscle. J Appl Physiol (1985) 66, 2454–2461. [DOI] [PubMed] [Google Scholar]
- Grocott MP, Martin DS, Levett DZ, McMorrow R, Windsor J & Montgomery HE (2009). Arterial blood gases and oxygen content in climbers on Mount Everest. N Engl J Med 360, 140–149. [DOI] [PubMed] [Google Scholar]
- Guilland JC & Klepping J (1985). Nutritional alterations at high altitude in man. Eur J Appl Physiol Occup Physiol 54, 517–523. [DOI] [PubMed] [Google Scholar]
- Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U & Schumacker PT (2005). Mitochondrial complex III is required for hypoxia‐induced ROS production and cellular oxygen sensing. Cell Metab 1, 401–408. [DOI] [PubMed] [Google Scholar]
- Guzy RD & Schumacker PT (2006). Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp Physiol 91, 807–819. [DOI] [PubMed] [Google Scholar]
- Heather LC, Cole MA, Tan JJ, Ambrose LJ, Pope S, Abd‐Jamil AH, Carter EE, Dodd MS, Yeoh KK, Schofield CJ & Clarke K (2012). Metabolic adaptation to chronic hypoxia in cardiac mitochondria. Basic Res Cardiol 107, 268. [DOI] [PubMed] [Google Scholar]
- Hepple RT, Hogan MC, Stary C, Bebout DE, Mathieu‐Costello O & Wagner PD (2000). Structural basis of muscle O2 diffusing capacity: evidence from muscle function in situ. J Appl Physiol (1985) 88, 560–566. [DOI] [PubMed] [Google Scholar]
- Hochachka PW, Clark CM, Holden JE, Stanley C, Ugurbil K & Menon RS (1996). 31P magnetic resonance spectroscopy of the Sherpa heart: a phosphocreatine/adenosine triphosphate signature of metabolic defense against hypobaric hypoxia. Proc Natl Acad Sci USA 93, 1215–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochachka PW, Stanley C, Merkt J & Sumar‐Kalinowski J (1983). Metabolic meaning of elevated levels of oxidative enzymes in high altitude adapted animals: an interpretive hypothesis. Respir Physiol 52, 303–313. [DOI] [PubMed] [Google Scholar]
- Hogan MC, Roca J, Wagner PD & West JB (1988). Limitation of maximal O2 uptake and performance by acute hypoxia in dog muscle in situ. J Appl Physiol (1985) 65, 815–821. [DOI] [PubMed] [Google Scholar]
- Hollander JM, Thapa D & Shepherd DL (2014). Physiological and structural differences in spatially distinct subpopulations of cardiac mitochondria: influence of cardiac pathologies. Am J Physiol Heart Circ Physiol 307, H1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway CJ, Montgomery HE, Murray AJ, Cochlin LE, Codreanu I, Hopwood N, Johnson AW, Rider OJ, Levett DZ, Tyler DJ, Francis JM, Neubauer S, Grocott MP, Clarke K & Caudwell Xtreme Everest Research Group (2011). Cardiac response to hypobaric hypoxia: persistent changes in cardiac mass, function, and energy metabolism after a trek to Mt. Everest Base Camp. FASEB J 25, 792–796. [DOI] [PubMed] [Google Scholar]
- Holloway CJ, Murray AJ, Mitchell K, Martin DS, Johnson AW, Cochlin LE, Codreanu I, Dhillon S, Rodway GW, Ashmore T, Levett DZ, Neubauer S, Montgomery HE, Grocott MP, Clarke K & Caudwell Xtreme Everest Investigators (2014). Oral coenzyme Q10 supplementation does not prevent cardiac alterations during a high altitude trek to everest base cAMP. High Alt Med Biol 15, 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppeler H & Billeter R (1991). Conditions for oxygen and substrate transport in muscles in exercising mammals. J Exp Biol 160, 263–283. [DOI] [PubMed] [Google Scholar]
- Hoppeler H, Howald H & Cerretelli P (1990. a). Human muscle structure after exposure to extreme altitude. Experientia 46, 1185–1187. [DOI] [PubMed] [Google Scholar]
- Hoppeler H, Kleinert E, Schlegel C, Claassen H, Howald H, Kayar SR & Cerretelli P (1990. b). Morphological adaptations of human skeletal muscle to chronic hypoxia. Int J Sports Med 11 Suppl 1, S3–9. [DOI] [PubMed] [Google Scholar]
- Hoppeler H & Vogt M (2001). Muscle tissue adaptations to hypoxia. J Exp Biol 204, 3133–3139. [DOI] [PubMed] [Google Scholar]
- Horscroft JA & Murray AJ (2014). Skeletal muscle energy metabolism in environmental hypoxia: climbing towards consensus. Extreme Physiol Med 3, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houston CS, Sutton JR, Cymerman A & Reeves JT (1987). Operation Everest II: man at extreme altitude. J Appl Physiol (1985) 63, 877–882. [DOI] [PubMed] [Google Scholar]
- Howald H & Hoppeler H (2003). Performing at extreme altitude: muscle cellular and subcellular adaptations. Eur J Appl Physiol 90, 360–364. [DOI] [PubMed] [Google Scholar]
- Howald H, Pette D, Simoneau JA, Uber A, Hoppeler H & Cerretelli P (1990). Effect of chronic hypoxia on muscle enzyme activities. Int J Sports Med 11 Suppl 1, S10–14. [DOI] [PubMed] [Google Scholar]
- Hurtado A, Rotta A, Merino C & Pons J (1937). Studies of myohemoglobin at high altitudes. Am J Med Sci 194, 708–713. [Google Scholar]
- Jacobs RA, Boushel R, Wright‐Paradis C, Calbet JA, Robach P, Gnaiger E & Lundby C (2013). Mitochondrial function in human skeletal muscle following high‐altitude exposure. Exp Physiol 98, 245–255. [DOI] [PubMed] [Google Scholar]
- Jacobs RA, Siebenmann C, Hug M, Toigo M, Meinild AK & Lundby C (2012). Twenty‐eight days at 3454‐m altitude diminishes respiratory capacity but enhances efficiency in human skeletal muscle mitochondria. FASEB J 26, 5192–5200. [DOI] [PubMed] [Google Scholar]
- Kayser B, Hoppeler H, Claassen H & Cerretelli P (1991). Muscle structure and performance capacity of Himalayan Sherpas. J Appl Physiol (1985) 70, 1938–1942. [DOI] [PubMed] [Google Scholar]
- Kayser B, Hoppeler H, Desplanches D, Marconi C, Broers B & Cerretelli P (1996). Muscle ultrastructure and biochemistry of lowland Tibetans. J Appl Physiol (1985) 81, 419–425. [DOI] [PubMed] [Google Scholar]
- Kim JW, Tchernyshyov I, Semenza GL & Dang CV (2006). HIF‐1‐mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3, 177–185. [DOI] [PubMed] [Google Scholar]
- Koh MY & Powis G (2012). Passing the baton: the HIF switch. Trends Biochem Sci 37, 364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latshang TD, Turk AJ, Hess T, Schoch OD, Bosch MM, Barthelmes D, Merz TM, Hefti U, Hefti JP, Maggiorini M & Bloch KE (2013). Acclimatization improves submaximal exercise economy at 5533 m. Scand J Med Sci Sports 23, 458–467. [DOI] [PubMed] [Google Scholar]
- Levett DZ, Fernandez BO, Riley HL, Martin DS, Mitchell K, Leckstrom CA, Ince C, Whipp BJ, Mythen MG, Montgomery HE, Grocott MP, Feelisch M & Caudwell Extreme Everest Research Group (2011). The role of nitrogen oxides in human adaptation to hypoxia. Sci Rep 1, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levett DZ, Radford EJ, Menassa DA, Graber EF, Morash AJ, Hoppeler H, Clarke K, Martin DS, Ferguson‐Smith AC, Montgomery HE, Grocott MP, Murray AJ & Caudwell Xtreme Everest Research Group (2012). Acclimatization of skeletal muscle mitochondria to high‐altitude hypoxia during an ascent of Everest. FASEB J 26, 1431–1441. [DOI] [PubMed] [Google Scholar]
- Levett DZ, Vigano A, Capitanio D, Vasso M, De Palma S, Moriggi M, Martin DS, Murray AJ, Cerretelli P, Grocott MP & Gelfi C (2015). Changes in muscle proteomics in the course of the Caudwell Research Expedition to Mt. Everest. Proteomics 15, 160–171. [DOI] [PubMed] [Google Scholar]
- Lundby C, Calbet JA, Sander M, van Hall G, Mazzeo RS, Stray‐Gundersen J, Stager JM, Chapman RF, Saltin B & Levine BD (2007). Exercise economy does not change after acclimatization to moderate to very high altitude. Scand J Med Sci Sports 17, 281–291. [DOI] [PubMed] [Google Scholar]
- Lundby C, Saltin B & van Hall G (2000). The ‘lactate paradox’, evidence for a transient change in the course of acclimatization to severe hypoxia in lowlanders. Acta Physiol Scand 170, 265–269. [DOI] [PubMed] [Google Scholar]
- MacDougall JD, Green HJ, Sutton JR, Coates G, Cymerman A, Young P & Houston CS (1991). Operation Everest II: structural adaptations in skeletal muscle in response to extreme simulated altitude. Acta Physiol Scand 142, 421–427. [DOI] [PubMed] [Google Scholar]
- Martin DS, Ince C, Goedhart P, Levett DZ, Grocott MP & Caudwell Xtreme Everest Research Group (2009). Abnormal blood flow in the sublingual microcirculation at high altitude. Eur J Appl Physiol 106, 473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogensen M, Bagger M, Pedersen PK, Fernstrom M & Sahlin K (2006). Cycling efficiency in humans is related to low UCP3 content and to type I fibres but not to mitochondrial efficiency. J Physiol 571, 669–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray AJ (2009). Metabolic adaptation of skeletal muscle to high altitude hypoxia: how new technologies could resolve the controversies. Genome Med 1, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray AJ (2014). Mitochondria at the extremes: pioneers, protectorates, protagonists. Extreme Physiol Med 3, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray AJ & Montgomery HE (2014). How wasting is saving: weight loss at altitude might result from an evolutionary adaptation. BioEssays 36, 721–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narravula S & Colgan SP (2001). Hypoxia‐inducible factor 1‐mediated inhibition of peroxisome proliferator‐activated receptor alpha expression during hypoxia. J Immunol 166, 7543–7548. [DOI] [PubMed] [Google Scholar]
- O'Hagan KA, Cocchiglia S, Zhdanov AV, Tambuwala MM, Cummins EP, Monfared M, Agbor TA, Garvey JF, Papkovsky DB, Taylor CT & Allan BB (2009). PGC‐1α is coupled to HIF‐1α‐dependent gene expression by increasing mitochondrial oxygen consumption in skeletal muscle cells. Proc Natl Acad Sci USA 106, 2188–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer JW, Tandler B & Hoppel CL (1977). Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem 252, 8731–8739. [PubMed] [Google Scholar]
- Peacock AJ (1998). ABC of oxygen: oxygen at high altitude. BMJ 317, 1063–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippi M & Sillau AH (1994). Oxidative capacity distribution in skeletal muscle fibers of the rat. J Exp Biol 189, 1–11. [DOI] [PubMed] [Google Scholar]
- Picard M, Taivassalo T, Gouspillou G & Hepple RT (2011). Mitochondria: isolation, structure and function. J Physiol 589, 4413–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regula KM, Ens K & Kirshenbaum LA (2002). Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia‐mediated cell death of ventricular myocytes. Circ Res 91, 226–231. [DOI] [PubMed] [Google Scholar]
- Reynafarje B (1962). Myoglobin content and enzymatic activity of muscle and altitude adaptation. J Appl Physiol 17, 301–305. [DOI] [PubMed] [Google Scholar]
- Robach P, Cairo G, Gelfi C, Bernuzzi F, Pilegaard H, Vigano A, Santambrogio P, Cerretelli P, Calbet JA, Moutereau S & Lundby C (2007). Strong iron demand during hypoxia‐induced erythropoiesis is associated with down‐regulation of iron‐related proteins and myoglobin in human skeletal muscle. Blood 109, 4724–4731. [DOI] [PubMed] [Google Scholar]
- Rose MS, Houston CS, Fulco CS, Coates G, Sutton JR & Cymerman A (1988). Operation Everest. II: Nutrition and body composition. J Appl Physiol (1985) 65, 2545–2551. [DOI] [PubMed] [Google Scholar]
- Schiffer TA, Ekblom B, Lundberg JO, Weitzberg E & Larsen FJ (2014). Dynamic regulation of metabolic efficiency explains tolerance to acute hypoxia in humans. FASEB J 28, 4303–4311. [DOI] [PubMed] [Google Scholar]
- Scott GR (2011). Elevated performance: the unique physiology of birds that fly at high altitudes. J Exp Biol 214, 2455–2462. [DOI] [PubMed] [Google Scholar]
- Scott GR, Richards JG & Milsom WK (2009). Control of respiration in flight muscle from the high‐altitude bar‐headed goose and low‐altitude birds. Am J Physiol Regul Integ Comp Physiol 297, R1066–1074. [DOI] [PubMed] [Google Scholar]
- Semenza GL (2007). Hypoxia‐inducible factor 1 (HIF‐1) pathway. Science's STKE 2007, cm8. [DOI] [PubMed] [Google Scholar]
- Semenza GL, Roth PH, Fang HM & Wang GL (1994). Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia‐inducible factor 1. J Biol Chem 269, 23757–23763. [PubMed] [Google Scholar]
- Sutton JR, Reeves JT, Wagner PD, Groves BM, Cymerman A, Malconian MK, Rock PB, Young PM, Walter SD & Houston CS (1988). Operation Everest II: oxygen transport during exercise at extreme simulated altitude. J Appl Physiol (1985) 64, 1309–1321. [DOI] [PubMed] [Google Scholar]
- Tsutsui H (2006). Mitochondrial oxidative stress and heart failure. Intern Med 45, 809–813. [DOI] [PubMed] [Google Scholar]
- van Hall G, Calbet JA, Sondergaard H & Saltin B (2001). The re‐establishment of the normal blood lactate response to exercise in humans after prolonged acclimatization to altitude. J Physiol 536, 963–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hall G, Lundby C, Araoz M, Calbet JA, Sander M & Saltin B (2009). The lactate paradox revisited in lowlanders during acclimatization to 4100 m and in high‐altitude natives. J Physiol 587, 1117–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan BE & Pace N (1956). Changes in myoglobin content of the high altitude acclimatized rat. Am J Physiol 185, 549–556. [DOI] [PubMed] [Google Scholar]
- Vigano A, Ripamonti M, De Palma S, Capitanio D, Vasso M, Wait R, Lundby C, Cerretelli P & Gelfi C (2008). Proteins modulation in human skeletal muscle in the early phase of adaptation to hypobaric hypoxia. Proteomics 8, 4668–4679. [DOI] [PubMed] [Google Scholar]
- Wagner PD (1996). Determinants of maximal oxygen transport and utilization. Ann Rev Physiol 58, 21–50. [DOI] [PubMed] [Google Scholar]
- Wagner PD (2000). Diffusive resistance to O2 transport in muscle. Acta Physiol Scand 168, 609–614. [DOI] [PubMed] [Google Scholar]
- Wagner PD (2010). The physiological basis of reduced in Operation Everest II. High Alt Med Biol 11, 209–215. [DOI] [PubMed] [Google Scholar]
- Ward M (1954). High altitude deterioration. Proc R Soc Lond B 143, 40–42. [DOI] [PubMed] [Google Scholar]
- Weibel ER, Taylor CR & Hoppeler H (1991). The concept of symmorphosis: a testable hypothesis of structure‐function relationship. Proc Natl Acad Sci USA 88, 10357–10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JB (1986). Lactate during exercise at extreme altitude. Fed Proc 45, 2953–2957. [PubMed] [Google Scholar]
- West JB, Hackett PH, Maret KH, Milledge JS, Peters RM, Jr , Pizzo CJ & Winslow RM (1983). Pulmonary gas exchange on the summit of Mount Everest. J Appl Physiol 55, R678–687. [DOI] [PubMed] [Google Scholar]
- West JB, Schoene RB & Milledge JS (2007). High Altitude Medicine And Physiology. Hodder Arnold, London. [Google Scholar]
- Westerterp KR & Kayser B (2006). Body mass regulation at altitude. Eur J Gastroenterol Hepatol 18, 1–3. [DOI] [PubMed] [Google Scholar]
- Zhang H, Bosch‐Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ & Semenza GL (2008). Mitochondrial autophagy is an HIF‐1‐dependent adaptive metabolic response to hypoxia. J Biol Chem 283, 10892–10903. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, Dang CV & Semenza GL (2007). HIF‐1 inhibits mitochondrial biogenesis and cellular respiration in VHL‐deficient renal cell carcinoma by repression of C‐MYC activity. Cancer Cell 11, 407–420. [DOI] [PubMed] [Google Scholar]