

Key points
Ablation of hypoxia signalling leads to respiratory distress syndrome (RDS). Administering vascular endothelial growth factor (VEGF) protects from RDS.
Reduced surfactant maturation in the chronically hypoxaemic placentally restricted (PR) fetus is associated with altered regulation of hypoxia signalling and may predispose to RDS.
We determined the effect of intratracheal VEGF administration on the expression of genes regulating vascularization, alveolarization, proliferation, inflammation, surfactant maturation and structural markers of lung maturation in the normally grown and PR fetus.
Although there were relatively few effects of VEGF on gene expression, there were positive effects on structural maturation in the normally grown and PR lung. There was no effect on fetal blood pressure or fetal breathing movements.
We have provided evidence that VEGF promotes structural fetal lung maturation and may result in synergistic effects if combined with current therapeutic treatments aiming to induce surfactant maturation and reduce the risk of RDS.
Abstract
Inhibition of hypoxia signalling leads to respiratory distress syndrome (RDS), whereas administration of vascular endothelial growth factor (VEGF), the most widely characterized hypoxia responsive factor, protects from RDS. In the lung of the chronically hypoxaemic placentally restricted (PR) fetus, there is altered regulation of hypoxia signalling. This leads to reduced surfactant maturation in late gestation and provides evidence for the increased risk of RDS in growth restricted neonates at birth. We evaluated the effect of recombinant human VEGF administration with respect to bypassing the endogenous regulation of hypoxia signalling in the lung of the normally grown and PR sheep fetus. There was no effect of VEGF administration on fetal blood pressure or fetal breathing movements. We examined the effect on the expression of genes regulating VEGF signalling (FLT1 and KDR), angiogenesis (ANGPT1, AQP1, ADM), alveolarization (MMP2, MMP9, TIMP1, COL1A1, ELN), proliferation (IGF1, IGF2, IGF1R, MKI67, PCNA), inflammation (CCL2, CCL4, IL1B, TNFA, TGFB1, IL10) and surfactant maturation (SFTP‐A, SFTP‐B, SFTP‐C, SFTP‐D, PCYT1A, LPCAT, LAMP3, ABCA3). Despite the effects of PR on the expression of genes regulating airway remodelling, inflammatory signalling and surfactant maturation, there were very few effects of VEGF administration on gene expression in the lung of both the normally grown and PR fetus. There were, however, positive effects of VEGF administration on percentage tissue, air space and numerical density of SFTP‐B positive alveolar epithelial cells in fetal lung tissue. These results provide evidence for the stimulatory effects of VEGF administration on structural maturation in the lung of both the normally grown and PR fetus.
Key points
Ablation of hypoxia signalling leads to respiratory distress syndrome (RDS). Administering vascular endothelial growth factor (VEGF) protects from RDS.
Reduced surfactant maturation in the chronically hypoxaemic placentally restricted (PR) fetus is associated with altered regulation of hypoxia signalling and may predispose to RDS.
We determined the effect of intratracheal VEGF administration on the expression of genes regulating vascularization, alveolarization, proliferation, inflammation, surfactant maturation and structural markers of lung maturation in the normally grown and PR fetus.
Although there were relatively few effects of VEGF on gene expression, there were positive effects on structural maturation in the normally grown and PR lung. There was no effect on fetal blood pressure or fetal breathing movements.
We have provided evidence that VEGF promotes structural fetal lung maturation and may result in synergistic effects if combined with current therapeutic treatments aiming to induce surfactant maturation and reduce the risk of RDS.
Abbreviations
- ACTB
β‐actin
- AEC
alveolar epithelial cell
- ADM
adrenomedullin
- ANGPT1
angiopoietin 1
- AQP
aquaporin
- BP
blood pressure
- CCL2
chemokine (C‐C motif) ligand 2
- CCL4
chemokine (C‐C motif) ligand 4
- COL1A1
collagen type I α1
- DBP
diastolic blood pressure
- ELN
elastin
- FBM
fetal breathing movements
- FLT1
fms‐related tyrosine kinase 1 (VEGF‐R1)
- GC
glucocorticoid
- Hct
haematocrit
- Hb
haemoglobin
- HR
heart rate
- IL1B
interleukin 1β
- IGF
insulin‐like growth factor
- IGF1R
insulin‐like growth factor receptor 1
- IL10
interleukin 10
- IUGR
intrauterine growth restriction
- KDR
kinase insert domain receptor (VEGF‐R2)
- LAMP3
lysosome‐associated membrane glycoprotein 3
- LPCAT
lysophosphatidylcholine acyltransferase 1
- MAP
mean arterial pressure
- MKI67
marker of proliferation KI‐67
- MNE
mean normalized expression
- MMP
matrix metallopeptidase
partial pressure of carbon dioxide
partial pressure of oxygen
- PCNA
proliferating cell nuclear antigen
- PCYT1A
phosphate cytidylyl transferase 1 choline α
- PPIA
peptidylprolyl isomerase
- PR
placental restriction
- qRT‐PCR
quantitative real time RT‐PCR
- RDS
respiratory distress syndrome
oxygen saturation
- SBP
systolic blood pressure
- SFTP
surfactant protein
- TGFB1
transforming growth factor β1
- TIMP
tissue inhibitor of matrix metallopeptidase
- TNF
tumour necrosis factor
- VEGF
vascular endothelial growth factor
- YWHAZ
tyrosine 3‐monooxygenase
Introduction
By contrast to the newborn and adult lung, which both function in the air‐breathing environment, fetal lung development occurs in an hypoxic environment (Gagnon, 2006). Hypoxia signalling is essential for normal vasculature and organ development in utero. The most widely characterized hypoxia responsive factor is vascular endothelial growth factor (VEGF), which is essential for embryonic vascular development, as demonstrated by lethality in the embryonic period after single allele VEGF deletion in mice (Carmeliet et al. 1996; Ferrara et al. 1996). During fetal lung development, VEGF is essential for normal airway branching, alveolarization and surfactant maturation (Jakkula et al. 2000; Brown et al. 2001). Inhibition of hypoxia signalling by hypoxia inducible factor (HIF)‐2α subunit knockout decreases VEGF expression (Compernolle et al. 2002), leading to respiratory distress syndrome (RDS). However, intra‐amniotic and postnatal intratracheal administration of VEGF protects newborn mice from RDS (Compernolle et al. 2002).
RDS is characterized by alveolar collapse resulting from reduced pulmonary surfactant, a complex mixture of lipids and proteins (SFTP‐A, ‐B, ‐C and ‐D), which functions to prevent the adherence of adjacent epithelial surfaces at low lung volumes (Avery & Mead, 1959; Creuwels et al. 1997; Pérez‐Gil, 2008). Altered VEGF homeostasis changes the regulation of both lipid and protein constituents of surfactant (Brown et al. 2001; Compernolle et al. 2002; Chen & Wang, 2007). Furthermore, VEGF is expressed highly in type II alveolar epithelial cells (AEC), the site of synthesis, storage and secretion of surfactant (Ng et al. 2001; Raoul et al. 2004; Breen et al. 2013). Hence, endogenous VEGF is an important molecule for normal respiratory development as a result of promoting epithelial proliferation in addition to surfactant maturation.
In the developing fetus, VEGF expression normally increases in late gestation and this expression declines significantly in the week after birth (Grover et al. 2003). VEGF is essential for normal structural lung development and is most highly expressed in the distal lung epithelium (Berse et al. 1992; Monacci et al. 1993; Shifren et al. 1994; Brown et al. 2001). In late gestation, when the lung surface area increases as a result of the rapid formation of alveoli, there is a concomitant rapid expansion of pulmonary vascular beds in close co‐ordination with alveolarization (Jakkula et al. 2000). This pulmonary alveolarization and vascularization maximizes the ability for gas exchange at the air‐liquid interface upon exposure to the air‐breathing environment at birth (Meyrick & Reid, 1982; Blanco et al. 1989; Burri, 2006). In addition to the regulation of structural lung maturation, VEGF is a local immunoregulator and induces the expression of the chemokines, cytokines and inflammatory mediators necessary for host pulmonary immune responses in vivo (Sow et al. 2009; Breen et al. 2013). This regulation by VEGF is essential for ensuring the successful transition to air‐breathing in extrauterine life and preventing pulmonary disease.
Disturbances of hypoxia signalling leading to downstream suppression of VEGF expression and further downstream signal transduction in utero may result in respiratory complications at birth (Lassus et al. 2001; Thébaud et al. 2005). These changes may result from exposure to an abnormal timing, as well as duration and severity, of hypoxic insults, which can be commonly associated with intrauterine growth restriction (IUGR) (Morrison, 2008). IUGR is associated with altered lung maturation, which may increase the risk of experiencing respiratory complications in both human and animal studies (Gagnon et al. 1999; McIntire et al. 1999; Bernstein et al. 2000; Maritz et al. 2001). In the fetal sheep, IUGR induced by placental restriction (PR) results in exposure to chronic hypoxaemia, increased expression of the prolyl hydroxylase domain (PHD) family of enzymes and reduced surfactant maturation in the lung (Orgeig et al. 2010; Orgeig et al. 2015). The PHDs normally function to limit hypoxia signalling by degradation of the HIF‐α subunit during periods of normoxaemia, although their expression has been shown to increase during periods of chronic hypoxaemia (Epstein et al. 2001; Ginouvès et al. 2008; Orgeig et al. 2015). Thus, altered regulation of hypoxia signalling by increased PHDs, despite the chronically hypoxaemic conditions experienced by the PR fetus, presents a potential mechanism leading to delayed surfactant maturation and an increased risk of RDS (Avery & Mead, 1959; Orgeig et al. 2010; Orgeig et al. 2015). In the present study, we have examined the effect of recombinant human VEGF administration, a key hypoxia signalling factor, with respect to bypassing the endogenous regulation of hypoxia signalling (e.g. altered regulation of hypoxia signalling by increased PHD expression in the PR fetus), on the molecular, structural and functional determinants of maturation in the lung of both the normally grown and PR fetus.
Methods
All experiments were approved by the University of South Australia Animal Ethics Committee and were performed in accordance with the guidelines of the Australian code of practice for the care and use of animals for scientific purposes. The experiments comply with the policies and regulations of The Journal of Physiology (Drummond, 2009).
Animals and surgery
Placental restriction
The uterus was incised and the majority of the endometrial caruncles (i.e. sites of placentation) were removed from the uterus in 13 randomly selected Merino ewes before conception, as described previously (Alexander, 1964; Robinson et al. 1979; Orgeig et al. 2010). The recovery of ewes from surgery was observed for 4–7 days. After a minimum of 10 weeks of recovery, these 13 ewes, in addition to 20 Control ewes, entered a mating programme.
Fetal surgery
From 119–120 days, the 33 pregnant ewes (Control, n = 20; PR, n = 13) were housed in individual pens in animal holding rooms, under a 12:12h light/dark cycle, and fed once daily with water ad libitum. At 123–124 days of gestation, general anaesthesia was induced in the ewe with i.v. diazepam (0.3 mg kg–1) and ketamine (5 mg kg–1) and maintained with 1–2% isoflurane in oxygen (Lyppards, Beverley, Australia). Vascular catheters were implanted in the ewe's jugular vein, a carotid artery and jugular vein of the fetus, and in the amniotic cavity, as described previously (Danielson et al. 2005). A non‐obstructive catheter was implanted anteriorly in the fetal trachea and did not extend past the carina to allow for administration of drug (saline or VEGF) into the lungs without preventing fetal breathing movements (FBM) (Morrison et al. 2001). Ewes received an i.m. injection of antibiotics (3.5 ml of norocillin; 150 mg ml–1 procaine penicillin and 112.5 mg ml–1 benzathine penicillin; Norbrook Laboratories Ltd, Gisborne, Australia) and 2 ml of 125 mg ml–1 dihydrostreptomycin in sterile saline (Lyppards) for 3 days after surgery. Antibiotics (500 mg; sodium ampicillin; Commonwealth Serum Laboratories, Melbourne, Australia) were administered intra‐amniotically to all fetal sheep daily for 4 days postoperatively. Ewes were allowed at least 4 days to recover from surgery prior to the experimental protocol.
Arterial blood gas measurements
Fetal arterial blood was collected daily after surgery to monitor fetal well‐being. Measurement of whole blood arterial partial pressure of oxygen (), arterial partial pressure of carbon dioxide (), pH, oxygen saturation (), haematocrit (Hct) and haemoglobin (Hb) content were measured using an ABL 80 FLEX (Radiometer, Copenhagen, Denmark) with the temperature corrected to 39 °C. All PR fetuses had a <17 mmHg. All Control fetuses had a mean gestational >17 mmHg (Butler et al. 2002; Danielson et al. 2005; Morrison, 2008).
Intratracheal infusion regimen
To avoid systemic effects and to restrict the effects of VEGF to the fetal lung, an intratracheal route of administration was chosen. The VEGF dose administered per day in the present study was based on those previously achieving significant effects on inflammatory regulation (Sow et al. 2009), pulmonary haemodynamics and structural lung maturation in a sheep model of persistent pulmonary hypertension (Grover et al. 2005), as well as hyperoxic lung injury in neonatal rats (Kunig et al. 2005). On 131 or 132 days of gestation, Control and PR ewes were randomly assigned to one of two infusion regimens, resulting in four treatment groups: Control (n = 11), Control + VEGF (n = 9), PR (n = 8) and PR + VEGF (n = 5). Human VEGF165 and sheep VEGF164 are 93.3 % identical at the amino acid level and intratracheal administration in newborn lambs induced changes in the regulation of inflammatory mediators in the lung (Sow et al. 2009). In the present study a 2.5 ml bolus dose of 50 μg of recombinant human VEGF (Life Technologies, Grand Island, NY, USA), diluted as recommended in 0.1% BSA (Sigma‐Aldrich, St Louis, MO, USA), and 1 × phosphate‐buffered saline (Sigma‐Aldrich) were administered intratracheally to the fetus on two occasions, 24 h apart, in the Control + VEGF (n = 9) and PR + VEGF (n = 5) groups. The Control (n = 11) and PR (n = 8) groups received a 2.5 ml saline bolus administered intratracheally to the fetus. Before administration of the bolus dose, some tracheal fluid was withdrawn into the catheter to ensure that the catheter was patent. Subsequent to the bolus infusion, the dead space of the catheter was cleared with infusion of 3 ml saline.
Determination of fetal lung liquid recombinant human VEGF concentration
For validation of the VEGF infusion regimen, fetal lung liquid fluid samples (n = 3) were obtained from the tracheal catheter 24 h after each infusion (+24 h and +48 h after the first infusion) and run in duplicate for determination of VEGF concentration using a commercially available solid phase sandwich colorimetric recombinant human VEGF enzyme‐linked immunosorbent assay kit (KHG0111; Life Technologies) in accordance with the manufacturer's instructions, as described previously (Kobata et al. 2008). The kit was used to detect the level of recombinant human VEGF present in fetal lung liquid as a result of intratracheal administration and does not detect endogenous sheep VEGF in lung liquid as indicated by levels less than the lower limit of detection (<5 pg ml–1) in undiluted samples of lung liquid obtained prior to the first VEGF infusion on day 1. Samples of lung liquid obtained after intratracheal VEGF administration were diluted to 1:50 with diluent buffer and run in duplicate to obtain a value within the limits of detection for the assay (<1500 pg ml–1). There was a total concentration of 0.020 ± 0.010 and 0.016 ± 0.011 μg ml–1 of recombinant human VEGF present in fetal lung liquid at +24 h and +48 h after the first infusion, respectively.
Physiological measurements
Fetal carotid artery, tracheal and amniotic catheters were connected to displacement transducers and a quad‐bridge amplifier (ADInstruments, Bella Vista, Australia) to record fetal blood pressure (BP), FBM and amniotic pressure, as described previously (Danielson et al. 2005; Poudel et al. 2015). All data were sampled at a rate of 400 Hz, digitized and recorded using LabChart 7 (ADInstruments). Fetal arterial BP, amniotic pressure and FBM were recorded from the day before infusion (130 or 131 days) until post mortem.
Post mortem tissue collection
On the day after the second fetal intratracheal infusion, ewes were humanely killed with an overdose of sodium pentobarbitone administered via the jugular vein (Virbac Pty Ltd, Peakhurst, Australia) and fetuses were delivered by hysterotomy. Fetal body and organ weights, crown rump length and abdominal circumference were recorded. Fetal lungs were then removed and dissected to remove surrounding connective tissue and fat before total lung weight was recorded. A section of lung tissue was snap frozen in liquid nitrogen and stored at ‐80°C for further molecular analysis: Control, n = 11 [6 female (F) + 5 male (M)]; Control + VEGF, n = 9 (3F + 6M); PR, n = 8 (4F + 4M); PR + VEGF, n = 5 (2F + 3M).
The upper left lung lobe was isolated from the lower left lung lobe with a suture to create a seal and then instillation fixed via cannulation of the left primary bronchus with 4 % paraformaldehyde (Jomar Bioscience, Welland, Australia) + 0.2 % gluteraldehyde (ProSciTech, Townsville, Australia) in 0.1 m phosphate buffer (VWR, Radnor, PA, USA) and distended at a constant pressure of 20 cm H2O. After 24 h in fixative, the lung lobe was removed and a slice of lung tissue was isolated before being processed and embedded in paraffin for further histological and immunohistochemical analysis: Control, n = 9 (5F + 4M); Control + VEGF, n = 7 (3F + 4M); PR, n = 7 (3F + 4M); PR + VEGF, n = 5 (2F + 3M).
Fetal BP and FBM analysis
Fetal systolic (SBP) and diastolic (DBP) blood pressure were calculated as the maximum and minimum pressure of the fetal arterial pressure, respectively, after subtraction of the intra‐amniotic pressure in LabChart 7 (ADInstruments). Fetal mean arterial pressure (MAP) was derived using the formula DBP +[ 1/3(SBP – DBP)] in LabChart 7 (Danielson et al. 2005; Dyer et al. 2009; Poudel et al. 2015). Heart rate (HR) was derived from the fetal arterial pressure signal using analysis tools in LabChart 7. Baseline fetal BP measures were determined as a mean of a 5 h period of recording on the day before infusion. On each day of infusion, the change (Δ) in fetal MAP and HR was measured for five consecutive 1 h epochs after infusion relative to the 30 min baseline recording before the infusion for each animal on each day (Control, n = 5; Control + VEGF, n = 6; PR, n = 4; PR + VEGF, n = 4). After subtraction of the intra‐amniotic pressure from the tracheal pressure recordings, the incidence, amplitude and frequency of FBM was determined (Control, n = 4; Control + VEGF, n = 6; PR, n = 4; PR + VEGF, n = 3). Repeated negative deflections in tracheal pressure >2 mmHg for longer than 30 s were classified as FBM (Bocking et al. 1988; Morrison et al. 1997). Baseline FBM measurements [incidence (%), amplitude (mmHg) and frequency (beats min–1)] were determined as the mean of five consecutive 1 h epochs (beginning between 09.00 h and 10.00 h) of recording on the day before infusion. On each day of infusion, FBM measurements were determined as the mean change for five consecutive 1 h epochs of recording after infusion on each day relative to the day before infusion (Bennet et al. 1990; Giussani et al. 1995; Morrison et al. 1997; Poudel et al. 2015).
Quantification of mRNA transcripts within the fetal lung
Total RNA extraction
All essential information regarding our procedure is included in accordance with the MIQE guidelines (Bustin et al. 2009). Total RNA was extracted from fetal lung samples (∼50 mg; Control, n = 11; Control + VEGF, n = 9; PR, n = 8; PR + VEGF, n = 5) using QIAzol Lysis Reagent Solution and Qiagen miRNeasy purification columns in accordance with the manufacturer's instructions (Qiagen, Victoria, Australia). Total RNA was quantified by spectrophotometric measurements at 260 and 280 nm using an Eppendorf BioPhotometer (Crown Scientific, Minto, Australia) and the 260/280 nm ratio results were checked for protein and DNA contamination. cDNA was synthesized using the Superscript III First Strand Synthesis System (Invitrogen, Carlsbad, CA, USA) with 2 μg of total RNA, random hexamers, dNTP, DTT and Superscript III in a final volume of 20 μl in a MJ Mini personal thermocycler (Bio‐Rad, Hercules, CA, USA). Controls containing either no RNA transcript or no Superscript III were used to test for reagent contamination and genomic DNA contamination, respectively (Gentili et al. 2009; McGillick et al. 2013).
Real‐time Quantitative RT‐PCR (qRT‐PCR)
The geNorm component of qbaseplus, version 2.0 (Biogazelle, Zwijnaarde, Belgium) was used to determine the most stable reference genes from a panel of candidate reference genes (Vandesompele et al. 2002) and the minimum number of reference genes required to calculate a stable normalization factor, as described previously (Soo et al. 2012; McGillick et al. 2013). For qRT‐PCR data output normalization, three stable reference genes, β‐actin (ACTB; U39357), peptidylprolyl isomerase A (PPIA; AY251270) (Passmore et al. 2009) and tyrosine 3‐monooxygenase (YWHAZ; AY970970) (McGillick et al. 2013), were run in parallel with target genes, as described previously (Soo et al. 2012; McGillick et al. 2013). Previously published or specifically designed (Table 1) primer sets were validated and optimized as described previously (Orgeig et al. 2010; McGillick et al. 2013). The expression of genes regulating VEGF signalling [Fms‐related tyrosine kinase 1 (VEGF‐R1), FLT1, NM_001191132.2; kinase insert domain receptor (VEGF‐R2), KDR, AF513909.1 (Botting et al. 2014)], angiogenesis [angiopoietin, ANGPT1, NM_001076797.1; aquaporin, AQP1, NM_001009194.1 (McGillick et al. 2013); adrenomedullin, ADM, NM_173888.3 (Botting et al. 2014)] and alveolarization [matrix metallopeptidase (MMP) 2, MMP2, NM_001166180.1; MMP9, FJ185130; tissue inhibitor of matrix metallopeptidase‐1, TIMP1, NM_001009319.2; collagen 1, COL1A1, FJ200442; elastin, ELN, DQ239624.1], in addition to genes regulating cellular proliferation [insulin like growth factor (IGF) 1, IGF1, XM_001251168.1; IGF2, NM_174087.3; IGF1 receptor, IGF1R, XM_606794.3 (Zhang et al. 2010); antigen KI‐67, MKI67, XM_004020367.1; proliferating cell nuclear antigen, PCNA, XM_004014340.1 (Orgeig et al. 2015)] were determined. Furthermore, the expression of chemokines [chemokine (C‐C motif) ligand 2, CCL2, NM_174006; chemokine (C‐C motif) ligand 4, CCL4, NM_001075147], pro‐inflammatory markers [interleukin‐1β, IL1B, NM_001009465.2 (Orgeig et al. 2015); tumour necrosis factor α, TNFA, X55152], anti‐inflammatory markers [transforming growth factor β, TGFB1, NM_001009400 (Orgeig et al. 2015); interleukin‐10, IL10, NM_001009327] and markers of surfactant maturation [SFTP‐A, AF211856; SFTP‐B, AF07544; SFTP‐C, AF076634; SFTP‐D, AJ133002 (Orgeig et al. 2010; McGillick et al. 2013); phosphate cytidylyl transferase 1, choline, α, PCYT1A, XM_004003005.1; lysophosphatidylcholine acyltransferase 1, LPCAT, XM_004017168.1; lysosomal‐associated membrane protein 3, LAMP3, XM_004003109.1 (Orgeig et al. 2015); ATP‐binding cassette, sub‐family A (ABC1), member 3, ABCA3, XM_004021123.1] were measured by qRT‐PCR using Fast SYBR® Green Master Mix (Applied Biosystems, Foster City, CA, USA) in a final volume of 6 μl on a ViiA7 Fast Real‐time PCR system (Applied Biosystems), as described previously (McGillick et al. 2013). Each qRT‐PCR well contained 3 μl of Fast SYBR Green Master Mix (2X), 2 μl of forward and reverse primer mixed with H2O to obtain final primer concentrations and 1 μl of diluted relevant cDNA. Each sample was run in triplicate for target genes and reference genes. The abundance of each transcript relative to the abundance of stable reference genes (Hellemans et al. 2007) was calculated using DataAssist, version 3.0 (Applied Biosystems) and is expressed as mRNA mean normalized expression (MNE) ± SEM (Soo et al. 2012; McGillick et al. 2013). Outliers were defined as values that were ± 2 SD from the mean for each treatment group and were removed from the analysis, and, consequently, sample sizes were not identical for all measurements in some cases. The specific number (n) for each individual parameter is also reported as appropriate.
Table 1.
qRT‐PCR primer sequences for designed target genes
| Primer name | Sequence 5’‐ to 3’ | Primer concentration (μm) | Accession number |
|---|---|---|---|
| ANGPT1 | AY881028.1 | ||
| Forward | TTGCCATAACCAGTCAGAG | 0.45 | |
| Reverse | AACCACCAGCCTCCTGTTA | 0.45 | |
| MMP2 | NM_001166180.1 | ||
| Forward | CTGATGGCGCCCATTTATAC | 0.45 | |
| Reverse | GATGAACCGGTCCTTGAAGA | 0.45 | |
| MMP9 | FJ185130 | ||
| Forward | TTTCCTCCTGGCTCAGGCATTCA | 0.45 | |
| Reverse | TTTCCGAAGTAGGTCGGGATCACA | 0.45 | |
| TIMP1 | NM_001009319.2 | ||
| Forward | ACTCCGAAGTCGTCATCAGG | 0.45 | |
| Reverse | CCAGCAGCATAGGTCTTGGT | 0.45 | |
| COL1A1 | FJ200442 | ||
| Forward | TTCACCTACAGCGTCACCTACGAT | 0.45 | |
| Reverse | ATGTCGAAGCCGAATTCCTGGTCT | 0.45 | |
| ELN | DQ239624.1 | ||
| Forward | TTGGGTCACACCATCTCTTG | 0.9 | |
| Reverse | AAGAGGGCTGTTCCCAATAC | 0.9 | |
| CCL2 | NM_174006 | ||
| Forward | AGTCACCAGCAGCAAGTGTCCTAA | 0.05 | |
| Reverse | GTGCTCAAGGCTTTGGAGTTTGGT | 0.05 | |
| CCL4 | NM_001075147 | ||
| Forward | TGCCAAGATCATGAAGCTCTGCGT | 0.45 | |
| Reverse | GCTGCTGGTCTCGAAGTAGTCATT | 0.45 | |
| TNFA | X55152 | ||
| Forward | ACACCATGAGCACCAAAAGC | 0.45 | |
| Reverse | AGGCACCAGCAACTTCTGGA | 0.45 | |
| IL10 | NM_001009327 | ||
| Forward | GTTGCCAAGCCTTGTCGGAAATGA | 0.45 | |
| Reverse | AGTTCACGTGCTCCTTGATGTCAG | 0.45 | |
| PCYT1A | XM_004003005.1 | ||
| Forward | GGGCAACAGAAGAAGATGGA | 0.45 | |
| Reverse | ACCCTGACATAGGGCTTACTA | 0.45 | |
| LPCAT | XM_004017168.1 | ||
| Forward | CAGAAGGCACGTGTACCAATAG | 0.9 | |
| Reverse | CGTGATGGTGTCCAGTTTGT | 0.9 | |
| ABCA3 | XM_004021123.1 | ||
| Forward | CCCTTACCCACCTTTCATCTC | 0.45 | |
| Reverse | CCTTCAGCTTCTTCTCCTTCTC | 0.45 |
Accession numbers refer to the published cDNA sequences from which the primer sequences were designed.
Determination of percent area occupied by tissue and air space in the fetal lung
In a subset of fetuses, fixed lung tissue sections of 5 μm thickness were baked at 60 °C for 1 h, deparaffinized and rehydrated (Control, n = 9; Control + VEGF, n = 7; PR, n = 7; PR + VEGF, n = 3). Subsequent to rehydration, progressive haematoxylin and eosin stains were carried out on all fetal lung tissue sections. Slides were visualized and photographed using AnalySIS Build 1243 software linked to a digital camera DP72 (Olympus Australia Pty Ltd, Notting Hill, Australia), which was connected to a BX53 Research Microscope (Olympus Australia Pty Ltd). All counting was undertaken by a single trained observer who was blinded to the experimental groups. An overlay grid counting frame with 375 evenly distributed points (1 cm2) was applied and ten non‐overlapping fields of view (200× magnification) for each tissue section were used in the analysis. The percentage of total area occupied by tissue and air space was calculated by the equation (Price et al. 1993):
Where P(tissue) and P(air space) represent the total number of points falling on lung tissue and air space, respectively, in all counting frames and P(tissue) + P(air space) represents the total number of points counted for each animal.
Quantification of SFTP‐B positive cells within the fetal lung
Immunohistochemistry to identify SFTP‐B positive cells in the alveolar epithelium
In a subset of fetuses (Control, n = 9; Control + VEGF, n = 7; PR, n = 7; PR + VEGF, n = 5), immunohistochemistry was performed using a rabbit anti‐human mature SFTP‐B antibody (WRAB‐48604; Seven Hills Bioreagents, Cincinnati, OH, USA), as described previously (McGillick et al. 2013; McGillick et al. 2014; Lock et al. 2015). Paraformaldehyde fixed, paraffin processed instillation fixed lung tissue sections of 5 μm thickness were baked at 60 °C for 1 h, deparaffinized and rehydrated. After rehydration, endogenous peroxide solution activity was blocked with 3 % hydrogen peroxide (Sigma‐Aldrich), and followed with heat‐induced antigen retrieval in citrate buffer (pH 6.0). Slides were incubated overnight with the aforementioned SFTP‐B antibody (1:500) at 4 °C. A negative control slide with primary antibody omitted was used to demonstrate that there was no non‐specific binding of the secondary antibody or reagent contamination (Neg 1). An additional negative control slide was included in which the primary antibody was replaced by rabbit serum (Sigma‐Aldrich) at the same protein concentration as the diluted primary antibody (1:500) (Neg 2). This negative control demonstrates that there is no non‐specific binding of immunoglobulins with cell and tissue components (Hewitt et al. 2014; Lock et al. 2015), thus highlighting primary antibody specificity in positive control slides in the alveolar epithelium observed in the present study. Negative controls were incubated overnight at 4 °C in parallel with test slides under the same experimental conditions. A Histostain‐Plus broad spectrum kit (Zymed Laboratories Inc., San Francisco, CA, USA) was utilized with horseradish peroxidase and 3,3‐diaminobenzidine chromagen (Metal Enhanced DAB Substrate Kit; Pierce Biotechnology, Rockford, IL, USA) for visualization of SFTP‐B positive staining cells. All sections were counterstained with Mayer's haematoxylin (Sigma‐Aldrich).
Quantitative assessment of SFTP‐B positive cells in the fetal lung
Sections were examined using Visiopharm new Computer Assisted Stereological Toolbox (NewCAST) software (Visiopharm, Hoersholm, Denmark), as described previously (McGillick et al. 2013; McGillick et al. 2014; Lock et al. 2015). Analysis was carried out by a trained individual who was blinded to treatment groups. Eighty counting frames (600× magnification) of the alveolar epithelium were randomly selected per tissue section. Point‐counting using an unbiased counting frame with an area of 25,000 μm2 was used to estimate the numerical density of SFTP‐B positive cells within the alveolar epithelium of fetal lung tissue sections. Using the four corners of the test frame, the reference space was estimated from the number of points falling on lung tissue in each field of view. Characteristic positively staining cuboidal‐shaped cells exhibiting cytoplasmic staining were present within the alveolar epithelium of tissue sections. The numerical density of SFTP‐B positive cells in the alveolar epithelium was expressed as SFTP‐B positive cells mm–2 of lung tissue and was obtained using the equation (Brüel et al. 2005; McGillick et al. 2013; McGillick et al. 2014; Lock et al. 2015):
where ΣQ¯ (SFTP‐B) represents the total number of SFTP‐B positive cells counted in all counting frames of one fetal lung tissue section and ΣP (Lung Tissue) represents the total number of points falling on lung tissue in each field of view. P is the number of points that were used to count the points included within the reference space (four corners per counting frame), where a is the total area of the counting frame.
Statistical analysis
Fetal blood gas data were expressed as the mean of the values collected each day from 3 days after surgery until the day of post mortem. All data are presented as the mean ± SEM. All statistical analyses were carried out using SPSS, version 21.0 (IBM Corp., Armonk, NY, USA). Data were analysed by two‐way ANOVA to determine the effect of treatment (Control vs. PR) and drug (Saline vs. VEGF). If an interaction was present, the Student's unpaired t test was used to determine if there was an effect of drug administration (relative to Control or PR saline infused).
Basal BP measures (SBP, DBP, MAP and HR) and FBM (incidence, amplitude and frequency) obtained on the day before infusion were analysed with the Student's unpaired t test (Control vs. PR). Physiological measures of fetal BP (ΔMAP and ΔHR) and FBM (Δincidence, Δamplitude and Δfrequency) on each day of infusion were analysed using STATA/IC 11 (StataCorp, College Station, TX, USA). A three‐way ANOVA with repeated measures was used to determine the effect of treatment (Control vs. PR), drug (Saline vs. VEGF) and time (repeated measure; infusion day 1 and infusion day 2). P < 0.05 was considered statistically significant.
Results
Fetal blood gas and organ growth
There was a significant effect of treatment, but not drug, on and (Table 2). and decreased in PR compared to Control fetuses (Table 2). In addition, there was a statistically significant reduction in pH in the PR fetuses compared to Controls (Table 2); however, this is probably the result of a relatively tight error between treatment groups. Furthermore, fetal pH is normal in the range pH 7.3–7.4 (Garite, 2012) and this finding is unlikely to be physiologically significant for the health of fetuses. There was no effect of treatment, drug or interaction between treatment and drug on , Hb and Hct (Table 2). Fetal body weight and abdominal circumference decreased in the PR compared to Control fetuses (Table 3). There was a significant effect of treatment and drug on fetal crown rump length, with reduced crown rump length in the PR fetuses and increased measurement in both the Control and PR fetuses receiving VEGF administration (Table 3). There was a treatment effect on relative brain weight, relative brain to liver weight ratio and relative lung weight, as demonstrated by an increased weight in the PR compared to the Control fetuses (Table 3).
Table 2.
Mean fetal blood gas parameters collected from Control and PR fetuses
| Control (n = 8) | Control + VEGF (n = 9) | PR (n = 7) | PR + VEGF (n = 5) | |
|---|---|---|---|---|
| (mmHg) | 21.9 ± 0.9 | 22.2 ± 0.9 | 14.7 ± 0.3† | 15.5 ± 0.6† |
| (mmHg) | 49.6 ± 1.4 | 50.3 ± 1.0 | 49.7 ± 0.8 | 54.3 ± 1.9 |
| (%) | 66.5 ± 5.6 | 62.7 ± 3.0 | 41.8 ± 2.0† | 44.3 ± 5.4† |
| pH | 7.378 ± 0.004 | 7.383 ± 0.009 | 7.374 ± 0.005† | 7.355 ± 0.005† |
| Hb (ml dl‐1) | 8.7 ± 0.6 | 9.6 ± 0.5 | 10.2 ± 0.6 | 9.9 ± 1.5 |
| Hct (%) | 28.4 ± 1.3 | 29.8 ± 1.4 | 31.3 ± 1.9 | 30.7 ± 4.4 |
Data (mean ± SEM) were analysed by two‐way ANOVA for treatment (Control vs. PR) and drug (Saline vs. VEGF). P < 0.05 was considered statistically significant. † P < 0.05 from Control fetuses (i.e. treatment effect).
Table 3.
Measures of fetal growth in the Control and PR fetuses
| Control (n = 11) | Control + VEGF (n = 9) | PR (n = 8) | PR + VEGF (n = 5) | |
|---|---|---|---|---|
| Gestational age at post mortem (days) | 132 ± 1 | 133 ± 1 | 133 ± 1 | 132± 1 |
| Fetal weight (kg) | 3.66 ± 0.11 | 3.68 ± 0.23 | 2.92 ± 0.30† | 3.21± 0.33† |
| Crown rump length (cm) | 50.3 ± 1.6 | 54.3 ± 0.6* | 46.8 ± 1.7† | 50.3 ± 2.1*, † |
| Abdominal circumference (cm) | 34.0 ± 0.7 | 35.0 ± 0.8 | 31.7 ± 1.6† | 32.4 ± 1.1† |
| Relative brain weight (g kg–1) | 15.1 ± 0.4 | 13.5 ± 0.4 | 17.3 ± 0.9† | 15.9 ± 1.5† |
| Relative brain to liver weight ratio | 0.63 ± 0.04 | 0.56 ± 0.04 | 0.74 ± 0.07† | 0.71 ± 0.07† |
| Relative lung weight (g kg–1) | 31.4 ± 1.6 | 33.0 ± 2.2 | 33.5 ± 1.7† | 39.3 ± 3.0† |
Data (mean ± SEM) were analysed by two‐way ANOVA for treatment (Control vs. PR) and drug (Saline vs. VEGF). P < 0.05 was considered statistically significant. † P < 0.05 from Control fetuses (i.e. treatment effect); *P < 0.05 from Control and PR (i.e. drug effect).
Fetal BP and FBM
There was no difference in basal BP measures between Control and PR fetuses on the day before infusion (SBP: Control, 41.84 ± 0.91 vs. PR, 39.66 ± 0.91 mmHg; DBP: Control, 28.20 ± 0.85 vs. PR, 27.79 ± 0.55 mmHg; MAP: Control, 32.87 ± 0.64 vs. PR, 31.75 ± 0.55 mmHg; HR: Control, 157.76 ± 4.60 vs. PR, 166.08 ± 3.52 beats min–1). There was no effect of treatment, drug, time or interaction between these effects on the change in the fetal MAP or HR measures from the basal level on each day of infusion (Table 4). There was no difference between Control and PR fetuses in the incidence (Control, 22.10 ± 2.56 vs. PR, 22.58 ± 5.57%), amplitude (Control, 4.10 ± 0.19 vs. PR, 3.95 ± 0.21 mmHg) or frequency (Control, 106.61 ± 8.00 vs. PR, 85.70 ± 6.25 beats min–1) of FBM during the 5 h period evaluated on the day before infusion. There was no effect of treatment, drug, time or interaction between these effects on the incidence, amplitude or frequency of FBM during the 5 h after infusion for each animal on each day of infusion relative to the day before infusion (Table 4).
Table 4.
Mean change (Δ) in fetal MAP and HR from baseline (30 min before infusion compared to 5 h after infusion) and incidence, amplitude and frequency of FBM from baseline (day before infusion compared to the first 5 h after infusion) on day 1 and day 2 in Control and PR fetuses
| Control | Control + VEGF | PR | PR + VEGF | |
|---|---|---|---|---|
| BP | (n = 5) | (n = 6) | (n = 4) | (n = 4) |
| ΔMAP (mmHg) | ||||
| Infusion day 1 | +0.07 ± 0.49 | +0.10 ± 0.31 | +0.23 ± 0.40 | +0.53 ± 0.29 |
| Infusion day 2 | –0.08 ± 0.15 | +0.73 ± 0.20 | –0.30 ± 0.63 | –0.84 ± 1.27 |
| ΔHR (beats min–1) | ||||
| Infusion day 1 | –4.53 ± 4.29 | –6.07 ± 3.26 | +2.73 ± 5.59 | +0.67 ± 5.49 |
| Infusion day 2 | –9.28 ± 2.91 | –5.52 ± 6.04 | –12.24 ± 7.32 | –3.79 ± 4.49 |
| FBM | (n = 4) | (n = 6) | (n = 4) | (n = 3) |
| ΔIncidence (%) | ||||
| Infusion day 1 | +6.42 ± 1.14 | –1.11 ± 4.77 | –2.00 ± 8.44 | +2.76 ± 3.13 |
| Infusion day 2 | –5.46 ± 5.06 | +13.96 ± 7.15 | +1.33 ± 14.76 | +11.08 ± 3.61 |
| ΔAmplitude (mmHg) | ||||
| Infusion day 1 | +0.72 ± 0.30 | –0.51 ± 0.36 | +0.08 ± 0.20 | –0.27 ± 0.37 |
| Infusion day 2 | +0.25 ± 0.80 | +0.05 ± 0.48 | +0.05 ± 0.14 | +0.64 ± 0.90 |
| ΔFrequency (beats min–1) | ||||
| Infusion day 1 | –11.96 ± 12.85 | +8.86 ± 7.73 | –0.63 ± 1.02 | +9.20 ± 16.69 |
| Infusion day 2 | +5.29 ± 30.36 | –11.39 ± 11.75 | +0.71 ± 7.65 | +4.57 ± 13.17 |
Data (mean ± SEM) were analysed by three‐way ANOVA for treatment (Control vs. PR), drug (Saline vs. VEGF) and time (infusion day 1 vs. infusion day 2) as a repeated measure. P < 0.05 was considered statistically significant.
Expression of genes regulating VEGF signalling and angiogenesis
There was no effect of treatment, drug or an interaction between the effects of treatment and drug on mRNA expression of FLT1, KDR, ANGPT1, AQP1 or ADM in the lung of Control and PR fetuses (Fig. 1).
Figure 1. No effect of placental restriction (PR) or VEGF on the expression of VEGF receptors or genes regulating vascularization .
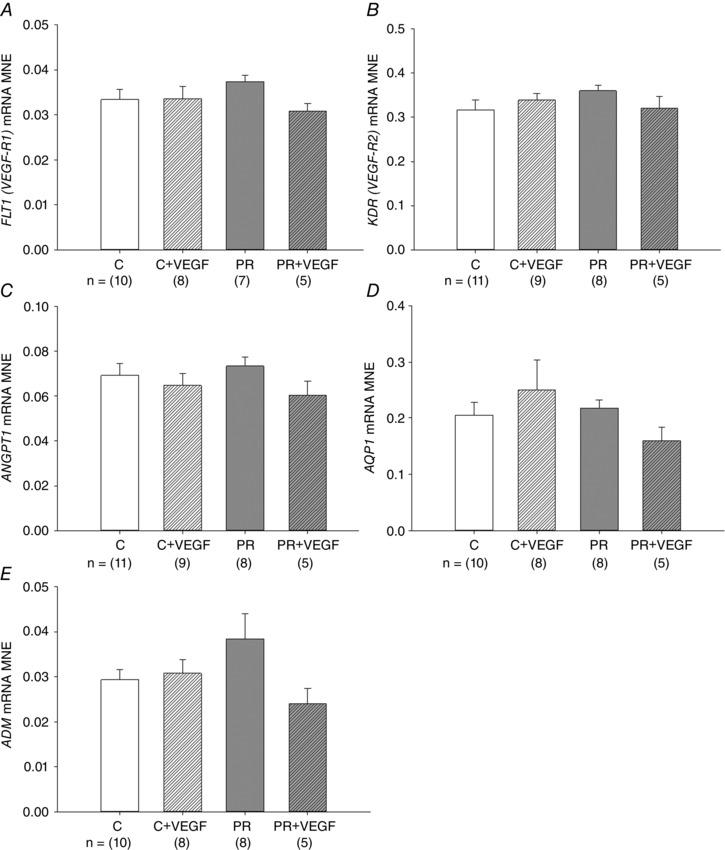
MNE of genes regulating VEGF signalling [FLT1 (VEGF‐R1; A), KDR (VEGF‐R2; B)] and angiogenesis [ANGPT1 (C), AQP1 (D) and ADM (E)] in the fetal lung. Control (C) (open bar); C + VEGF (open hashed bar); PR (grey bar); PR + VEGF (grey hashed bar). The sample size for each group is indicated below the individual graphs. Data (mean ± SEM) were analysed by two‐way ANOVA for treatment (Control vs. PR) and drug (Saline vs. VEGF). P < 0.05.
Expression of genes regulating alveolarization and airway remodelling
There was a significant effect of treatment on TIMP1 and COL1A1 mRNA expression, with increased expression in the lung of the PR fetuses (Fig. 2 C and D). There was no effect of treatment, drug or an interaction between treatment and drug on MMP2 or ELN mRNA expression (Fig. 2 A and E); however, there was a significant effect of drug leading to increased MMP9 mRNA expression (Fig. 2 B).
Figure 2. Effect of placental restriction (PR) and VEGF on the expression of genes involved in structural regulation of lung maturation .
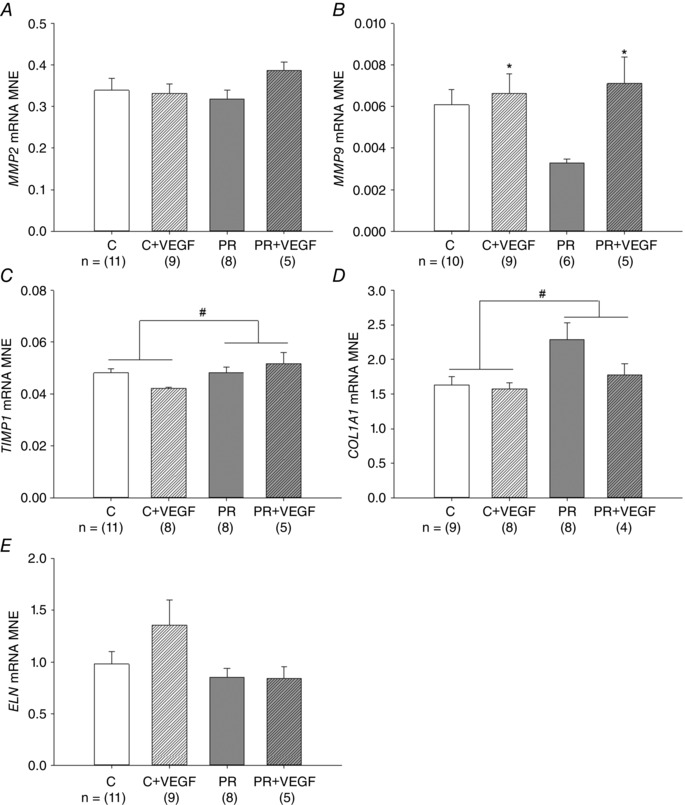
MNE of genes regulating alveolarization and airway remodelling [MMP2 (A), MMP9 (B), TIMP1 (C), COL1A1 (D) and ELN (E)] in the fetal lung. Control (C) (open bar); C + VEGF (open hashed bar); PR (grey bar); PR + VEGF (grey hashed bar). The sample size for each group is indicated below the individual graphs. Data (mean ± SEM) were analysed by two‐way ANOVA for treatment (Control vs. PR) and drug (Saline vs. VEGF). # P < 0.05 from C fetuses (i.e. treatment effect); *P < 0.05 from C or PR (i.e. drug effect).
Expression of genes regulating cellular proliferation
There was no effect of treatment, drug or interaction between these effects on lung IGF1, IGF1R, MKI67 or PCNA mRNA expression (Fig. 3 A, C, D and E). There was a significant effect of drug administration on IGF2 mRNA expression, leading to reduced expression in the lung of fetuses receiving VEGF treatment (Fig. 3 B).
Figure 3. Effect of VEGF on markers of cellular proliferation .
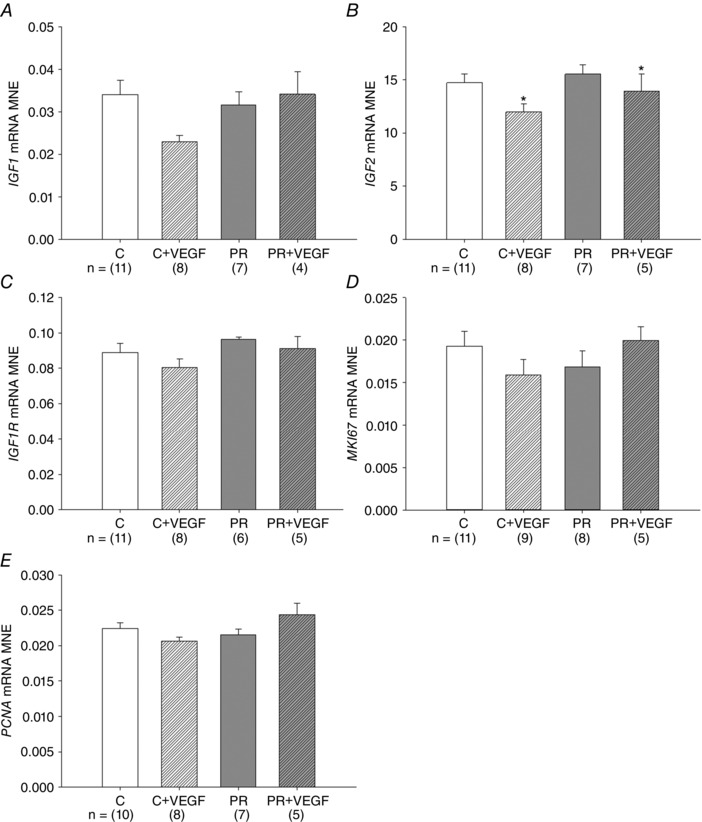
MNE of genes regulating cellular proliferation [IGF1 (A), IGF2 (B), IGF1R (C), MK167 (D) and PCNA (E)] in the fetal lung. Control (C) (open bar); C + VEGF (open hashed bar); PR (grey bar); PR + VEGF (grey hashed bar). The sample size for each group is indicated below the individual graphs. Data (mean ± SEM) were analysed by two‐way ANOVA for treatment (Control vs. PR) and drug (Saline vs. VEGF). *P < 0.05 from C or PR (i.e. drug effect).
Expression of chemokines, pro‐inflammatory and anti‐inflammatory markers
There was no effect of treatment, drug or an interaction between the effects of treatment and drug on mRNA expression of CCL4, TNFA, TGFB1 and IL10 (Fig. 4 B, D, E and F). There was, however, a significant effect of treatment leading to reduced mRNA expression of CCL2 and IL1B in the PR fetal lung (Fig. 4 A and C).
Figure 4. Effect of placental restriction (PR) but not VEGF on the expression of inflammatory markers .
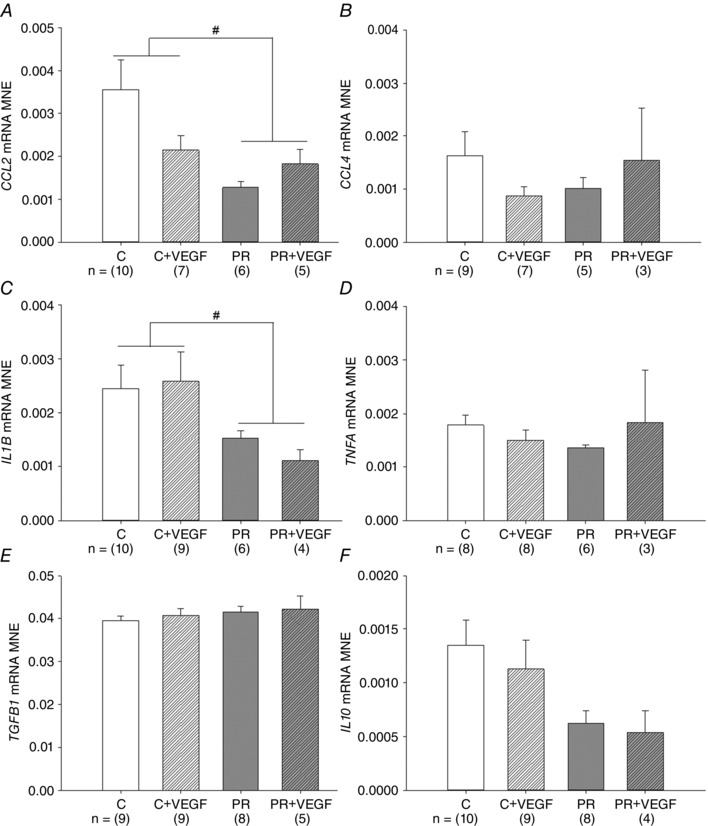
MNE of chemokines [CCL2 (A) and CCL4 (B)], pro‐inflamatory markers [IL1B (C) and TNFA (D)] and anti‐inflammatory markers [TGFB1 (E) and IL10 (F)] in the fetal lung. Control (C) (open bar); C + VEGF (open hashed bar); PR (grey bar); PR + VEGF (grey hashed bar). The sample size for each group is indicated below the individual graphs. Data (mean ± SEM) were analysed by two‐way ANOVA for treatment (Control vs. PR) and drug (Saline vs. VEGF). # P < 0.05 from C fetuses (i.e. treatment effect).
Surfactant system maturation & lung structural morphometry
There was a significant effect of treatment on surfactant protein maturation, such that SFTP‐A, SFTP‐B and SFTP‐C mRNA expression was lower in the PR compared to Control fetuses (Fig. 5 A to C). There was no effect of drug on lung SFTP‐A, ‐B, ‐C or ‐D mRNA expression (Fig. 5 A to D). There was no effect of treatment, drug or interaction between the effects of treatment and drug on lung PCYT1A, LPCAT, ABCA3 or LAMP3 mRNA expression (Fig. 5 E to H). There was an interaction between the effect of treatment and the effect of drug on the total percentage of tissue and air space in the fetal lung (P < 0.05), such that there was a significant decrease in the percentage of tissue and increase in the percentage of air space after VEGF administration in the lung of PR (P < 0.05) but not Control (P = 0.06) fetuses (Fig. 6 E and F). There was a significant effect of drug, but not treatment, resulting in an increased numerical density of SFTP‐B positive cells in the alveolar epithelium of the fetal lung after VEGF administration (Fig. 7 G).
Figure 5. Effect of placental restriction (PR) but not VEGF on markers of surfactant maturation .
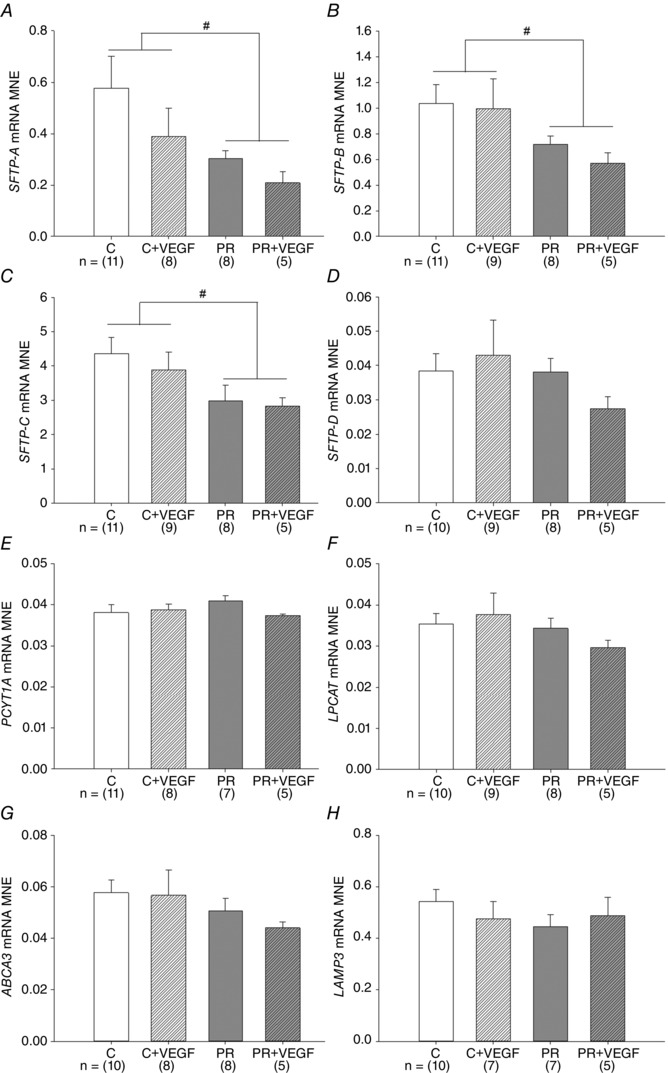
MNE of genes regulating surfactant maturation [SFTP‐A (A), SFTP‐B (B), SFTP‐C (C), SFTP‐D (D), PCYT1A (E), LPCAT (F), ABCA3 (G) and LAMP3 (H)] in the fetal lung. Control (C) (open bar); C + VEGF (open hashed bar); PR (grey bar); PR + VEGF (grey hashed bar). The sample size for each group is indicated below the individual graphs. Data (mean ± SEM) were analysed by two‐way ANOVA for treatment (Control vs. PR) and drug (Saline vs. VEGF). # P < 0.05 from C fetuses (i.e. treatment effect).
Figure 6. Evaluation of total percentage area occupied by tissue and air space in the fetal lung .
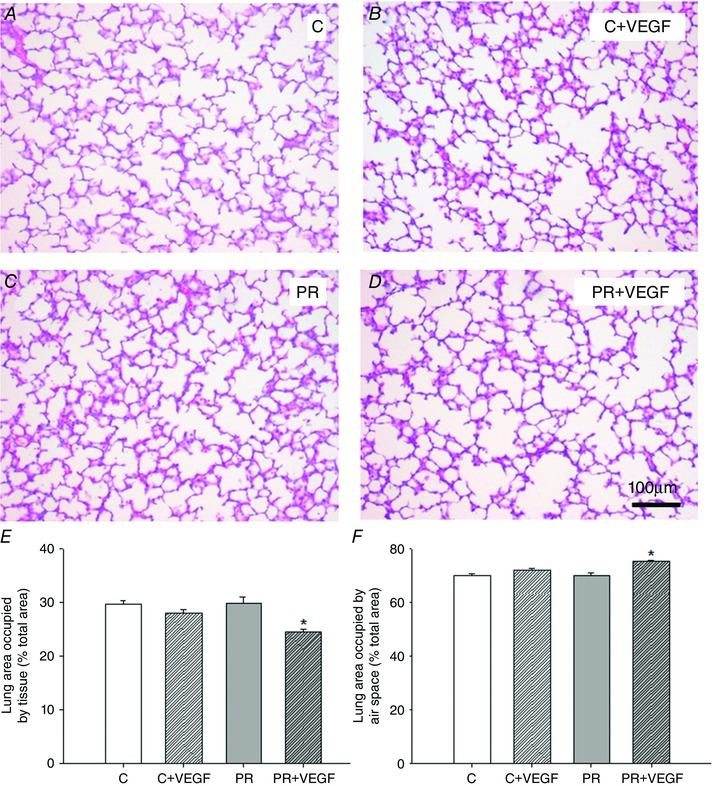
Micrographs of hematoxylin and eosin staining in the lung tissue of the Control (C) (A), C + VEGF (B), PR (C) and PR + VEGF (D) fetuses (100× magnification, scale bar = 100 μm). Percentage of total area occupied by tissue (E) and air space (F) in the fetal lung determined by point counting in the C (n = 9, open bar), C + VEGF (n = 7, open hashed bar), PR (n = 7, grey bar) and PR + VEGF (n = 3, grey hashed bar) fetuses. VEGF administration significantly decreased the total percentage of tissue and increased the total percentage of air space in the PR lung, but not the Control lung (P = 0.06). Data (mean ± SEM) were analysed by two‐way ANOVA for treatment (Control vs. PR) and drug (Saline vs. VEGF). *P < 0.05 from Control or PR (i.e. drug effect).
Figure 7. VEGF administration increases the numerical density of SFTP‐B positive cells in the alveolar epithelium in the Control and PR fetal lung .
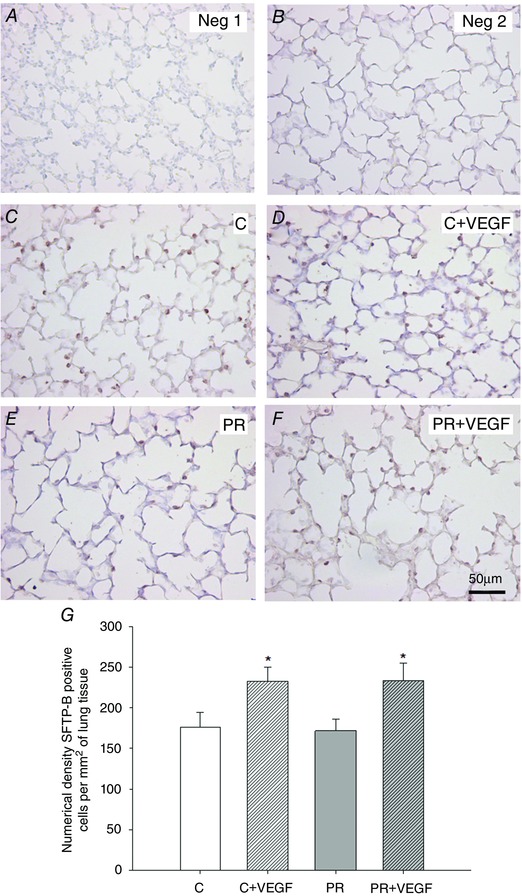
Micrographs demonstrating no primary antibody negative control (Neg 1) (A), 1:500 rabbit serum negative control (Neg 2) (B) and SFTP‐B immunoreactivity (indicated by brown intracellular precipitate) identified by immunohistochemistry in the Control (C) (C), C + VEGF (D), PR (E) and PR + VEGF (F) fetal lung (200× magnification, scale bar = 50 μm). Numerical density of SFTP‐B positive staining cells present in the alveolar epithelium (G) in the C (n = 9, open bar), C + VEGF (n = 7, open hashed bar), PR (n = 7, grey bar) and PR + VEGF (n = 5, grey hashed bar) fetal lung. Data (mean ± SEM) were analysed by two‐way ANOVA for treatment (Control vs PR) and drug (Saline vs VEGF). *P < 0.05 from Control or PR (i.e. drug effect).
Discussion
In the present study, we have provided evidence for increased structural maturation of the fetal lung after intratracheal recombinant human VEGF administration in the normally grown and PR fetus. However, there were only modest effects of VEGF on molecular determinants of VEGF signalling, angiogenesis, alveolarization, cellular proliferation, inflammation and surfactant maturation in the late gestation sheep fetus. Physiologically, there was no effect of either PR or VEGF on fetal BP and FBM. Importantly, intratracheal VEGF administration had positive effects on percentage of tissue, airspace and numerical density of SFTP‐B positive cells present in the alveolar epithelium. These structural effects may be regulated by an increased expression of extracellular matrix remodelling factor MMP9 in the fetal lung. The present study provides further in vivo evidence for the promotion of fetal lung development by VEGF, which may be a potential treatment target for delayed development under the clinical conditions associated with altered hypoxia signalling in utero.
In the present study, we have utilized the model of PR in sheep characterized by chronic fetal hypoxaemia, altered regulation of hypoxia signalling by increased PHD expression and reduced surfactant maturation (Orgeig et al. 2010; Orgeig et al. 2015). To understand the mechanisms contributing to delayed lung maturation in response to altered hypoxia signalling in the fetal lung, we administered recombinant human VEGF, the most widely characterized hypoxia responsive factor. Intra‐amniotic VEGF administration in mice protects from RDS (Compernolle et al. 2002) and improves lung mechanics in fetal rabbits after intratracheal administration (Debeer et al. 2010). In addition, intra‐amniotic VEGF administration promotes surfactant maturation in the late gestation fetal rat lung (Chen & Wang, 2007). VEGF also improves the severity of pulmonary vascular remodelling in the late gestation sheep fetus when administered to the circulation via the pulmonary artery (Grover et al. 2005). Similarly, positive effects of VEGF treatment have been observed on the pulmonary inflammatory response when administered postnatally directly into the lung via a bronchoscope (Sow et al. 2009). Moreover, i.m. administration in rats enhances vessel growth and alveolarization (Kunig et al. 2005). As a result of the systemic nature of VEGF action, for the present study, an intratracheal administration route was chosen to isolate the effects of recombinant human VEGF administration to the developing fetal lung. In the present study, the dose administered was based on those previously demonstrated to have significant effects on lung development in vivo (Grover et al. 2005; Kunig et al. 2005; Sow et al. 2009). Because of its similarity to sheep VEGF at the amino acid level, recombinant human VEGF administration induces changes in the sheep lung (Sow et al. 2009). In the present study, we have provided evidence suggesting that there is detectable VEGF in the fetal lung liquid at 24 and 48 h after the first intratracheal administration of VEGF. Although some studies have demonstrated outcomes after longer durations of exposure to VEGF, including I.M. treatment for 7 days (Kunig et al. 2005) or i.v. treatment for 14 days (Grover et al. 2005), positive effects on lung maturation and respiratory outcomes have also been observed after acute exposure to exogenous VEGF (Compernolle et al. 2002; Sow et al. 2009). For example, there was increased pulmonary maturation in late gestation mice delivered 1 day after intra‐amniotic VEGF administration, resulting in VEGF uptake in the fetal lung because of FBM (Compernolle et al. 2002). In lambs evaluated 16, 24 and 32 h after intratracheal delivery of VEGF, effects on inflammatory mediators were observed; however, the largest magnitude effects in the lung were observed 32 h after VEGF administration (Sow et al. 2009). The infusion regimen chosen for the present study was two doses administered 24 h apart, which parallels clinical practice where glucocorticoids (GCs) are administered to women at risk of preterm delivery to promote fetal lung maturation. Because there is evidence to suggest that the threat of RDS persists despite antenatal GC administration in IUGR infants (Elimian et al. 1999; Torrance et al. 2009; Orgeig & Morrison, 2010), identification of an additional factor in combination with the positive effects of antenatal GCs on fetal lung development may promote the successful transition to air‐breathing at birth in both normally grown and IUGR infants.
Subsequent to administration, VEGF remains restricted to the alveolar compartment, with only minor transfer into the interstitium and circulation (Kaner & Crystal, 2001). Subsequent to postnatal intratracheal administration of VEGF (1 μg) in newborn mice, less than 0.1% of the VEGF recovered in the fetal plasma after 1 h and no VEGF was detectable 3 h after administration (Compernolle et al. 2002). In the present study, we have provided evidence for the presence of small detectable levels of recombinant human VEGF in fetal lung liquid 24 and 48 h after the first infusion. Furthermore, we have demonstrated that there is no effect of intratracheal VEGF administration on fetal cardiovascular status because there was no change in BP measures as a result of the drug. Moreover, there was no effect of VEGF on the incidence, amplitude or frequency of FBM. Although there was no effect of VEGF on relative lung weight, there was a significant effect of treatment leading to an increased relative lung weight in the PR fetuses. Although this is in contrast to the findings of a previous study evaluating the molecular regulation of lung maturation in the PR fetus (Orgeig et al. 2010), there is other evidence supporting altered lung indices relative to body weight in the PR fetus (Lipsett et al. 2006), possibly as a result of a sparing of lung growth with increasing somatic growth restriction.
Despite the angiogenic properties of VEGF, we found no effect on the expression of its receptors FLT1 (VEGF‐R1) and KDR (VEGF‐R2), a marker of endothelial cells (AQP1), regulator of vascular tone (ANGPT1) or vascular permeability (ADM). This is consistent with a previous study in mice where there was no stimulation of angiogenesis in the alveolar septa after intra‐amniotic and intratracheal VEGF administration (Compernolle et al. 2002). Because of the important developmental link between vascularization and alveolarization, we examined the expression of genes regulating structural lung remodelling (Jakkula et al. 2000). Alveolarization is a dynamic process of structural regulation of the immature pulmonary mesenchyme into structural units of gas exchange, the alveoli, and the associated capillary network (Burri, 2006). Within lung tissue, collagen and elastin provide the structural components of the respiratory architecture (Willet et al. 1999). Airway remodelling is under the control of the MMP degrading enzymes, which are essential for normal developmental regulation of maturation and maintenance to protect from injury and lung disease in extrauterine life (Lagente et al. 2005). To maintain homeostatic balance, MMP function is regulated by TIMP activity to limit the collagen degradation (Vincenti, 2001). Within the lung of the PR fetus, we observed increased TIMP1 expression, an inhibitor of MMP9 and COL1A1, and a substrate of MMP9 activity. This increased expression may be a result of increased structural reorganization potentially because of smaller alveoli, as previously reported in the lung of the PR fetus (Lipsett et al. 2006). Interestingly, VEGF administration led to increased MMP9 expression in the lung of both the normally grown and PR fetus. MMP9, a key regulatory enzyme of the extracellular martix, is involved in the degradation of various structural proteins, including collagen and elastin (Vincenti, 2001). This finding provides molecular evidence for altered structural regulation/reorganization in the fetal lung after intratracheal VEGF administration.
There were relatively few effects of VEGF on the expression of genes that regulate surfactant maturation or cellular proliferation at the molecular level. There was reduced IL1B expression in the PR fetuses at 133 days of gestation in this cohort, which reflects the same pattern of change observed at 133 days of gestation in a previous study evaluating the effect of age and treatment on outcomes in the PR fetal lung (Orgeig et al. 2015). This finding of reduced IL1B correlates with reduced surfactant maturation in the PR fetus because SFTP‐A is a regulator of IL1B (Kremlev & Phelps, 1994). Moreover, there was no effect of PR on IGF1. Interestingly, however, there was reduced IGF2 expression after VEGF administration in both the Control and PR fetus. On balance, there was no effect of PR on IGF1R expression, suggesting that there are few effects on downstream signalling.
We evaluated the effect of VEGF administration on the expression of pro‐ and anti‐inflammatory markers in the lung of both the Control and PR fetus. This provides important information regarding the regulation of respiratory immune capacity. The immunoregulatory role of VEGF in the lung has been previously evaluated in response to immunological challenge postnatally in the fetal sheep lung (Meyerholz et al. 2006; Meyerholz et al. 2007); however, in the present study, the direct intratracheal VEGF administration in the fetal lung provides information about the regulation of immunity in preparation for exposure to the air‐breathing environment at birth. Sow et al. (2009) have provided evidence for dynamic regulation of inflammatory mediators depending on the dose of VEGF administration in the newborn lamb lung. In the present study, despite a negative effect of PR treatment on IL1B and CCL2 expression, there was no effect of VEGF administration on the panel of chemokine, pro‐ and anti‐inflammatory markers evaluated in the Control and PR fetal lung. The limited immunomodulatory response of the fetal lung to VEGF administration observed in the present study may be a result of differences in exposure time, the route of administration and age at the time of drug administration compared to other studies. For example, despite our administered dose being similar to the higher dose of recombinant human VEGF doses evaluated by Sow et al. (2009), administration in the neonatal lung may exert more pronounced effects as a result of the stimulus that the continued threat of air‐breathing poses to pulmonary immunity compared to the fluid‐filled fetal lung in utero.
Surfactant maturation is dynamically regulated by exposure to hypoxia and is under the control of hypoxia responsive genes, including VEGF (Tuder et al. 1995; Brown et al. 2001; Ito et al. 2011). VEGF administration directly promotes surfactant protein production and increases surfactant lipid synthesis (Brown et al. 2001; Compernolle et al. 2002; Raoul et al. 2004; Chen & Wang, 2007). Although, in some cases, this positive effect on surfactant maturation has been observed only at the highest doses of VEGF administered in these studies. For example, Chen & Wang (2007) demonstrated increased SFTP‐B and SFTP‐D expression after intra‐amniotic administration of high dose (5 μg) but not low dose (2.5 μg) VEGF. Interestingly, there was no effect of either dose on total phospholipid or the major surfactant phospholipid (i.e phosphatidylcholine) content (Chen & Wang, 2007). In isolated fetal rat type II AECs, there was no effect on SFTP expression at 50 ng ml–1, although there was an increase in both SFTP‐B gene and protein expression after incubation with VEGF at 100 ng ml–1 (Raoul et al. 2004). Furthermore, freshly isolated adult rat type II AECs incubated with 200 ng ml–1 VEGF had increased SFTP‐B and SFTP‐C expression (Compernolle et al. 2002). Interestingly, intra‐amniotic administration of 0.5 μg of VEGF increases the number of glycogen containing AECs in fetal mouse lungs and this may lead to upregulation of glycogen conversion into surfactant lipids, thus aiding in the successful transition to air‐breathing at birth (Compernolle et al. 2002). In the present study, we found no significant effect of VEGF administration on expression of the four SFTP genes in the Control or PR fetus. Furthermore, we did not find any significant effect of treatment or VEGF administration on the expression of genes including PCYT1A (rate‐limiting enzyme involved in surfactant phosphatidylcholine synthesis), LPCAT (regulator of surfactant lipid dipalmitoyl phosphatidylcholine synthesis from phosphatidylcholine), ABCA3 (surfactant lipid transporter) or LAMP3 (expressed in lamellar bodies in type II AECs), all of which play important roles in regulating surfactant lipid processing and transport (Stahlman et al. 2007; Agassandian & Mallampalli, 2013).
There was no effect of PR on structural determinants of the fetal lung, including the numerical density of SFTP‐B positive cells in the alveolar epithelium. This is an interesting finding because one proposed explanation for the reduced surfactant maturation in the PR fetal lung had been a reduction in the proliferation of type II AECs responsible for the synthesis and secretion of surfactant (Orgeig et al. 2010; Orgeig et al. 2015). Because the methodology for evaluating numerical density employs quantitation of the number of SFTP‐B positive staining cells rather than the intensity of staining obtained, the immunohistochemistry results cannot be quantified as changes in SFTP protein expression. Therefore, in combination with the molecular analysis of surfactant maturation, we gain insight into the effects of treatments with respect to both the cellular number and molecular capacity of the fetal lung to produce surfactant (McGillick et al. 2013; McGillick et al. 2014; Lock et al. 2015). Hence, the findings of the present study indicate that there is a reduced functional capacity of the mature type II AECs in the PR fetal lung to produce surfactant rather than an overall change to the number of cells able to produce surfactant.
Despite finding relatively few effects on the molecular regulation of lung maturation, we have observed a positive impact of VEGF administration on structural regulation in the fetal lung. This is consistent with previous studies demonstrating positive effects of VEGF on epithelium volume density and cellular proliferation in human fetal lung tissue (Brown et al. 2001). In addition, intratracheal VEGF administration after delivery increases the total aerated area in the lung of preterm mice (Compernolle et al. 2002). Moreover, inhibition of hypoxia signalling by HIF‐α knockout leads to decreased SFTP‐D positive cells in the neonatal mouse lung (Compernolle et al. 2002), whereas direct blocking of VEGF signalling in newborn rats reduces alveolar counts and postnatal alveolarization (Le Cras et al. 2002). In the PR fetal lung, VEGF administration resulted in a significant decrease in the total percentage of tissue and an increase in the total percentage of air space present in the lung. This is suggestive of increased alveolarization or increased alveolar size after the administration of VEGF. Although changes to the percentage of tissue and air space in the Control lung receiving VEGF administration compared to the respective Control did not reach statistical significance (P = 0.06), this provides further evidence for the ability of VEGF administration to modulate structural regulation with respect to remodelling the fetal lung. Furthermore, VEGF administration increased the numerical density of SFTP‐B positive cells in the alveolar epithelium of lung tissue in both the Control and PR fetus. This provides evidence for an increase in the number of cells that produce surfactant (i.e. a potential increased functional capacity of the lung after VEGF administration). Despite a potential increase in the functional capacity of the lung to produce surfactant, VEGF administration did not stimulate maturation of the surfactant system at the molecular level. It is plausible that a higher dose of VEGF or combination with an additional agent (e.g. GCs) known to promote molecular lung maturation may have further beneficial effects on the developing lung. Thus, co‐treatment may promote molecular surfactant maturation in addition to the positive structural effects observed after VEGF administration in the fetal lung. This has the potential to accelerate preparation for a successful transition to the air‐breathing environment and minimize the risk of experiencing respiratory complications at birth.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
SO and JLM were responsible for the conception and design of the experiments. EVM, SO and JLM were each involved in the experimentation and sample/data acquisition. EVM, SO and JLM were involved in the analysis and interpretation of the data. EVM, SO and JLM drafted the article and all authors contributed to the final version. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was funded by an NHMRC Project Grant (APP1030853) to JLM and SO. JLM was funded by a South Australian Cardiovascular Research Network Fellowship (CR10A4988) and an NHMRC Career Development Fellowship (APP1066916).
Translational perspective
In the present study, we evaluated the effect of intratracheal vascular endothelial growth factor (VEGF) administration on molecular and structural regulators of lung development, fetal blood pressure and fetal breathing movements in the normally grown and placentally restricted (PR) fetus. We have provided evidence of positive effects on structural regulation of lung morphometry after intratracheal VEGF administration in the lung of the normally grown and PR sheep fetus during late gestation. Despite relatively few effects of VEGF administration on the molecular regulation of lung development, it is possible that combination treatment with an additional agent to promote surfactant maturation at the molecular level may provide synergistic effects to promote lung maturation in combination with the structural maturation achieved with VEGF administration alone. This may prove beneficial with respect to increasing the functional capacity of the fetal lung to produce surfactant and promote the successful transition to air‐breathing. Overall, the present study has provided understanding of VEGF signalling in the lung of the normally grown and PR fetus and demonstrated positive effects on structural markers of lung architecture. This may provide a target to promote lung maturation aiming to reduce respiratory complications resulting from structural immaturity of the fetal lung at birth.
Acknowledgements
We acknowledge the assistance of Stacey Holman in surgical procedures and expert post‐surgical care of the ewe and her fetus. We also acknowledge members of the Early Origins of Adult Health Research Group for their assistance in sample collection.
References
- Agassandian M & Mallampalli RK (2013). Surfactant phospholipid metabolism. Biochim Biophys Acta – Mol Cell Biol L 1831, 612–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander G (1964). Studies on the placenta of the sheep (Ovis aries L.). Effect of surgical reduction in the number of caruncles. J Reprod Fertil 7, 307–322. [DOI] [PubMed] [Google Scholar]
- Avery ME & Mead J (1959). Surface properties in relation to atelectasis and hyaline membrane disease. AMA J Dis Child 97, 517–523. [DOI] [PubMed] [Google Scholar]
- Bennet L, Johnston BM, Vale W & Gluckman P (1990). The effects of corticotrophin‐releasing factor and two antagonists on breathing movements in fetal sheep. J Physiol 421, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein IM, Horbar JD, Badger GJ, Ohlsson A & Golan A (2000). Morbidity and mortality among very‐low‐birth‐weight neonates with intrauterine growth restriction. Am J Obstet Gynecol 182, 198–206. [DOI] [PubMed] [Google Scholar]
- Berse B, Brown LF, Van De Water L, Dvorak HF & Senger DR (1992). Vascular permeability factor (vascular endothelial growth factor) gene is expressed differentially in normal tissues, macrophages, and tumors. Mol Biol Cell 3, 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco LN, Massaro GD & Massaro D (1989). Alveolar dimensions and number: developmental and hormonal regulation. Am J Physiol Lung Cell Mol Physiol 257, L240–L247. [DOI] [PubMed] [Google Scholar]
- Bocking AD, Gagnon R, Milne KM & White SE (1988). Behavioral activity during prolonged hypoxemia in fetal sheep. J Appl Physiol 65, 2420–2426. [DOI] [PubMed] [Google Scholar]
- Botting KJ, McMillen IC, Forbes H, Nyengaard JR & Morrison JL (2014). Chronic hypoxemia in late gestation decreases cardiomyocyte number but does not change expression of hypoxia‐responsive genes. J Am Heart Assoc 3, e000531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breen EC, Malloy JL, Tang K, Xia F, Fu Z, Hancock RE, Overhage J, Wagner PD & Spragg RG (2013). Impaired pulmonary defense against Pseudomonas aeruginosa in VEGF gene inactivated mouse lung. J Cell Physiol 228, 371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KRS, England KM, Goss KL, Snyder JM & Acarregui MJ (2001). VEGF induces airway epithelial cell proliferation in human fetal lung in vitro. Am J Physiol Lung Cell Mol Physiol 281, L1001–L1010. [DOI] [PubMed] [Google Scholar]
- Brüel A, Oxlund H & Nyengaard JR (2005). The total length of myocytes and capillaries, and total number of myocyte nuclei in the rat heart are time‐dependently increased by growth hormone. Growth Horm IGF Res 15, 256–264. [DOI] [PubMed] [Google Scholar]
- Burri PH (2006). Structural aspects of postnatal lung development – alveolar formation and growth. Biol Neonate 89, 313–322. [DOI] [PubMed] [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW & Shipley GL (2009). The MIQE guidelines: minimum information for publication of quantitative real‐time PCR experiments. Clin Chem 55, 611–622. [DOI] [PubMed] [Google Scholar]
- Butler TG, Schwartz J & McMillen IC (2002). Differential effects of the early and late intrauterine environment on corticotrophic cell development. J Clin Invest 110, 783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K & Eberhardt C (1996). Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 380, 435–439. [DOI] [PubMed] [Google Scholar]
- Chen CM & Wang LF (2007). High‐dose vascular endothelial growth factor increases surfactant protein gene expressions in preterm rat lung. Early Hum Dev 83, 581–584. [DOI] [PubMed] [Google Scholar]
- Compernolle V, Brusselmans K, Acker T, Hoet P, Tjwa M, Beck H, Plaisance S, Dor Y, Keshet E & Lupu F (2002). Loss of HIF‐2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med 8, 702–710. [DOI] [PubMed] [Google Scholar]
- Creuwels L, Van Golde L & Haagsman H (1997). The pulmonary surfactant system: biochemical and clinical aspects. Lung 175, 1–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielson L, McMillen IC, Dyer JL & Morrison JL (2005). Restriction of placental growth results in greater hypotensive response to α‐adrenergic blockade in fetal sheep during late gestation. J Physiol 563, 611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debeer A, Sbragia L, Vrancken K, Hendriks A, Roubliova X, Jani J, Naulaers G, Carmeliet P & Deprest J (2010). Antenatal fetal VEGF therapy to promote pulmonary maturation in a preterm rabbit model. Early Hum Dev 86, 99–105. [DOI] [PubMed] [Google Scholar]
- Drummond G (2009). Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol 587, 713–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer J, McMillen IC, Warnes KE & Morrison JL (2009). No evidence for an enhanced role of endothelial nitric oxide in the maintenance of arterial blood pressure in the IUGR sheep fetus. Placenta 30, 705–710. [DOI] [PubMed] [Google Scholar]
- Elimian A, Verma U, Canterino J, Shah J, Visintainer P & Tejani N (1999). Effectiveness of antenatal steroids in obstetric subgroups. Obstet Gynecol 93, 174. [DOI] [PubMed] [Google Scholar]
- Epstein ACR, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI & Dhanda A (2001). C. elegans EGL‐9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107, 43–54. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Carver Moore K, Chen H, Dowd M, Lu L, O'Shea KS, Powell Braxton L, Hillan KJ & Moore MW (1996). Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 380, 439–442. [DOI] [PubMed] [Google Scholar]
- Gagnon R (2006). An obstetric point of view on fetal adaptation and reprogramming. NeoReviews 7, e189‐e194. [Google Scholar]
- Gagnon R, Langridge J, Inchley K, Murotsuki J & Possmayer F (1999). Changes in surfactant‐associated protein mRNA profile in growth‐restricted fetal sheep. Am J Physiol Lung Cell Mol Physiol 276, L459–L465. [DOI] [PubMed] [Google Scholar]
- Garite T (2012). Intrapartum fetal evaluation In Obstetrics: Normal and Problem Pregnancies, Chapter 16, 6th edn, ed. Gabbe SG, Niebyl JR, Galan HL, Jauniaux ER, Landon MB, Simpson JL. & Driscoll DA. (eds). Saunders Elsevier, Philadelphia, PA. [Google Scholar]
- Gentili S, Morrison JL & McMillen IC (2009). Intrauterine growth restriction and differential patterns of hepatic growth and expression of IGF1, PCK2, and HSDL1 mRNA in the sheep fetus in late gestation. Biol Reprod 80, 1121–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginouvès A, Ilc K, Macías N, Pouysségur J & Berra E (2008). PHDs overactivation during chronic hypoxia ‘desensitizes’ HIFα and protects cells from necrosis. Proc Natl Acad Sci USA 105, 4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giussani D, Moore P, Bennet L, Spencer J & Hanson M (1995). Alpha 1‐ and alpha 2‐adrenoreceptor actions of phentolamine and prazosin on breathing movements in fetal sheep in utero. J Physiol 486, 249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover TR, Parker TA, Markham NE & Abman SH (2005). rhVEGF treatment preserves pulmonary vascular reactivity and structure in an experimental model of pulmonary hypertension in fetal sheep. Am J Physiol Lung Cell Mol Physiol 289, L315–L321. [DOI] [PubMed] [Google Scholar]
- Grover TR, Parker TA, Zenge JP, Markham NE, Kinsella JP & Abman SH (2003). Intrauterine hypertension decreases lung VEGF expression and VEGF inhibition causes pulmonary hypertension in the ovine fetus. Am J Physiol Lung Cell Mol Physiol 284, L508–L517. [DOI] [PubMed] [Google Scholar]
- Hellemans J, Mortier G, De Paepe A, Speleman F & Vandesompele J (2007). qBase relative quantification framework and software for management and automated analysis of real‐time quantitative PCR data. Genome Biol 8, R19.11–R19.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt SM, Baskin DG, Frevert CW, Stahl WL & Rosa‐Molinar E (2014). Controls for immunohistochemistry. The Histochemical Society's Standards of Practice for Validation of Immunohistochemical Assays. J Histochem Cytochem 62, 693–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Ahmad A, Kewley E & Mason R (2011). Hypoxia‐Inducible factor regulates expression of surfactant protein in alveolar type II cells in vitro. Am J Respir Cell Mol Biol 45, 938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakkula M, Le Cras TD, Gebb S, Hirth KP, Tuder RM, Voelkel NF & Abman SH (2000). Inhibition of angiogenesis decreases alveolarization in the developing rat lung. Am J Physiol Lung Cell Mol Physiol 279, L600–L607. [DOI] [PubMed] [Google Scholar]
- Kaner RJ & Crystal RG (2001). Compartmentalization of vascular endothelial growth factor to the epithelial surface of the human lung. Mol Med 7, 240. [PMC free article] [PubMed] [Google Scholar]
- Kobata R, Tsukahara H, Ohshima Y, Ohta N, Tokuriki S, Tamura S & Mayumi M (2008). High levels of growth factors in human breast milk. Early Hum Dev 84, 67–69. [DOI] [PubMed] [Google Scholar]
- Kremlev SG & Phelps DS (1994). Surfactant protein A stimulation of inflammatory cytokine and immunoglobulin production. Am J Physiol Lung Cell Mol Physiol 267, L712–L719. [DOI] [PubMed] [Google Scholar]
- Kunig AM, Balasubramaniam V, Markham NE, Morgan D, Montgomery G, Grover TR & Abman SH (2005). Recombinant human VEGF treatment enhances alveolarization after hyperoxic lung injury in neonatal rats. Am J Physiol Lung Cell Mol Physiol 289, L529–L535. [DOI] [PubMed] [Google Scholar]
- Lagente V, Manoury B, Nenan S, Le Quement C, Martin‐Chouly C & Boichot E (2005). Role of matrix metalloproteinases in the development of airway inflammation and remodeling. Braz J Med Biol Res 38, 1521–1530. [DOI] [PubMed] [Google Scholar]
- Lassus P, Turanlahti M, HEIKKILÄ PI, Andersson LC, Nupponen I, Sarnesto A & Andersson S (2001). Pulmonary vascular endothelial growth factor and Flt‐1 in fetuses, in acute and chronic lung disease, and in persistent pulmonary hypertension of the newborn. Am J Respir Crit Care Med 164, 1981–1987. [DOI] [PubMed] [Google Scholar]
- Le Cras TD, Markham NE, Tuder RM, Voelkel NF & Abman SH (2002). Treatment of newborn rats with a VEGF receptor inhibitor causes pulmonary hypertension and abnormal lung structure. Am J Physiol Lung Cell Mol Physiol 283, L555–L562. [DOI] [PubMed] [Google Scholar]
- Lipsett J, Tamblyn M, Madigan K, Roberts P, Cool JC, Runciman SI, McMillen IC, Robinson J & Owens JA (2006). Restricted fetal growth and lung development: a morphometric analysis of pulmonary structure. Pediatr Pulmonol 41, 1138–1145. [DOI] [PubMed] [Google Scholar]
- Lock MC, McGillick EV, Orgeig S, Zhang S, McMillen IC & Morrison JL (2015). Mature surfactant protein‐B expression by immunohistochemistry as a marker for surfactant system development in the fetal sheep lung. J Histochem Cytochem 63, 866–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maritz G, Cock M, Louey S, Joyce B, Albuquerque C & Harding R (2001). Effects of fetal growth restriction on lung development before and after birth: a morphometric analysis. Pediatr Pulmonol 32, 201–210. [DOI] [PubMed] [Google Scholar]
- McGillick EV, Morrison JL, McMillen IC & Orgeig S (2014). Intrafetal glucose infusion alters glucocorticoid signaling and reduces surfactant protein mRNA expression in the lung of the late‐gestation sheep fetus. Am J Physiol Regul Integr Comp Physiol 307, R538–R545. [DOI] [PubMed] [Google Scholar]
- McGillick EV, Orgeig S, McMillen IC & Morrison JL (2013). The fetal sheep lung does not respond to cortisol infusion during the late canalicular phase of development. Physiol Rep 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntire DD, Bloom SL, Casey BM & Leveno KJ (1999). Birth weight in relation to morbidity and mortality among newborn infants. N Engl J Med 340, 1234–1238. [DOI] [PubMed] [Google Scholar]
- Meyerholz DK, Gallup JM, Lazic T, De Macedo MM, Lehmkuhl HD & Ackermann MR (2007). Pretreatment with recombinant human vascular endothelial growth factor reduces virus replication and inflammation in a perinatal lamb model of respiratory syncytial virus infection. Viral Immunol 20, 188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerholz DK, Grubor B, Lazic T, Gallup JM, De Macedo MM, McCray P & Ackermann MR (2006). Monocytic/macrophagic pneumonitis after intrabronchial deposition of vascular endothelial growth factor in neonatal lambs. Vet Pathol 43, 689–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyrick B & Reid L (1982). Pulmonary arterial and alveolar development in normal postnatal rat lung. Am Rev Respir Dis 125, 468–473. [DOI] [PubMed] [Google Scholar]
- Monacci WT, Merrill MJ & Oldfield EH (1993). Expression of vascular permeability factor/vascular endothelial growth factor in normal rat tissues. Am J Physiol Cell Physiol 264, C995–C995. [DOI] [PubMed] [Google Scholar]
- Morrison JL (2008). Sheep models of intrauterine growth restriction: fetal adaptations and consequences. Clin Exp Pharmacol Physiol 35, 730–743. [DOI] [PubMed] [Google Scholar]
- Morrison JL, Carmichael L, Homan J & Richardson BS (1997). The effects of ‘sleep promoting agents' on behavioural state in the ovine fetus. Dev Brain Res 103, 1–8. [DOI] [PubMed] [Google Scholar]
- Morrison JL, Chien C, Gruber N, Rurak D & Riggs W (2001). Fetal behavioural state changes following maternal fluoxetine infusion in sheep. Brain Res Dev Brain Res 131, 47–56. [DOI] [PubMed] [Google Scholar]
- Ng YS, Rohan R, Sunday M, Demello D & d'Amore P (2001). Differential expression of VEGF isoforms in mouse during development and in the adult. Dev Dyn 220, 112–121. [DOI] [PubMed] [Google Scholar]
- Orgeig S, Crittenden TA, Marchant C, McMillen IC & Morrison JL (2010). Intrauterine growth restriction delays surfactant protein maturation in the sheep fetus. Am J Physiol Lung Cell Mol Physiol 298, L575–L583. [DOI] [PubMed] [Google Scholar]
- Orgeig S, McGillick EV, Botting KJ, Zhang S, McMillen IC & Morrison JL (2015). Increased lung prolyl hydroxylase and decreased glucocorticoid receptor are related to decreased surfactant protein in the growth restricted sheep fetus. Am J Physiol Lung Cell Mol Physiol 309, L84–L97. [DOI] [PubMed] [Google Scholar]
- Orgeig S & Morrison JL (2010). Does the intrauterine growth‐restricted fetus benefit from antenatal glucocorticoids? Expert Rev Obstet Gynecol 5, 149–152. [Google Scholar]
- Passmore M, Nataatmadja M & Fraser JF (2009). Selection of reference genes for normalisation of real‐time RT‐PCR in brain‐stem death injury in Ovis aries . BMC Mol Biol 10, 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez‐Gil J (2008). Structure of pulmonary surfactant membranes and films: the role of proteins and lipid‐protein interactions. Biochim Biophys Acta 1778, 1676–1695. [DOI] [PubMed] [Google Scholar]
- Poudel R, McMillen IC, Dunn SL, Zhang S & Morrison JL (2015). Impact of chronic hypoxemia on blood flow to the brain, heart, and adrenal gland in the late‐gestation IUGR sheep fetus. Am J Physiol Regul Integr Comp Physiol 308, R151–R162. [DOI] [PubMed] [Google Scholar]
- Price LT, Chen Y & Frank L (1993). Epidermal growth factor increases antioxidant enzyme and surfactant system development during hyperoxia and protects fetal rat lungs in vitro from hyperoxic toxicity. Pediatr Res 34, 577–585. [DOI] [PubMed] [Google Scholar]
- Raoul W, Chailley‐Heu B, Barlier‐Mur AM, Delacourt C, Maître B & Bourbon JR (2004). Effects of vascular endothelial growth factor on isolated fetal alveolar type II cells. Am J Physiol Lung Cell Mol Physiol 286, L1293–L1301. [DOI] [PubMed] [Google Scholar]
- Robinson JS, Kingston EJ, Jones CT & Thorburn GD (1979). Studies on experimental growth retardation in sheep. The effect of removal of a endometrial caruncles on fetal size and metabolism. J Dev Physiol 1, 379–398. [PubMed] [Google Scholar]
- Shifren J, Doldi N, Ferrara N, Mesiano S & Jaffe RB (1994). In the human fetus, vascular endothelial growth factor is expressed in epithelial cells and myocytes, but not vascular endothelium: implications for mode of action. J Clin Endocrinol Metabol 79, 316–322. [DOI] [PubMed] [Google Scholar]
- Soo PS, Hiscock J, Botting KJ, Roberts CT, Davey AK & Morrison JL (2012). Maternal undernutrition reduces P‐glycoprotein in guinea pig placenta and developing brain in late gestation. Reprod Toxicol 33, 374–381. [DOI] [PubMed] [Google Scholar]
- Sow FB, Gallup JM, Meyerholz DK & Ackermann MR (2009). Gene profiling studies in the neonatal ovine lung show enhancing effects of VEGF on the immune response. Dev Comp Immunol 33, 761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahlman MT, Besnard V, Wert SE, Weaver TE, Dingle S, Xu Y, von Zychlin K, Olson SJ & Whitsett JA (2007). Expression of ABCA3 in developing lung and other tissues. J Histochem Cytochem 55, 71–83. [DOI] [PubMed] [Google Scholar]
- Thébaud B, Ladha F, Michelakis ED, Sawicka M, Thurston G, Eaton F, Hashimoto K, Harry G, Haromy A & Korbutt G (2005). Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia‐induced lung injury evidence that angiogenesis participates in alveolarization. Circulation 112, 2477–2486. [DOI] [PubMed] [Google Scholar]
- Torrance HL, Derks JB, Scherjon SA, Wijnberger LD & Visser GHA (2009). Is antenatal steroid treatment effective in preterm IUGR fetuses? Acta Obstet Gynecol Scand 88, 1068–1073. [DOI] [PubMed] [Google Scholar]
- Tuder RM, Flook BE & Voelkel NF (1995). Increased gene expression for VEGF and the VEGF receptors KDR/Flk and Flt in lungs exposed to acute or to chronic hypoxia. Modulation of gene expression by nitric oxide. J Clin Invest 95, 1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A & Speleman F (2002). Accurate normalization of real‐time quantitative RT‐PCR data by geometric averaging of multiple internal control genes. Genome Biol 3, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincenti MP (2001). The matrix metalloproteinase (MMP) and tissue inhibitor of metalloproteinase (TIMP) genes In Matrix Metalloproteinase Protocols, Ian M Clark. (ed.) pp. 121–148. Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- Willet KE, McMenamin P, Pinkerton KE, Ikegami M, Jobe AH, Gurrin L & Sly PD (1999). Lung morphometry and collagen and elastin content: changes during normal development and after prenatal hormone exposure in sheep. Pediatr Res 45, 615–625. [DOI] [PubMed] [Google Scholar]
- Zhang S, Rattanatray L, MacLaughlin SM, Cropley JE, Suter CM, Molloy L, Kleemann D, Walker SK, Muhlhausler BS & Morrison JL (2010). Periconceptional undernutrition in normal and overweight ewes leads to increased adrenal growth and epigenetic changes in adrenal IGF2/H19 gene in offspring. FASEB J 24, 2772–2782. [DOI] [PubMed] [Google Scholar]