

Key points
Asphyxia at the time of birth is a significant cause of death or disability in newborns. There is very limited treatment available for these newborns.
Autologous umbilical cord blood (UBC) mononuclear cells reduce clinical markers of brain damage following perinatal asphyxia.
Autologous UBC mononuclear cells reduce neuroinflammation and neuronal apoptosis within the brain following perinatal asphyxia.
Autologous UBC mononuclear cells administered 12 h after perinatal asphyxia are neuroprotective, and a well‐tolerated and feasible treatment for infants following hypoxic ischaemic encephalopathy.
Abstract
Perinatal asphyxia is a significant cause of death or long‐term neurodevelopmental impairment. Hypothermia, currently the only effective treatment, leads to modest improvements, but new therapeutic strategies are required. Umbilical cord blood (UCB) mononuclear cells have potent anti‐inflammatory properties and may reduce neuropathology. This study examined whether autologous UCB mononuclear cells were neuroprotective when administered to newborn lambs at 12 h after birth asphyxia. At caesarean section, birth asphyxia was induced by clamping the umbilical cord until mean arterial blood pressure decreased to 18–20 mmHg. Asphyxia (n = 20) or control (n = 11) lambs were resuscitated and maintained, with magnetic resonance spectroscropy (MRS) performed at 12 and 72 h, and were then killed at 72 h. Cord blood was collected once the cord was clamped, and mononuclear cells were isolated and labelled fluorescently and administered to control (n = 3) or asphyxia (n = 8) lambs. Asphyxia induced a significant increase in cellular apoptosis (caspase‐3 immunopositive) within all brain regions examined, including cortex, hippocampus, thalamus, striatum and subcortical white matter (P < 0.01 vs. control). Additionally, asphyxia induced significant and widespread astrogliosis and increased inflammatory cells (activated microglia and macrophages). The administration of UCB mononuclear cells (asphyxia+UCB) significantly decreased neuronal apoptosis, astrogliosis and inflammation (P < 0.05 vs. asphyxia alone). Asphyxia+UCB lambs also demonstrated decreased brain metabolites lactate:choline (P = 0.01) and lactate:N‐acetylaspartate (P < 0.01) from 12 to 72 h, detected using MRS. Autologous UCB mononuclear cell treatment restores normal brain metabolism following perinatal asphyxia, and reduces brain inflammation, astrogliosis and neuronal apoptosis, supporting its use as a neuroprotective therapy following asphyxia.
Key points
Asphyxia at the time of birth is a significant cause of death or disability in newborns. There is very limited treatment available for these newborns.
Autologous umbilical cord blood (UBC) mononuclear cells reduce clinical markers of brain damage following perinatal asphyxia.
Autologous UBC mononuclear cells reduce neuroinflammation and neuronal apoptosis within the brain following perinatal asphyxia.
Autologous UBC mononuclear cells administered 12 h after perinatal asphyxia are neuroprotective, and a well‐tolerated and feasible treatment for infants following hypoxic ischaemic encephalopathy.
Abbreviations
- CSF
cerebrospinal fluid
- GFAP
glial fibrillary acidic protein
- HIE
hypoxic ischaemic encephalopathy
- HLA
human leukocyte antigen
- HR
heart rate
- IBA‐1
ionised calcium‐binding adapter molecule 1
- IL
interleukin
- MAP
mean arterial pressure
- MRI
magnetic resonance imaging
- MRS
magnetic resonance spectroscopy
- NAA
N‐acetylaspartate
- TNF
tumour necrosis factor
- UCB
umbilical cord blood
Introduction
Acute asphyxia at the time of birth remains a significant cause of perinatal death or long‐term disability. Worldwide, perinatal asphyxia accounts for nearly one in four neonatal deaths, while an estimated 1 million or more infants who survive perinatal asphyxia each year will be diagnosed with cerebral palsy and/or serious cognitive and other developmental disabilities (Lawn et al. 2007; Edwards et al. 2010). When prolonged and severe, perinatal asphyxia results in hypoxic ischaemic encephalopathy (HIE). In high‐income countries, babies with HIE are treated with hypothermia, although meta‐analyses show that it confers only modest improvements in survival and neurodevelopmental outcomes (Edwards et al. 2010; Jacobs et al. 2013). Additionally, for hypothermia to be efficacious, it must commence within the first 6 h after birth, which may limit its clinical application (Drury et al. 2010). New therapeutic strategies that extend the treatment window and mitigate the known injurious pathways leading to brain injury are required to improve outcomes subsequent to perinatal asphyxia.
Perinatal asphyxia and reperfusion of cerebral tissue leads to complex cellular cascades that can ultimately result in widespread cell death. Animal studies and magnetic resonance imaging (MRI) in humans have revealed that perinatal asphyxia induces brain injury that evolves over time and can be divided into different phases. The primary phase describes the period of acute asphyxial insult, initiating mitochondrial dysfunction, excitotoxicity and the programming of cell death (apoptotic) pathways within the brain. A latent phase follows reperfusion, characterised initially by apparent recovery of metabolic processes only to be followed by a secondary phase of cell death about 6 h later. Events that occur during this secondary phase are considered critical for intervention because, even when the primary insult has been very severe, most neuronal death is initiated during the secondary phase (Inder & Volpe, 2000; Volpe, 2012). The secondary phase leads to up‐regulation of brain inflammatory pathways including the production of pro‐inflammatory cytokines, interleukins (IL) ‐6 and ‐8. Inflammatory cytokines are expressed within the brain and cerebrospinal fluid (CSF) within hours following HIE and are associated with subsequent development of cerebral palsy (Dammann & O'Shea, 2008). Thus, there is a critical window of timing in which preventative treatment could be employed.
Stem cell therapies are being investigated for the early treatment of developmental brain injury, such as in preterm birth or perinatal asphyxia (Pimentel‐Coelho & Mendez‐Otero, 2010; Li et al. 2014). Several primary sources of stem cells have been used to investigate their neuroprotective benefits – umbilical cord blood (UCB) is a particularly promising source of stem cells that is very appealing for use in prevention of perinatal brain injury (Bennet et al. 2012). UCB can be readily collected at birth and contains large numbers of mononuclear cells with a heterogeneous population of stem/progenitor cells, it has low immunogenicity, it can be administered allogeneically with minimal human leukocyte antigen (HLA) matching, and UBC cells also demonstrate high plasticity (Bennet et al. 2012; Castillo‐Meléndez et al. 2013; Li et al. 2014). Human UCB mononuclear cells have been investigated as a neuroprotective treatment in neonatal rats exposed to hypoxia–ischaemia (Meier et al. 2006; Pimentel‐Coelho & Mendez‐Otero, 2010; Geißler et al. 2011), where they have been shown to decrease cell death and induce functional motor and behavioural improvements.
The current study was undertaken to examine whether autologous UCB mononuclear cells demonstrate anti‐inflammatory and neuroprotective benefits in a large animal model of perinatal asphyxia and subsequent encephalopathy. We have shown previously that perinatal (birth) asphyxia in term lambs mimics the clinical characteristics of human HIE, inducing brain anaerobic metabolism with increased brain lactate, astrocyte activation and neuronal death (Aridas et al. 2014). This animal model offers the ability to collect, process and administer autologous UCB mononuclear cells, over a time period that could reasonably be replicated in human babies following birth asphyxia while also allowing the ability to track the labelled cells within the brain, and to examine brain histopathology following treatment. We hypothesised that autologous UCB mononuclear cells administered 12 h after perinatal asphyxia would reduce brain inflammation and neuronal cell death.
Methods
Ethical approval
Experiments complied with the National Health and Medical Research Council of Australia guidelines for the care and use of animals for scientific purposes and were approved by Monash Medical Centre Animal Ethics Committee A.
Surgery
Near‐term twin pregnant ewes (Merino cross Border Leicester) at 139–141 days of gestation (term gestation ∼145 days) underwent sterile surgery induced with sodium thiopentone and were maintained under general anesthesia with 1–2.5% isoflurane. The head of the fetus remained in utero, while the fetal body was exposed to allow catherisation of a femoral artery and vein, and a brachial artery. The fetus was then returned into the amniotic cavity. The femoral artery catheter was used for continuous physiological data recording (Powerlab SP, ADInstruments Pty Ltd, Bella Vista NSW Australia), commencing immediately after the catheter was inserted. Ten minutes of in utero recordings were made prior to the hypoxia–ischaemia insult.
Hypoxia–ischaemia and resuscitation
Once the fetus was instrumented, it was randomly allocated to one of four experimental groups using serially numbered opaque envelopes: control, control+UCB, asphyxia and asphyxia+UCB. In all fetal lambs, the umbilical cord was exposed, while the head and upper body of the fetus remained in utero, and an umbilical cord clamp was placed 50 mm from the fetus. In control animals, the umbilical cord was immediately cut and the lamb delivered and resuscitated. Lambs undergoing asphyxia remained in utero until mean arterial blood pressure (MAP) decreased to 18–20 mmHg. The cord was then cut, and the lamb delivered and resuscitated as described below. We have previously shown that this protocol produces a clinically applicable degree of severe birth asphyxia and HIE (Aridas et al. 2014).
All lambs were resuscitated according to the Australian and New Zealand Resuscitation Council guidelines for neonatal resuscitation (www.resus.org.au). Briefly, immediately after cutting the cord and delivery, lambs were dried and provided with tactile stimulation. All lambs were intubated and received positive pressure mechanical ventilation (NeoPuff, Fisher and Paykel Healthcare, Aukland, New Zealand; 30 cmH2O positive inspiratory pressure (PIP); 5–8 cmH2O positive end expiratory pressure (PEEP), 10 l min–1 of room air, 30 breaths min–1). Assisted ventilation was commenced following initial resuscitation as required (Babylog 8000+, Draeger, Lübeck, Germany; volume guarantee 5 ml kg−1, PEEP 5–7 cmH2O, 30 breaths min–1). Parameters were altered to maintain target oxygen saturation between 85 and 90% within the first 10 min of birth, and thereafter > 90%. Assisted ventilation was ceased if lambs began to spontaneously breath > 50% of the time. Blood samples were collected for assessment of blood gases, biochemistry and inflammatory markers at regular time points: in utero, during asphyxia (5 and 8 min), and postnatally at 30 min, and 1, 2, 12, 24, 48 and 72 h, with additional time points for animals treated with UCB mononuclear cells at 11 h 45 min, and 12 h 15 min.
UCB collection and processing
Where lambs were allocated into groups to receive autologous cord blood mononuclear cell administration, UCB was collected immediately after the umbilical cord was clamped. UCB was collected from accessible arteries and veins, immediately distal to the cord clamp. An 18‐gauge needle was used to collect blood into heparin pre‐coated 20 ml syringes. On average, 80 ± 8 ml of UCB was collected from each lamb. UCB was then stored in falcon tubes at room temperature until cell isolation and labelling.
Five hours before administration to the lambs, the mononuclear cell layer of the UCB was isolated. Briefly, UCB was centrifuged at 1000 g for 12 min, with no brake. The mononuclear cells were separated and washed in 20 ml PBS and centrifuged at 1000 g for 5 min to isolate a cell pellet. Red blood cell lysis buffer (ammonium chloride, potassium bicarbonate and EDTA dissolved in double distilled water) was added for 2 min to remove excess red blood cells. Cell counts and viability were determined using trypan blue exclusion dye (Gibco, Waltham, MA, USA) and counted with a haemocytometer. The reaction was stopped with excess media (16.5% fetal bovine serum and Alpha‐MEM; Gibco) and centrifuged at 500 g for 5 min, and the supernatant then aspirated. The isolated mononuclear cells used for administration to lambs were re‐suspended in media and labelled by incubating them with a fluorescent‐iron cell tag (Iodex FITC) in a 10 cm Petri dish for 4 h at 37°C. The mononuclear cells were harvested and then centrifuged (300 g for 5 min), the supernatant was aspirated and re‐suspended in 3 ml PBS and cells were re‐counted using a haemocytometer. Cells were then kept on ice until administration.
UCB mononuclear cell administration
At 12 h after birth, control+UCB and asphyxia+UCB lambs were administered ∼100 million UCB mononuclear cells. Cells were administered slowly over 2 min via the brachial artery catheter followed by 3 ml of saline to flush the line. Prior to, during and following UCB mononuclear cell administration, physiological parameters [MAP, heart rate (HR), pH, base excess, bicarbonate, lactate, and ] were assessed to determine the safety of acute administration of UCB mononuclear cells.
Maintenance and neuro‐behavioural milestones
Lambs were provided with maintenance fluid (10% glucose; 40 ml kg–1 day–1) to maintain glucose levels > 3.5 mmol l–1 until oral intake was established. Once alert and extubated, lambs with good physical tone and suckling reflex were offered sheep milk formula orally (Sheep Milk Replacer, Wombaroo Food Products, Glen Osmond, SA, Australia).
Lambs are precocious and therefore normal lambs achieve a range of clinically measurable behavioural milestones in the hours to days after birth that can provide an index of wellbeing, and motor and cognitive function. Tone, suckling ability, time taken to attempt to stand, attainment of standing position for > 5 s (Miller et al. 2014) and seizure activity were assessed, using a procedure modified to reflect HIE stages. Overt clinical seizures were assessed by clinical personnel (A.M., F.W. and M.C.F.) and defined as repetitive eye movements, ‘lip smacking’, neck arching, ‘running’ leg movements and apnoeic episodes, as described for human seizures (Volpe, 2001). Overt clinical seizures were treated with 20 mg kg−1 phenobarbitone, administered by intravenous infusion in saline over 30 min.
Magnetic resonance spectroscopy (MRS)
MRS analysis of a region of interest within the lamb brain was undertaken at approximately 12 h 30 min (30 min after UCB mononuclear cell administration) and again at 72 h after birth, as previously described (Aridas et al. 2014). Briefly, scans were carried out, under light sedation (0.1 mg kg−1, Domitor, Pfizer Australia, Sydney, Australia), in a 3 T Siemens Vario scanner with the head of the lamb placed in an eight‐channel knee coil. The deep grey matter within the brain was the primary area of interest and was assessed by using a 2 cm3 voxel incorporating the basal ganglia, thalamus and hippocampus. For each metabolite, lactate, choline and N‐acetylaspartate (NAA), relative concentrations were measured via a computer algorithm of peak area under the MRS curve. The ratios of the three metabolites of interest (NAA:choline, lactate:choline, lactate:NAA) were calculated at 12 and 72 h time points.
Cytokine assay
Cytokine concentrations of IL‐1β, IL‐6, IL‐10 and tumour necrosis factor‐α (TNF‐α) were assessed in plasma and CSF using a Flex Set Cytokine Bead Array Kit (BD Biosciences, Australia), following the manufacturer's instructions.
Brain pathology
Lambs were killed immediately after the 72 h MR via an overdose of pentobarbitone. CSF was collected and the brain removed and weighed. The left‐brain hemisphere was divided into regions of interest and frozen for analysis at a later date. The right brain hemisphere was cut coronally into 5 mm slices from the frontal cortex and slices were immersion fixed in 4% paraformaldehyde for 48 h and then embedded in paraffin wax for histological analysis.
Cellular apoptosis was assessed using immunostaining for activated caspase‐3 (R&D Systems, Minneapolis, MN, USA). Brain inflammatory cell (macrophages and microglia) counts were assessed using ionised calcium‐binding adapter molecule 1 (IBA‐1; Wako Pure Chemical Industries, Osaka, Japan). Astrocytes were assessed (cell number and morphology) using immunohistochemistry for glial fibrillary acidic protein (GFAP; Sigma‐Aldrich Co. LLC, Castle Hill, NSW, Australia).
For immunostaining, brain blocks containing the hippocampus, striatum and thalamus were sectioned at 10 μm. Two serial sections were placed on SuperFrost Plus glass slides and dewaxed in xylene and rehydrated through a series of ethanol dilutions. Sections underwent blockade of endogenous peroxidase activity by heating in citric acid buffer three times for 5 min each followed by a 20 min incubation in citric acid buffer and a subsequent incubation in 3% hydrogen peroxide in 50% methanol for a further 20 min. Non‐specific binding was blocked by animal serum (caspase‐3: 5% normal goat serum and 1% BSA; IBA‐1: 10% normal goat serum; GFAP: 5% normal rabbit serum) in 0.3% Triton X‐100 PBS for 45 min. Slides were then incubated in primary antibody solution (caspase‐3: 1:1000 in 5% normal goat serum, R&D Systems; IBA‐1: 1:500, Wako Pure Chemical Industries; GFAP: 1:400, Sigma‐Aldrich) in 0.2% Triton X‐100 PBS at 4°C overnight. Sections were then incubated in a 1:200 dilution of secondary antibody [caspase‐3 and IBA‐1: biotinylated goat anti‐rabbit IgG antibody (Vector Laboratories, Burlingame, CA, USA); GFAP: biotinylated rabbit anti‐mouse IgG antibody (Wako Pure Chemical Industries)] in PBS for 1 h. Staining was visualised using 3,3’‐diaminobenzidine (Pierce Biotechnology, Rockford, IL, USA). Following immunostaining, sections were cover slipped using aqueous mounting medium (Dako Australia, Campbellfield, Vic., Australia) after drying with DEPX (Merck, Kilsyth, Vic., Australia). Immunopositive cells were counted by light microscopy (Olympus, Tokyo, Japan).
Two serial sections of each brain slice were examined. Within each region of interest, the average number of positive cells was counted across three fields of view. Results were then averaged across all animals in each group. The brain regions of interest were the hippocampus (CA1 and dentate gyrus), mid‐temporal cortex (pleomorphic layer), striatum (internal capsule) thalamic nuclei (pretectal nucleus) and sub‐cortical white matter.
Double label immunofluorescence was used to identify apoptotic neurons using mouse monoclonal anti‐NeuN (1:600; Merck‐Millipore) and rabbit polyclonal anti‐human/mouse activated caspase‐3 (1:1000; R&D Systems). Immunoreactivity was visualised with Alexa Fluor 594 goat anti‐mouse (red, 1:1000; Invitrogen, Mount Waverley, Vic., Australia) for NeuN and Alexa Fluor 488 goat anti‐rabbit (green, 1:1000; Invitrogen) for caspase‐3. Double‐label fluorescence images were visualised with fluorescence (red and green filters) using a Nikon C1 Digital Eclipse Modular Confocal Microscope System (Nikon Instruments, Tokyo, Japan) and photographs weretaken (magnification: ×200 for NeuN/lcaspase‐3 double labelling) using the NIS elements software (Nikon Instruments). Images were analysed using fluorescence microscopy (cortex and hippocampus, CA1) using a semi‐quantitative assessment utilising the following scoring system: 1–5 cells, 6–15 cells, 15–25 cells and > 25 cells immunopositive for both NeuN and caspase‐3 per field of view within the cortex and hippocampus.
Statistics
Data are presented as mean ± SEM. Blood gas parameters were analysed using two‐way (time and group) repeated measures ANOVA. One‐way ANOVA with Tukey's multiple comparison post‐hoc analysis was used for cell counts, cytokine assay data analysis, clinical assessments and brain metabolites obtained via MRS. Correlation analysis utilised the Pearson correlation coefficient. Values were considered statistically significant at P < 0.05.
Results
Asphyxia and resuscitation
Thirty‐one newborn lambs were studied. Twenty animals were exposed to asphyxia (asphyxia, n = 12; asphyxia+UCB, n = 8), and 11 were control animals (control, n = 8; control+UCB, n = 3). The sex of each animal was noted but there were insufficient numbers of each sex to allow any differences between sex to be explored. Fetal physiological parameters including MAP, HR, blood O2, CO2, pH and lactate in utero were not different between groups (Fig. 1, Table 1). The mean duration of asphyxia (10 min 7 s) was not different between groups; (asphyxia: 9 min 52 s ± 25 s; asphyxia+UCB: 10 min 34 s ± 25 s; P = 0.3). In all asphyxic animals, cord occlusion caused an initial transient hypertension and subsequent hypotension to a nadir (∼12–14 mmHg) prior to resuscitation with no differences between the asphyxia and asphyxia+UCB groups (P = 0.3). Heart rate fell progressively to 80–90 bpm in asphyxia animals prior to resuscitation (P = 0.1; Fig. 1). In response to asphyxia, we observed a mixed metabolic–respiratory acidosis with disturbance in pH, lactate, base excess, and compared to control (Table 1). These parameters returned to control levels within 4 h after birth, as follows: pH and by 30 min, by 1 h, base excess and bicarbonate by 2 h, and lactate by 4 h. A secondary significant disturbance in blood parameters was also observed for lactate (8–24 h), base excess (4–24 h), bicarbonate (8–24 h) and (12 h). There was no difference in birth weight between groups (control: 4.1 ± 0.3 kg; control+UCB: 4.3 ± 0.9 kg; asphyxia: 3.5 ± 0.9 kg; asphyxia+UCB: 3.7 ± 0.5 kg; P = 0.45).
Figure 1. Physiological parameters in utero, during the asphyxia, and 8 h ex utero .
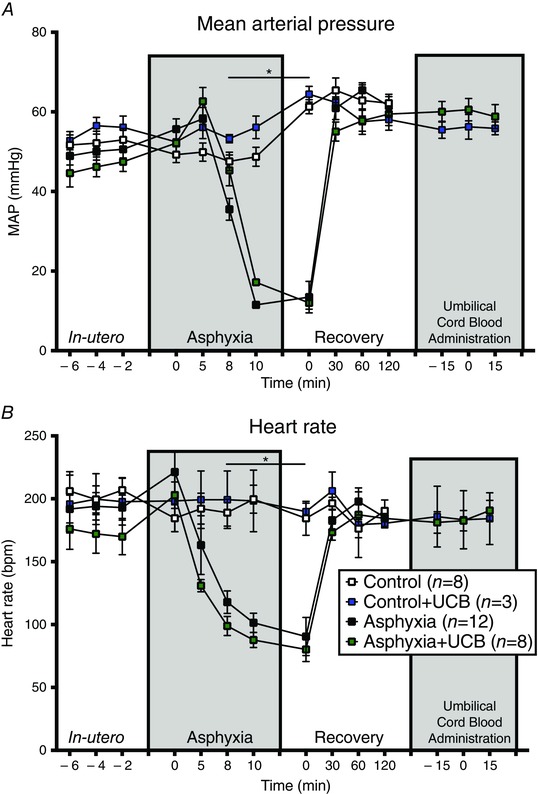
In utero there was no difference seen between groups in MAP or HR. Once asphyxia was commenced, a transient hypertension was seen followed by a gradual decrease in blood pressure until the cessation of the asphyxia when blood pressure dropped to below 20 mmHg. Following delivery and resuscitation, MAP increased and stabilised. HR showed a continual decrease following asphyxia commencement and stabilised at about 80–90 bpm before delivery and resuscitation resulted in a return to levels of approximately 200 bpm.
Table 1.
Physiological parameters in utero, during the asphyxia (8 min), during the recovery phase following asphyxia (10 min, 30 min, 1 h, 2 h, 4 h and 8 h), during umbilical cord administration (−15 min, 12 h, +15 m) and during the maintenance phase (24 h, 48 h, 72 h)
| Recovery | UCB administration | Maintenance | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Treatment | In utero | Asphyxia | 10 min | 30 min | 1 h | 2 h | 4 h | 8 h | −15 min | 12 h | +15 min | 24 h | 48 h | 72 h | |
| pH | |||||||||||||||
| Control | 7.3 ± 0.0 | — | 7.2 ± 0.0 | 7.2 ± 0.0 | 7.2 ± 0.0 | 7.3 ± 0.0 | 7.3 ± 0.0 | 7.3 ± 0.0 | — | 7.3 ± 0.0 | — | 7.4 ± 0.0 | 7.4 ± 0.0 | 7.4 ± 0.0 | |
| Control+UCB | 7.3 ± 0.0 | — | 7.2 ± 0.0 | 7.2 ± 0.0 | 7.3 ± 0.0 | 7.3 ± 0.0 | 7.3 ± 0.0 | 7.3 ± 0.0 | 7.3 ± 0.0 | 7.4 ± 0.0 | 7.3 ± 0.0 | 7.4 ± 0.0 | 7.4 ± 0.0 | 7.4 ± 0.0 | |
| Asphyxia | 7.2 ± 0.0 | 6.9 ± 0.0* | 7.1 ± 0.0* | 7.2 ± 0.0 | 7.2 ± 0.0 | 7.3 ± 0.0 | 7.3 ± 0.0 | 7.3 ± 0.0 | — | 7.4 ± 0.0 | — | 7.4 ± 0.0 | 7.4 ± 0.0 | 7.4 ± 0.0 | |
| Asphyxia+UCB | 7.3 ± 0.0 | 6.9 ± 0.0* | 7.1 ± 0.0* | 7.2 ± 0.0 | 7.3 ± 0.0 | 7.3 ± 0.0 | 7.3 ± 0.0 | 7.3 ± 0.0 | 7.3 ± 0.0 | 7.3 ± 0.0 | 7.3 ± 0.0 | 7.4 ± 0.0 | 7.4 ± 0.0 | 7.4 ± 0.0 | |
| Lactate | |||||||||||||||
| Control | 2.2 ± 0.2 | — | 3.8 ± 0.7 | 3.4 ± 0.5 | 2.3 ± 0.4 | 2.1 ± 0.2 | 3.8 ± 0.5 | 4.1 ± 0.6 | — | 4.7 ± 0.5 | — | 3.0 ± 0.2 | 2.3 ± 0.2 | 2.0 ± 0.2 | |
| Control+UCB | 2.6 ± 0.1 | — | 2.6 ± 0.7 | 2.1 ± 06 | 1.9 ± 0.4 | 1.1 ± 0.1 | 1.9 ± 0.1 | 4.0 ± 0.1 | 4.0 ± 0.1 | 4.0 ± 0.0 | 3.6 ± 0.4 | 3.6 ± 0.3 | 2.3 ± 0.3 | 1.8 ± 0.3 | |
| Asphyxia | 3.4 ± 0.6 | 9.4 ± 0.6* | 7.4 ± 0.5* | 7.3 ± 0.6* | 6.1 ± 0.7* | 5.1 ± 0.5* | 4.7 ± 0.5 | 7.2 ± 0.6* | — | 8.5 ± 0.8* | — | 5.0 ± 0.5* | 2.3 ± 0.2 | 2.5 ± 0.3 | |
| Asphyxia+UCB | 3.0 ± 0.4 | 9.4 ± 0.5* | 7.5 ± 0.5* | 7.3 ± 0.4* | 5.5 ± 0.7* | 3.7 ± 0.3* | 3.2 ± 0.3 | 6.2 ± 0.8* | 7.0 ± 1.0 | 7.0 ± 0.8* | 5.9 ± 0.7 | 5.8 ± 0.6* | 3.8 ± 0.5 | 2.1 ± 0.2 | |
| Base excess | |||||||||||||||
| Control | −2.6 ± 2.0 | — | −7.3 ± 1.3 | −5.6 ± 0.9 | −4.9 ± 0.9 | −3.0 ± 0.9 | −4.8 ± 1.4 | −3.3 ± 0.9 | — | −2.5 ± 0.6 | — | 3.1 ± 1.0 | 4.6 ± 1.2 | 4.3 ± 1.3 | |
| Control+UCB | −1.5 ± 0.6 | — | −3.9 ± 0.9 | −1.7 ± 0.9 | −0.5 ± 1.5 | −0.9 ± 1.1 | −1.4 ± 1.0 | −3.7 ± 1.0 | −3.7 ± 1.0 | −1.9 ± 0.8 | −3.8 ± 0.0 | 0.3 ± 0.7 | 4.2 ± 0.6 | 2.1 ± 0.4 | |
| Asphyxia | −2.6 ± 1.2 | −13.4 ± 0.7* | −14.0 ± 1.3* | −11.0 ± 1.0* | −8.4 ± 1.1* | −4.9 ± 1.0 | −4.6 ± 1.2 | −8.4 ± 1.5* | — | −7.6 ± 1.2* | — | −0.4 ± 1.0 | 4.3 ± 1.3 | 2.8 ± 1.4 | |
| Asphyxia+UCB | 0.1 ± 0.4 | −11.6 ± 0.6* | −12.2 ± 1.0* | −7.1 ± 0.7* | −3.9 ± 0.9 | −1.9 ± 0.5 | −0.4 ± 0.4* | −5.0 ± 1.0 | −4.9 ± 0.9 | −5.8 ± 0.8 | −6.7 ± 1.2 | −2.3 ± 1.3* | 3.9 ± 0.8 | 4.7 ± 0.7 | |
| Bicarbonate | |||||||||||||||
| Control | 24 ± 2 | — | 21 ± 1 | 24 ± 1 | 24 ± 1 | 23 ± 1 | 21 ± 2 | 22 ± 1 | — | 24 ± 1 | — | 27 ± 1 | 29 ± 1 | 28 ± 1 | |
| Control+UCB | 23 ± 2 | — | 24 ± 1 | 27 ± 1 | 25 ± 1 | 25 ± 1 | 24 ± 1 | 21 ± 1 | 21 ± 1 | 22 ± 1 | 22 ± 0 | 24 ± 1 | 27 ± 1 | 26 ± 0 | |
| Asphyxia | 26 ± 1 | 24 ± 1 | 16 ± 1* | 17 ± 1* | 19 ± 1* | 21 ± 1 | 21 ± 1 | 19 ± 1* | — | 24 ± 1 | — | 24 ± 1 | 29 ± 1 | 27 ± 1 | |
| Asphyxia+UCB | 28 ± 0 | 25 ± 1 | 18 ± 1* | 20 ± 1* | 23 ± 1 | 24 ± 0 | 24 ± 0 | 20 ± 1 | 22 ± 1 | 21 ± 1* | 19 ± 1 | 22 ± 1* | 28 ± 1 | 28 ± 1 | |
|
|
|||||||||||||||
| Control | 27 ± 1 | — | 37 ± 6 | 42 ± 7 | 47 ± 5 | 37 ± 4 | 49 ± 3 | 61 ± 10 | — | 50 ± 6 | — | 58 ± 5 | 70 ± 4 | 74 ± 4 | |
| Control+UCB | 32 ± 2 | — | 36 ± 9 | 41 ± 7 | 72 ± 16 | 55 ± 7 | 77 ± 5 | 57 ± 13 | 60 ± 10 | 65 ± 5 | 53 ± 0 | 76 ± 9 | 81 ± 10 | 88 ± 12 | |
| Asphyxia | 26 ± 1 | 6.8 ± 1.8* | 57 ± 6* | 54 ± 5 | 58 ± 7 | 63 ± 8 | 68 ± 5 | 71 ± 6 | — | 68 ± 6* | — | 78 ± 6 | 72 ± 5 | 81 ± 6 | |
| Asphyxia+UCB | 25 ± 2 | 6.1 ± 1.2* | 69 ± 11* | 58 ± 7 | 50 ± 8 | 43 ± 3 | 56 ± 6 | 55 ± 5 | 55 ± 5 | 70 ± 10* | 81 ± 9 | 90 ± 6 | 75 ± 9 | 82 ± 4 | |
|
|
|||||||||||||||
| Control | 57 ± 5 | — | 58 ± 5 | 71 ± 7 | 65 ± 9 | 52 ± 3 | 50 ± 5 | 49 ± 2 | — | 41 ± 1 | — | 41 ± 1 | 47 ± 2 | 46 ± 2 | |
| Control+UCB | 74 ± 5 | — | 63 ± 12 | 65 ± 1 | 48 ± 4 | 51 ± 2 | 48 ± 1 | 42 ± 2 | 42 ± 2 | 39 ± 1 | 42 ± 3 | 41 ± 2 | 43 ± 1 | 42 ± 1 | |
| Asphyxia | 69 ± 1 | 139 ± 6* | 60 ± 6 | 52 ± 2* | 51 ± 5 | 50 ± 4 | 44 ± 2 | 42 ± 4 | — | 42 ± 4 | — | 42 ± 4 | 47 ± 3 | 42 ± 1 | |
| Asphyxia+UCB | 63 ± 6 | 140 ± 5* | 61 ± 7 | 50 ± 2* | 53 ± 4 | 58 ± 2 | 47 ± 2 | 41 ± 2 | 45 ± 3 | 43 ± 1 | 40 ± 2 | 40 ± 1 | 42 ± 1 | 44 ± 1 | |
In utero there is no difference between groups. The asphyxia insult causes a derangement in pH, lactate, base excess, and by 8 min into the asphyxia. Bicarbonate is decreased by 10 min following birth. In asphyxia groups these factors return to control levels within 4 h after birth (pH: 30 min; lactate: 4 h; base excess: 2 h; bicarbonate: 2 h; : 30 min; : 1 h). A second derangement is seen between 4 and 24 h in some groups (lactate: 8–24 h; base excess: 4–24 h; bicarbonate: 8–24 h; : 12 h). There was no significant change in parameters with UCB administration. All factors showed significant differences for time and group.
*Versus Control±UCB (P < 0.05).
UCB administration
Lambs in the control+UCB and asphyxia+UCB groups received autologous UCB mononuclear cell administration. Cell viability of the mononuclear cells prior to administration was > 80% for all animals. Lambs received 153 ± 12 million cells each (control+UCB: 174 ± 12 million; asphyxia+UCB: 140 ± 15 million; P = 0.2).
Figure 1 and Table 1 summarise MAP, HR and blood parameters before and after cell administration. At the time of UCB mononuclear cell administration, lactate was increased and base excess decreased in asphyxia+UCB compared to control+UCB (lactate: P = 0.03; base excess: P = 0.04). No other parameters were different between groups. Over time, improved in asphyxia+UCB animals (P = 0.04; Table 1).
Neuro‐behavioural milestones
The ability of the lambs to attain normal clinical behavioural milestones was scored at 12 h (immediately prior to UCB mononuclear cell administration) and 72 h after birth. At 12 h, all asphyxia lambs (asphyxia alone and asphyxia+UCB) were grouped, and compared to all control lambs (control and control+UCB), as cell administration had not occurred in any animals at this time. At 12 h, all control lambs (11/11) were standing, displayed normal tone and were suckling from a bottle, whereas asphyxia lambs showed deficits in these outcomes (Table 2), with 50% (10/20) unable to stand (P < 0.01) and 50% having abnormal tone (hyper‐ or hypotonic; P < 0.01). By 12 h, seizures were observed in 30% of asphyxia lambs (6/20) compared to no seizures observed in control lambs (P = 0.06). At 72 h, after the administration of cells to a subset of control and asphyxia lambs, all asphyxia+UCB lambs were able to stand (8/8), all but one had normal tone (7/8) and a strong suckle reflex was observed in 88% (7/8). In the asphyxia alone group, 75% (9/12) of lambs were able to stand, 58% (7/12) had normal tone and 66% (8/12) had a strong suckle reflex.
Table 2.
Attainment of neuro‐behavioural milestones at 12 and 72 h postnatally
| Able to stand (>5 s) | Normal tone | Good suckle reflex | ||||||
|---|---|---|---|---|---|---|---|---|
| n | 12 h | 72 h | 12 h | 72 h | 12 h | 72 h | Seizures | |
| Control | 8 | 100% | 100% | 100% | 100% | 100% | 100% | 0% |
| Control+UCB | 3 | 100% | 100% | 100% | ||||
| Asphyxia | 12 | 50% | 75% | 50% | 58% | 55% | 66% | 30% |
| Asphyxia+UCB | 8 | 100% | 75% | 88% | ||||
| P | <0.01 | 0.15 | <0.01 | 0.13 | 0.01 | 0.12 | 0.06 | |
At 12 h, prior to UCB administration parameters were assessed. At this time, asphyxia led to significant derangement in all parameters when compared to control animals. However, at 72 h due to small sample sizes there was no significant difference.
In control lambs, 4/8 were able to drink formula without requiring glucose infusion and all (8/8) established feeding by the end of the experimental period, on average within 1.5 h of birth. Control+UCB animals were similar, 2/3 requiring glucose infusion and all (3/3) establishing feeding within 2 h of birth. All asphyxia animals required glucose infusion. Three of 12 asphyxia animals did not establish feeding and required maintenance fluids over the duration of the experimental period. Those asphyxia animals that did establish feeding took an average of 13 h to commence formula feeding. All asphyxia+UCB animals (8/8) required glucose infusion initially, and all established feeding at an average of 13.5 h.
MRS
Assessment of brain metabolites was undertaken at 12 h 30 min (after cell administration) and at 72 h. At 12 h 30 min, there were no significant differences between experimental groups for NAA:choline, lactate:choline and lactate:NAA (Fig. 2). At 72 h, NAA:choline was significantly elevated in the control lambs compared to 12 h (P < 0.01) and, between groups, NAA:choline was significantly reduced in asphyxia lambs compared to controls (control: 0.81 ± 0.15; asphyxia: 0.57 ± 0.16; P = 0.02; Fig. 2 A), while NAA:choline in asphyxia+UCB lambs was not different to control lambs. Interestingly, in lambs treated with UCB mononuclear cells immediately after the first MRS, we observed a significant decrease in brain lactate (lactate:choline; P = 0.01; Fig. 2 B) and a significant improvement in the ratio of lactate:NAA (P < 0.01, Fig. 2 C). No other parameter showed differences over time.
Figure 2. MRS parameters at 12 and 72 h postnatally .
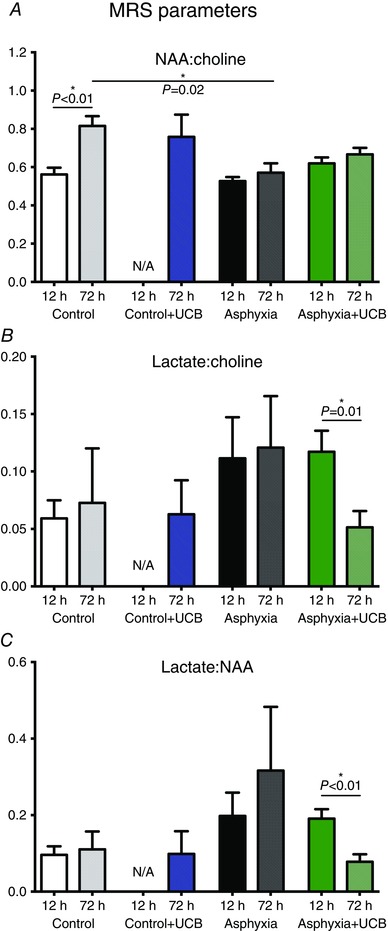
Each control group is represented by results at 12 and 72 h. A, NAA:choline ratio between groups. There is a significant increase between the 12 and 72 h results of control animals. There is a significant difference between control and asphyxia at 72 h. B, a significant decrease in lactate:choline ratio of asphyxia+UCB animals between 12 and 72 h. C, as in B, a significant decrease in lactate:NAA ratio in the asphyxia+UCB animals between 12 and 72 h.
Cytokine assay
There were no significant changes in circulating inflammatory cytokine concentrations (IL‐6, IL‐10, IL1‐β or TNF‐α) in any group measured in plasma. In CSF collected at 72 h, the concentration of IL1‐β was increased to 60 ± 29 pg ml−1 in asphyxia animals but was undetectable in control±UCB and asphyxia+UCB animals. TNF‐α, IL‐10 and IL‐6 were not detectable in any group. There was an > 85% recovery of the standards with an intra‐assay coefficients of variation of: IL‐6, 163.67%; IL‐10, 120.74%; IL1‐β, 157.11%; and TNF‐α, 115.71%.
Brain histopathology
Asphyxia was associated with significant and widespread apoptosis, as evidenced by increased activated caspase‐3, in the brains of untreated lambs, specifically within the dentate gyrus and the CA1 region of the hippocampus, subcortical white matter, the internal capsule, thalamic nuclei and the pleomorphic layer of the cortex (Fig. 3 C, Table 3).
Figure 3. Representative photomicrographs of the thalamic nuclei stained for apoptosis, inflammation and astrogliosis .
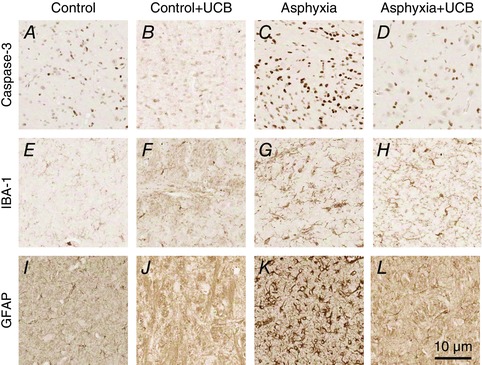
Apoptosis: control (A), control+UCB (B), asphyxia (C) and asphyxia+UCB (D); inflammation: control (E), control+UCB (F), asphyxia (G) and asphyxia+UCB (H); and astrogliosis: control (I), control+UCB (J), asphyxia (K) and asphyxia+UCB (L). Asphyxia resulted in increased apoptosis (activated caspase‐3), inflammation (IBA‐1) and astrogliosis (GFAP) compared to controls. UCB administration to asphyxia animals resulted in significantly reduced numbers of apoptotic cells, inflammatory cells and astrocytes. UCB administration to control animals did not result in any change from control only.
Table 3.
Mean cell counts (cells mm2) of immunohistochemistry for apoptosis (activated caspase‐3; Cas‐3), inflammation (ionised calcium‐binding adapter molecule 1; IBA‐1) and astrogliosis (glial fibrillary acidic protein; GFAP)
| Control | Control+UCB | Asphyxia | Asphyxia+UCB | P | |
|---|---|---|---|---|---|
| Activated Cas‐3 | |||||
| Hippo: CA1 | 330 ± 80* | 258 ± 103 | 708 ± 85†, ‡ | 197 ± 84* | < 0.01 |
| Hippo: DG | 827 ± 160* | 506 ± 185* | 1401 ± 72†, §, ‡ | 559 ± 131* | < 0.01 |
| SCWM | 156 ± 56* | 80 ± 71* | 537 ± 67†, §, ‡ | 206 ± 114* | 0.02 |
| Striat: IC | 169 ± 83* | 49 ± 38* | 438 ± 56†, §, ‡ | 62 ± 18* | < 0.01 |
| Thal: PrT | 192 ± 78 | 159 ± 84 | 372 ± 47‡ | 49 ± 14* | < 0.01 |
| Cortex: PM | 158 ± 66* | 126 ± 32 | 462 ± 87†, ‡ | 163 ± 35* | <0.01 |
| IBA‐1 | |||||
| Hippo:CA1 | 55 ± 18* | 65 ± 26* | 356 ± 57† § ‡ | 122 ± 53* | < 0.01 |
| Hippo: DG | 119 ± 46* | 84 ± 24* | 387 ± 53†, §, ‡ | 162 ± 67* | < 0.01 |
| SCWM | 139 ± 43* | 146 ± 33 | 319 ± 44† | 168 ± 42 | 0.02 |
| Striat: IC | 177 ± 35* | 179 ± 74 | 332 ± 27†, ‡ | 186 ± 53* | < 0.01 |
| Thal: PrT | 44 ± 27* | 125 ± 30* | 449 ± 57†, §, ‡ | 179 ± 42* | < 0.01 |
| Cortex: PM | 72 ± 22 | 122 ± 44 | 227 ± 49 | 101 ± 56 | 0.08 |
| GFAP | |||||
| Hippo: CA1 | 256 ± 83* | 174 ± 79* | 701 ± 57†, §, ‡ | 226 ± 103* | < 0.01 |
| Hippo: DG | 437 ± 129 | 217 ± 124 | 758 ± 55 | 367 ± 138 | < 0.01 |
| SCWM | 326 ± 83 | 189 ± 179* | 609 ± 91§, ‡ | 167 ± 106* | 0.01 |
| Striat: IC | 415 ± 127 | 244 ± 137 | 660 ± 45‡ | 219 ± 79* | < 0.01 |
| Thal: PrT | 410 ± 111* | 415 ± 189 | 760 ± 66†, ‡ | 334 ± 101* | < 0.01 |
| Cortex: PM | 130 ± 67* | 67 ± 23 | 479 ± 79†, ‡ | 74 ± 44* | < 0.01 |
There was a significant increase in apoptosis, inflammation and astrogliosis in all regions examined except for inflammation in the cortex. Hippo: CA1, hippocampus: CA1; Hippo: DG, hippcampus: dentate gyrus; SCWM, subcortical white matter; Striat: IC, striatum: internal capsule; Thal: PrT, thalamus: pretectal nucleus; Cortex: PM, cortex: pleomorphic layer.
P < 0.05: † vs. control; § vs. control+UCB; *vs. asphyxia; ‡ vs. asphyxia+UCB.
There was a significant increase in the number of inflammatory cells, particularly activated microglia and macrophages, within the dentate gyrus and the CA1 region of the hippocampus, subcortical white matter, the internal capsule and thalamic nuclei of asphyxia animals (Fig. 3 G). Morphologically, these inflammatory cells predominately had the appearance of activated microglia with a subset of infiltrating macrophages, indicating widespread inflammation in these regions (Fig. 3 G). There was a strong correlation between the number of inflammatory cells and the number of apoptotic cells within the CA1 region of the hippocampus in all groups (r 2: 0.96; P < 0.01).
The number and morphology of astrocytes was examined by immunostaining for GFAP. We found an increase in the number of astrocytes within all brain regions assessed (see above) in asphyxia brains compared to control brains (Fig. 3 K vs. I). Morphologically, astrocytes in the asphyxia alone lambs were distinct from normal resting astrocytes observed within control brains (Fig. 3 I) showing a reactive gliosis characterised by increased staining intensity of the cell soma and increased number and thickness of processes (Fig. 3 K). Cell counts for apoptosis, inflammation and astrogliosis were not different in control+UCB lambs compared to control lambs. Treatment with UCB mononuclear cells (asphyxia+UCB) significantly decreased the number of inflammatory cells when compared to asphyxia alone animals (P < 0.05), apoptosis (P < 0.05) and astrogliosis (P < 0.05) within the hippocampus, thalamus and internal capsule brain regions (Table 3).
Double label immunohistochemistry was performed to determine whether the apoptosis observed was present in neurons (NeuN immunopositive cells). The control+UCB group was excluded from this analysis as no significant differences in caspase‐3 immunoreactivity were observed between this and the control group in the single label. An increase in activated caspase‐3 immunofluorescence was observed in the cortex and hippocampus (CA1) of asphyxia lambs compared to control lambs (Fig. 4 D and E). In asphyxia+UCB lambs, the number of NeuN immunopositive cells co‐localised with caspase‐3 immunofluorescence was similar to that seen in control lambs (Fig. 4 I and G, respectively). The cytoarchitecture is shown at both high (Fig. 4 G–I) and low (Fig. 4 J–L) power. The CA1 region in asphyxia lambs was altered with disorganised neurons seen in comparison to control (Fig. 4 H and K compared to Fig. 4 G and J, respectively). This abnormality in the hippocampus was not observed in asphyxia+UCB lambs (Fig. 4 I and L). Semi‐quantification of the immunofluorescence revealed a low density (< 15 cells per field in the cortex and hippocampus) of co‐localisation between caspase‐3 and NeuN in control and asphyxia+UCB brains, while the asphyxia alone group demonstrated high levels of co‐localisation (>25 NeuN positive cells co‐localised with caspase‐3 per field; Fig. 4 G, J, I and K vs. Fig. 4 H and L, respectively).
Figure 4. Confocal images of brains at 72 h postnatally (60 h after receiving UCB administration) .
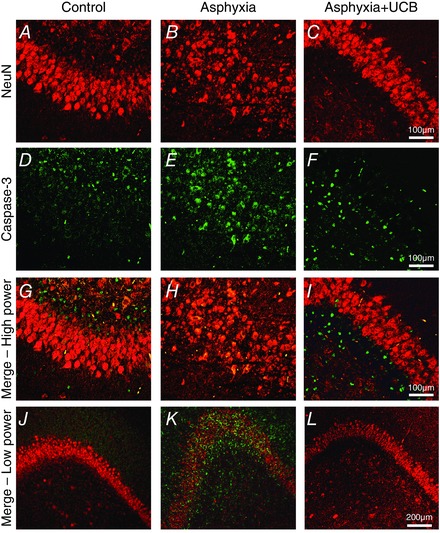
The CA1 of control brains are shown in A, D and G with NeuN (red), caspase‐3 (green) and double fluorescence (merge), respectively. Asphyxia causes neuronal disorganisation in CA1 (B), and increased caspase‐3 immunofluorescence (E), with a large number of NeuN‐positive cells co‐localising with caspase‐3 (H). Asphyxia lambs treated with UCB displayed normal CA1 cytoarchitecture (C), reduced levels of apoptosis (caspase‐3 immunofluorescence, F) and decreased neuronal apoptosis (I). Low power merged images show neuronal disorganisation in asphyxia (K) compared to control (J) and asphyxia+UCB (L).
Labelled UCB mononuclear cells were widely but sparsely distributed within the brains of animals that received UCB, principally in the hippocampus, cortex and the subcortical white matter, corresponding to brain regions that showed significant neuropathology in response to asphyxia (Fig. 5).
Figure 5. Fluorescence microscopy of brains 72 h postnatally (60 h after receiving UCB administration) .
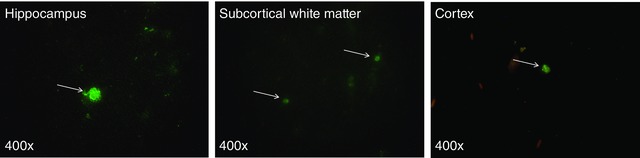
White arrows highlight UCB cells labelled with a fluorescent‐iron cell tag.
Discussion
This study set out to examine whether autologous UCB mononuclear cell administration at 12 h after perinatal (birth) asphyxia provided neuroprotective benefit. We chose to administer the cells at 12 h after birth as this represents a time course that could be readily achieved for clinical collection, processing and re‐administration of autologous UCB mononuclear cells, and could extend the therapeutic window for commencing treatment that is currently 6 h for hypothermia. The results of this study demonstrate that autologous UCB mononuclear cell administration at 12 h after birth reduces brain inflammation, improves brain metabolic activity and prevents neuronal apoptosis in response to perinatal asphyxia. Furthermore, intravenous UCB mononuclear cell administration did not lead to any significant physiological changes that would preclude its use as a safe therapeutic strategy.
Neuroinflammation is a key response following a severe asphyxic insult (Wolfberg et al. 2007; Dammann & O'Shea, 2008). Pro‐inflammatory cytokines, including IL‐6 and Il‐8, are expressed within the brain and CSF following HIE (Dammann & O'Shea, 2008), and excess levels of IL‐6/‐8 lead to activation of resident CNS macrophages and microglia, and induce lactate production within the basal ganglia (Bartha et al. 2004). In this study, the administration of autologous UCB mononuclear cells at 12 h after perinatal asphyxia decreased inflammation and apoptosis. During the secondary phase of insult following perinatal asphyxia, the neuroinflammatory response is considered critical – it is beneficial, acting to recruit inflammatory cells that produce growth factors involved in neurorepair and regeneration. However, when brain inflammatory processes are prolonged or severe, they exacerbate damage by stimulating an influx of cytokines, chemokines and further inflammatory mediators released from activated microglia (Stoll et al. 2002). In turn, inflammatory cytokines within the brain activate the pro‐apoptotic pathways to induce programmed cell death and neuronal apoptosis (Hu et al. 1997). Consistent with this, in asphyxia lambs we found a strong correlation between the presence of inflammatory cells within the hippocampus and the extent of apoptosis. Hence, the anti‐inflammatory properties of UCB mononuclear cell administration following perinatal asphyxia are likely to be a key mechanism of neuroprotection. A variety of anti‐inflammatory cells present within the mononuclear cell fraction may be active in this process, including regulatory T cells, mesenchymal stem cells and monocyte derived suppressor cells (Castillo‐Meléndez et al. 2013; Li et al. 2014).
Selective neuronal cell loss is the most common brain injury observed in infants with HIE, reflecting widespread degeneration of neurons in the grey matter of the cerebral cortex, hippocampus, basal ganglia and thalamus (Folkerth, 2005; Volpe, 2012). Neuronal populations of the cerebral cortex and hippocampus are said to be particularly vulnerable to hypoxic injury in the full‐term human neonate (Volpe, 1997). In this study we used a clinically relevant model of birth asphyxia in term lambs to demonstrate brain injury consistent with newborn HIE (Aridas et al. 2014). We showed cellular apoptosis within the hippocampus, striatum, thalamus, cortical grey matter and subcortical white matter. Not surprisingly, double‐label immunostaining demonstrated that the principal cell type affected by apoptosis was neurons. Both necrotic and apoptotic cell death result from a severe hypoxic insult, with Nakajima et al. (2000) demonstrating in hypoxic–ischaemic newborn rats that the time course for apoptosis peaks between 12 h and 7 days after insult. Thus, the administration of UCB mononuclear cells at 12 h after perinatal asphyxia is timely, reducing brain inflammation and the upregulation of the apoptotic cell cascade towards cell death. Consistent with findings in human neonates, we observed an increase in neuronal apoptosis in the asphyxia group compared to control lambs, but this was prevented in asphyxiated lambs treated with UCB mononuclear cells, indicating that UCB mononuclear cell therapy results in neuroprotection. Complementing our findings, UCB mononuclear cells were found to be neuroprotective in a human in vitro model of neuronal hypoxia, by reducing caspase‐3‐mediated apoptosis (Hau et al. 2008).
Whilst we were mainly interested in neuronal response in this study, glial cells are also involved in injury and repair following perinatal asphyxia. We assessed the number and morphology of astrocytes within our brain regions of interest and found significant astrogliosis in response to perinatal asphyxia with an increased number of astrocytes and with a ‘reactive’ morphology. The administration of UCB at 12 h significantly decreased astrogliosis at 72 h. Reactive astrocytes may be neuroprotective, with the release of neurotrophic factors, although when astrogliosis is severe or prolonged, they can exacerbate neurodegeneration via increasing inflammation, oxidative stress and/or cerebrovascular dysregulation (Avila‐Muñoz & Arias, 2014). Thus, decreasing astrogliosis with UCB administration may be a critical neuroprotective mechanism within the young brain. Evidence to date supports that cord blood cells may provide neuroprotective benefit due to actions on a range of complementary biochemical pathways that become dysregulated in response to perinatal asphyxia.
Several identified sources of stem cells have been examined for their neuroprotective benefits. In the current study we administered autologous UCB mononuclear cells, obtained from the umbilical cord of each lamb immediately after cord clamping. This mononuclear cell population contains a heterogeneous mix of haematopoietic, endothelial and mesenchymal progenitor and lymphocytic and monocytic cell types, which all probably contribute towards anti‐inflammatory and neuroprotective benefits. Other studies in rodents using a postnatal model of term hypoxia–ischaemia have shown that xenotransplanted (human) UCB mononuclear cells have the ability to provide a neuroprotective benefit (Meier et al. 2006; Pimentel‐Coelho & Mendez‐Otero, 2010; Geißler et al. 2011). In postnatal day 7 rats exposed to hypoxia–ischaemia, intraperitoneal administration of human UCB mononuclear cells (3 or 24 h after insult) significantly decreased apoptotic and necrotic cell death within the brain, and resulted in functional motor and behavioural improvements (Pimentel‐Coelho et al. 2010; Geißler et al. 2011). Human cells tracked to the lesion site within the young rat brain but did not differentiate into a neuronal phenotype (Meier et al. 2006; Geißler et al. 2011). Conversely, a similar experimental design reported no neuroprotective actions of UCB mononuclear cells in postnatal rat hypoxic–ischaemic injury when cells were injected intravenously at 24 h after insult (de Paula et al. 2009).
Within hours after birth, the number of stem and progenitor cells within the newborn circulation is decreased precipitously, indicative of a loss of cells with potential therapeutic benefit (Li et al. 1999). In non‐asphyxiated human newborns, haematopoetic progenitors (CD34+ cells) show a significant decrease from levels measured at 2 h postnatally to a nadir within 48 h, with the most marked decrease observed within 4 h of birth (Li et al. 1999). This shift in the proportion of circulating stem/progenitor cells soon after birth may place the asphyxic newborn at risk over a critical period in which encephalopathy is developing. In the current study, we administered the UCB mononuclear cells at 12 h after perinatal asphyxia, via a brachial artery to maximise the chance of the cells accessing the brain. However, it is more likely that the cells circulated for some time and ‘homed’ to sites of damage, as, when we collected a small blood sample onto a slide from the femoral artery at 1 h after cell administration in two asphyxia+UCB lambs, we were able to detect fluorescent cells on a blood smear, confirming that UCB mononuclear cells continued to circulate following administration. We chose to administer autologous cord blood cells at 12 h after birth to replicate what could be readily translatable in the clinical situation of a severely asphyxiated newborn, and the time required to diagnose encephalopathy and to process and isolate the cord blood cells. In support for the use of cord blood cells in the clinical situation, Cotten et al. (2014) have shown that autologous UCB collection, preparation and intravenous infusion is feasible and well‐tolerated in clinical practice in infants with HIE. Thus, together these findings indicate that UCB mononuclear cell treatment to HIE infants is feasible and well‐tolerated and the cells probably act via anti‐inflammatory and anti‐apoptotic actions to induce neuroprotection.
MRS provides a non‐biased assessment of the biochemistry of the neonatal brain. We did not observe any differences in brain metabolite concentrations between experimental groups at 12 h after birth. However at 72 h, the NAA:choline ratio was significantly reduced in asphyxia lambs compared to control lambs, indicative of a loss of functional neurons within the basal ganglia, thalamus and hippocampus (the area of interest for the MRS). It was also interesting to note that, where the ratio of lactate:NAA progressively increased (albeit not to significance) in asphyxia alone lambs from 12 to 72 h, brain lactate:NAA decreased significantly in the asphyxia lambs treated with UCB mononuclear cells immediately after the 12 h MRS. Similarly, asphyxia+UCB lambs showed a significant decrease in brain lactate:choline concentration between 12 and 72 h, indicating that UCB mononuclear cell administration was able to reduce and reverse brain lactate induced by anaerobic glycolysis and thereby restore normal brain metabolism over this relatively short period of time. In the clinic, MRS is the most sensitive marker available for the assessment of brain injury, and provides the clinician and parents with crucial prognostic information. MRS is most commonly undertaken 5–7 days following perinatal asphyxia in human neonates (Barkovich & Hallam, 1997), with an increased ratio of lactate:NAA considered a reliable biomarker of HIE and a predictor of death or disability at age 1 year (Amess et al. 1999; Azzopardi & Edwards, 2010). We have also previously shown that changes in brain biochemistry obtained using MRS at 72 h correlated closely with necrotic cell death in those same brain areas of interest (Aridas et al. 2014). Our data provide compelling evidence for UCB mononuclear cells to potentially repair in addition to prevent injury following an asphyxic insult at birth. This improvement in brain metabolism was not observed in asphyxia alone lambs.
The animal model, stem cell procedures and timing of treatment used in this study were undertaken with clinical translation in mind. We acknowledge, however, that this study has two important limitations. First, we did not use hypothermia as a therapy in any of our animals. Hypothermia is now standard clinical care for the term asphyxiated infant with HIE. Having shown the potential utility of UCB cells as a neuroprotectant it would now be important to examine the use of UCB as an adjuvant therapy to hypothermia. However, hypothermia is not a suitable treatment for all asphyxiated neonates, including those born preterm, with congenital malformations, coagulopathies and severe haemorrhage (Jacobs et al. 2013). Furthermore, the therapeutic window for commencing hypothermia is relatively short (< 6 h) and it is not shown to be effective in infants with the most severe encephalopathy (Gluckman et al. 2005). Thus, future experiments should assess UCB administration as an adjuvant therapy with cooling, or compare UCB with the benefits of cooling in a strict experimental paradigm. Second, our lambs were only kept alive for 72 h after the asphyxial insult, allowing us to explore short‐ to medium‐term histopathological effects of the UCB infusions. However, it meant that we were unable to explore long‐term benefits of UCB mononuclear cell therapy, for both function and structure. A major challenge of such longer term studies in our model is simply the cost of keeping the asphyxiated lambs alive longer than 72 h, as they require neonatal intensive‐care unit‐like care 24 h a day. Indeed, we wonder whether longer term experimental studies are merited or whether, given our promising results and good safety data, it would be more ethical to proceed to clinical trials and explore longer term functional outcomes in babies.
In summary, this study demonstrates that autologous UCB mononuclear cells are very effective as a neuroprotective therapy for asphyxiated neonates. The administration of UCB mononuclear cells 12 h after perinatal asphyxia reduced neuroinflammation, astrogliosis and neuronal apoptosis within the basal ganglia, thalamus, hippocampus, cortex and subcortical white matter, and restored normal brain metabolic activity, assessed using MRS within the basal ganglia, thalamus and hippocampus. The results from this study extend the potential therapeutic window of opportunity for treatment to 12 h after birth, where current diagnosis of HIE and treatment with hypothermia must commence within 6 h. Our results also demonstrate the critical nature of brain inflammation contributing to neuronal cell death following perinatal asphyxia, and the ability of UCB mononuclear cells to protect against inflammatory‐mediated cell death. UCB mononuclear cell administration has been shown to be a feasible and well‐tolerated treatment in infants and children with suspected or established brain injury, and the results of the current study show UCB mononuclear cells, administered at 12 h after birth, act during the secondary phase of encephalopathy to reduce metabolic disturbance and brain inflammatory response, which in turn prevents the apoptotic cascade of neuronal cell death. These findings support the use of UCB mononuclear cell administration in this high‐risk group of asphyxiated newborns, for the prevention of long‐term cognitive and developmental disabilities such as cerebral palsy.
Additional information
Competing interests
None of the authors has any conflict of interest related to this work.
Author contributions
Conception and design was provided by: JDSA, CAM, TY, MD, MCF, FW, EMW, GJ, SLM. Financial support provided to: MCF, EMW, GJ, SLM. Administrative support: JDSA, YP, GJ, SLM. Provision of study materials or patients: JDSA, CAM, MCBP, TY, AES, IN, YP, FW, MCM, KB, GJ, SLM. Collection and assembly of data: JDSA, CAM, MCBP, TY, AES, IN, YP, MD, FW, AM, MCM, GJ, SLM. Data analysis and interpretation: JDSA, CAM, MCBP, TY, AES, MD, MCF, FW, AM, MCM, GJ, SLM. Manuscript writing: JDSA, CAM, MCBP, TY, AES, MCF, AM, MCM, EMW, GJ, SLM. Final approval of manuscript: all authors.
Funding
This study was supported by the National Health and Medical Research Council (NHMRC) of Australia Grant No: 1048039, Australian Research Council Future Fellowship (SLM), Inner Wheel Australia, and the Victorian Government's Operational Infrastructure Support Program.
Acknowledgements
The authors gratefully acknowledge the expert technical assistance of Jan Loose, Monique Mortale, Lesley Wiadrowski, David Shipp, Richard McIntyre and Patricia Heidmann.
References
- Amess PN, Penrice J, Wylezinska M, Lorek A, Townsend J, Wyatt JS, Amiel‐Tison C, Cady EB & Stewart A (1999). Early brain proton magnetic resonance spectroscopy and neonatal neurology related to neurodevelopmental outcome at 1 year in term infants after presumed hypoxic‐ischaemic brain injury. Dev Med Child Neurol 41, 436–445. [PubMed] [Google Scholar]
- Aridas JDS, Yawno T, Sutherland AE, Nitsos I, Ditchfield M, Wong FY, Fahey MC, Malhotra A, Wallace EM, Jenkin G & Miller SL (2014). Detecting brain injury in neonatal hypoxic ischemic encephalopathy: closing the gap between experimental and clinical research. Exp Neurol 261, 281–290. [DOI] [PubMed] [Google Scholar]
- Avila‐Muñoz E & Arias C (2014). When astrocytes become harmful: functional and inflammatory responses that contribute to Alzheimer's disease. Ageing Res Rev 18, 29–40. [DOI] [PubMed] [Google Scholar]
- Azzopardi D & Edwards AD (2010). Magnetic resonance biomarkers of neuroprotective effects in infants with hypoxic ischemic encephalopathy. Semin Fetal Neonatal Med 15, 261–269. [DOI] [PubMed] [Google Scholar]
- Barkovich AJ & Hallam D (1997). Neuroimaging in perinatal hypoxic‐ischemic injury. Ment Retard Dev Disabil Res Rev 3: 28–41. [Google Scholar]
- Bartha AI, Foster‐Barber A, Miller SP, Vigneron DB, Glidden DV, Barkovich AJ & Ferriero DM (2004). Neonatal encephalopathy: association of cytokines with MR spectroscopy and outcome. Pediatr Res 56, 960–966. [DOI] [PubMed] [Google Scholar]
- Bennet L, Tan S, Van den Heuij L, Derrick M, Groenendaal F, van Bel F, Juul S, Back SA, Northington F, Robertson NJ, Mallard C & Gunn AJ (2012). Cell therapy for neonatal hypoxia‐ischemia and cerebral palsy. Ann Neurol 71, 589–600. [DOI] [PubMed] [Google Scholar]
- Castillo‐Meléndez M, Yawno T, Jenkin G & Miller SL (2013). Stem cell therapy to protect and repair the developing brain: a review of mechanisms of action of cord blood and amnion epithelial derived cells. Front Neurosci 7, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotten CM, Murtha AP, Goldberg RN, Grotegut CA, Smith PB, Goldstein RF, Fisher KA, Gustafson KE, Waters‐Pick B, Swamy GK, Rattray B, Tan S & Kurtzberg J (2014). Feasibility of autologous cord blood cells for infants with hypoxic‐ischemic encephalopathy. J Pediatr 164, 973–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dammann O & O'Shea TM (2008). Cytokines and perinatal brain damage. Clin Perinatol 35, 643–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Paula S, Vitola AS, Greggio S, de Paula D, Mello PB, Lubianca JM, Xavier LL, Fiori HH & Dacosta JC (2009). Hemispheric brain injury and behavioral deficits induced by severe neonatal hypoxia‐ischemia in rats are not attenuated by intravenous administration of human umbilical cord blood cells. Pediatr Res 65, 631–635. [DOI] [PubMed] [Google Scholar]
- Drury PP, Bennet L & Gunn AJ (2010). Mechanisms of hypothermic neuroprotection. Semin Fetal Neonatal Med 15, 287–292. [DOI] [PubMed] [Google Scholar]
- Edwards AD, Brocklehurst P, Gunn AJ, Halliday H, Juszczak E, Levene M, Strohm B, Thoresen M, Whitelaw A & Azzopardi D (2010). Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: synthesis and meta‐analysis of trial data. BMJ 340, c363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkerth RD (2005). Neuropathologic substrate of cerebral palsy. J Child Neurol 20, 940–949. [DOI] [PubMed] [Google Scholar]
- Geißler M, Dinse HR, Neuhoff S, Kreikemeier K & Meier C (2011). Human umbilical cord blood cells restore brain damage induced changes in rat somatosensory cortex. PLoS ONE 6, e20194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluckman PD, Wyatt JS, Azzopardi D, Ballard R, Edwards AD, Ferriero DM, Polin RA, Robertson CM, Thoresen M, Whitelaw A & Gunn AJ (2005). Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet 365, 663–670. [DOI] [PubMed] [Google Scholar]
- Hau S, Reich DM, Scholz M, Naumann W, Emmrich F, Kamprad M & Boltze J (2008). Evidence for neuroprotective properties of human umbilical cord blood cells after neuronal hypoxia in vitro . BMC Neurosci 9, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Peterson PK & Chao CC (1997). Cytokine‐mediated neuronal apoptosis. Neurochem Int 30, 427–431. [DOI] [PubMed] [Google Scholar]
- Inder TE & Volpe JJ (2000). Mechanisms of perinatal brain injury. Semin Neonatol 5, 3–16. [DOI] [PubMed] [Google Scholar]
- Jacobs SE, Berg M, Hunt R, Tarnow‐Mordi WO, Inder TE, Davis PG. Cooling for newborns with hypoxic ischaemic encephalopathy Cochrane Database Systematic Reviews 2013, Issue 1. Art No.: CD003311. DOI:10.1002/14651858.CD003311.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawn JE, Manandhar A, Haws RA & Darmstadt GL (2007). Reducing one million child deaths from birth asphyxia–a survey of health systems gaps and priorities. Health Res Policy Syst 5, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, McDonald CA, Fahey MC, Jenkin G & Miller SL (2014). Could cord blood cell therapy reduce preterm brain injury? Front Neurol 5, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Liu J, Fok TF, Yau F.W., Wong A, Li CK, Yang M, So KW, Chik KW, Tsang KS, Shing MM & Yuen PM (1999). Human neonatal blood: stem cell content, kinetics of CD34+ cell decline and ex vivo expansion capacity. Br J Haematol 104, 178–185. [DOI] [PubMed] [Google Scholar]
- Meier C, Middelanis J, Wasielewski B, Neuhoff S, Roth‐Haerer A, Gantert M, Dinse HR, Dermietzel R & Jensen A (2006). Spastic paresis after perinatal brain damage in rats is reduced by human cord blood mononuclear cells. Pediatr Res 59, 244–249. [DOI] [PubMed] [Google Scholar]
- Miller SL, Yawno T, Alers NO, Castillo‐Meléndez M, Supramaniam VG, VanZyl N, Sabaretnam T, Loose JM, Drummond GR, Walker DW, Jenkin G & Wallace EM (2014). Antenatal antioxidant treatment with melatonin to decrease newborn neurodevelopmental deficits and brain injury caused by fetal growth restriction. J Pineal Res 56, 283–294. [DOI] [PubMed] [Google Scholar]
- Nakajima W, Ishida A, Lange MS, Gabrielson KL, Wilson MA, Martin LJ, Blue ME & Johnston MV (2000). Apoptosis has a prolonged role in the neurodegeneration after hypoxic ischemia in the newborn rat. J Neurosci 20, 7994–8004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimentel‐Coelho PM & Mendez‐Otero R (2010). Cell therapy for neonatal hypoxic‐ischemic encephalopathy. Stem Cells Dev 19, 299–310. [DOI] [PubMed] [Google Scholar]
- Pimentel‐Coelho PM, Magalhães ES, Lopes LM, deAzevedo LC, Santiago MF & Mendez‐Otero R (2010). Human cord blood transplantation in a neonatal rat model of hypoxic‐ischemic brain damage: functional outcome related to neuroprotection in the striatum. Stem Cells Dev 19, 351–358. [DOI] [PubMed] [Google Scholar]
- Stoll G, Jander S & Schroeter M (2002). Detrimental and beneficial effects of injury‐induced inflammation and cytokine expression in the nervous system. Adv Exp Med Biol 513, 87–113. [DOI] [PubMed] [Google Scholar]
- Volpe JJ (1997). Overview: Perinatal and neonatal brain injury. Ment Retard Dev Disabil Res Rev 3, 1–2. [DOI] [PubMed] [Google Scholar]
- Volpe JJ (2001). Perinatal brain injury: from pathogenesis to neuroprotection. Ment Retard Dev Disabil Res Rev 7, 56–64. [DOI] [PubMed] [Google Scholar]
- Volpe JJ (2012). Neonatal encephalopathy: an inadequate term for hypoxic‐ischemic encephalopathy. Ann Neurol 72, 156–166. [DOI] [PubMed] [Google Scholar]
- Wolfberg AJ, Dammann O & Gressens P (2007). Anti‐inflammatory and immunomodulatory strategies to protect the perinatal brain. Semin Fetal Neonatal Med 12, 296–302. [DOI] [PubMed] [Google Scholar]