

Key Clinical Message
Maternal uniparental disomy of chromosome 14 (upd(14)mat) is responsible for a Prader–Willi‐like syndrome with precocious puberty. Although upd(14) is often hypothesized to result from trisomy rescue mechanism, T14 cell lines are usually not found with postnatal cytogenetic investigations. We report the coexistence of both chromosomal abnormalities in a 15‐year‐old girl.
Keywords: Abnormal skin pigmentation pattern, genomic imprinting, intellectual disability, maternal uniparental disomy of chromosome 14, Prader–Willi‐like phenotype, precocious puberty, trisomy 14
Introduction
Maternal uniparental disomy of chromosome 14 (upd(14)mat) is a rare chromosomal disease in which both chromosomes 14 are inherited from the mother, instead of one copy from each parent. The main clinical features of upd(14)mat, overlapping with those observed in Prader–Willi syndrome (PWS), are prenatal and postnatal growth retardation, hypotonia, feeding difficulties in the neonatal period with a catch‐up usually noted after 4 years and a progressive overweight after 6 years, joint hyperlaxity, motor delay, and early‐onset puberty. Learning difficulties are inconstant and mild dysmorphic features may be observed such as broad and tall forehead, short nose, small and large hands, and clinodactyly of the fifth fingers 1, 2, 3.
Complete trisomy 14 (T14) mosaicism, on the other hand, is a very rare chromosomal condition associating failure to thrive, hypotonia, developmental delay, intellectual disability, congenital heart and genitourinary abnormalities, and abnormal skin pigmentation pattern. Patients with T14 mosaicism usually present with craniofacial dysmorphic features such as broad nose, hypertelorism, ear abnormalities, micrognatia, and cleft or highly arched palate 4.
Although the phenotypic spectrum of upd(14)mat is thought to be mainly the consequence of fetal/placental T14 mosaicism and genomic imprinting, the coexistence of mosaic T14 and upd(14)mat in a living patient has, to our knowledge, not previously been described.
Materials and Methods
Cytogenetic analyses were performed on blood sample, hyperpigmented and normal skin, urine and saliva samples.
DNA was analyzed using a custom Aligent CGH/SNP array according manufacturer recommended procedures. A total of 31,174 CGH probes and 26,415 SNP probes spreaded along autosomes, and X and Y chromosomes were selected for this custom array. Analysis was performed using cytogenomics software (Aligent, Santa Clara, CA). Complementary FISH analysis was performed using IGH probes for 14q32 region.
Results
Clinical report
A 15‐year‐old Guinean girl, fourth child of healthy and nonconsanguineous parents, was referred to our clinical genetics center in a context of short stature, obesity, hyperextensible small joints, intellectual disability, and precocious puberty. Written informed consent was obtained from the patient's mother for publication of the medical history and the photographs.
The proband was born at term, after an uneventful pregnancy, with low birth weight (2 kg, −3 SD) but normal height (48 cm, −0.8 SD). The neonatal period was marked by hypotonia and poor suction. She developed early feeding difficulties but despite her low birth weight and feeding problems, she developed a progressive overweight after the age of 2 years.
Psychomotor development was delayed with walking acquisition at 5 years and severe speech delay. The girl also presented with moderate intellectual impairment and severe learning difficulties.
Precocious puberty with breast development and menarche was noted before the age of 8 years.
On clinical examination at the age of 15 years, the girl weighed 54.7 kg (+0.33 SD) for a height of 129 cm (−4.7 SD), giving a body mass index at 32.87 kg/m² (+5.5 SD).
She showed mild facial dysmorphic features with broad and depressed nasal bridge, short nose and up‐slanting palpebral fissures. We also noted marked truncal obesity, hyperlaxity of metacarpophalangeal and interphalangeal joints with camptodactyly of the last interphalangeal joints, and irregular skin hyperpigmentation following Blaschko lines (Fig. 1).
Figure 1.
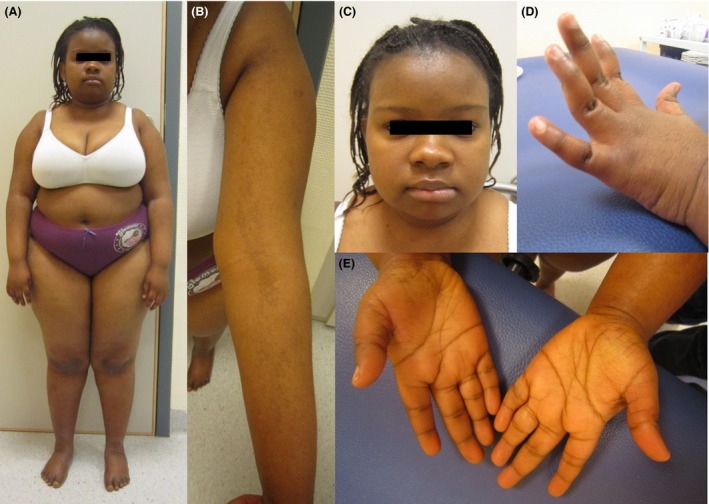
Proband's clinical phenotype: short stature and obesity (A), irregular skin hyperpigmentation following Blaschko lines (B), mild dysmorphic features with round face and short palpebral fissures (C), hyperlaxity of metacarpophalangeal and proximal interphalangeal joints, and camptodactyly of the distal interphalangeal joints (D), clinodactyly of the fifth fingers (E).
Blood sample analysis revealed an increased LDL‐cholesterol level (Total cholesterol 224 mg/dL, LDL‐cholesterol 146 mg/dL), but endocrine tests revealed no specific abnormalities (low IGF‐1 level in a context of obesity, normal cortisol level, normal oral glucose tolerance test, and HOMA testing). Bone X‐ray identified a 3‐year bone age advance, as expected with precocious puberty, but no signs of skeletal dysplasia nor subcutaneous calcifications.
Brain MRI and ophthalmologic assessments were normal.
Echocardiography showed mitral myxoid degeneration of the mitral valve with mild mitral insufficiency but without valve prolapse.
Electron microscopy analysis of biopsied skin fragments revealed abnormal collagen fibers with a wide variability of fiber caliber and the presence of a large proportion of cauliflower‐like fibers, mainly seen in the hyperpigmented skin sample. Elastic fibers were abnormally fragmented and frayed (Fig. 2).
Figure 2.
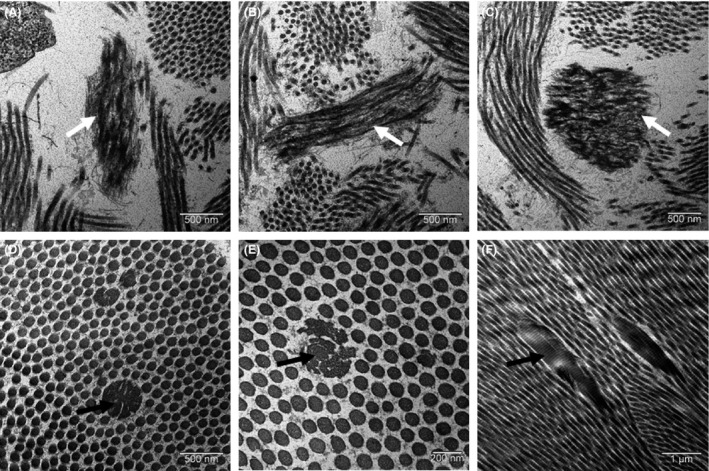
Electron microscopy analysis of skin biopsy: fragmented and frayed elastic fibers (A, B, and C, white arrows), abnormal collagen fibers (black arrows) with a wide variability of fiber caliber and the presence of cauliflower‐like fibers (D, E, F).
Genetic findings
Molecular genetic evaluations first excluded McCune Albright syndrome (normal Sanger sequencing of GNAS1 gene) and Prader–Willi syndrome (normal 15q11‐q13 methylation profile).
DNAarray – oligo ISCA 180k (Agilent) and FISH analysis revealed T14 mosaicism in blood (31%), urine (42%), and saliva (49%) (Fig. 3 and Table 1). The detection of trisomic cells in hyperpigmented and normal skin biopsies was not significant (2.5% and 2%, respectively).
Figure 3.
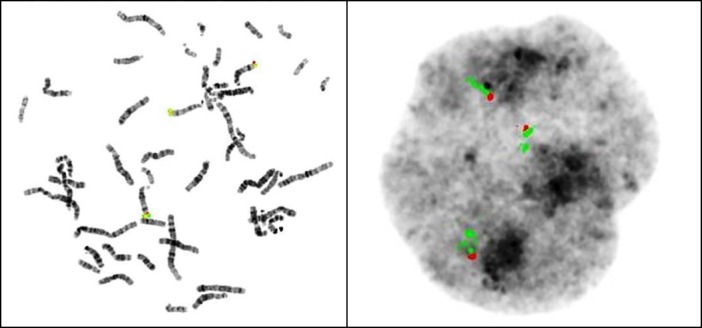
Cytogenetic analysis in blood: FISH with DNA IGH probes for 14q32 showing T14.
Table 1.
Proportion of trisomic 14 cells in analyzed tissues
| Tissue | % of T14 cells |
|---|---|
| Blood | 31 |
| Hyperpigmented skin | 2.5 |
| Normal skin | 2 |
| Urine | 42 |
| Saliva | 49 |
Genome‐wide single nucleotide polymorphism (SNP) array (Agilent) analysis of a skin biopsy showed proximal and distal iso‐upd(14)mat and central hetero‐upd(14)mat (Fig. 4).
Figure 4.
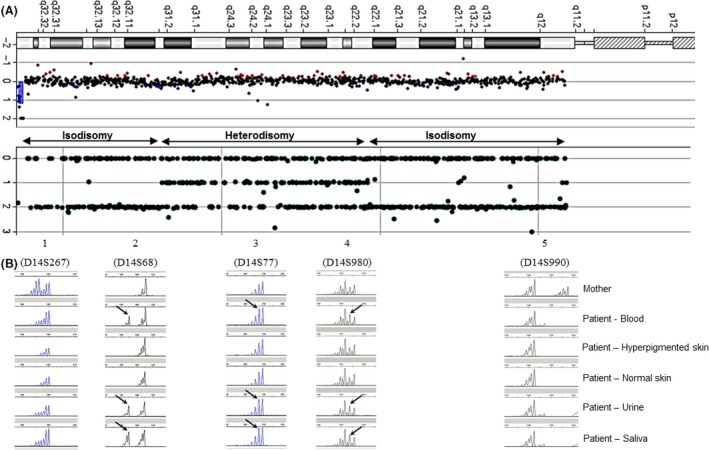
(A) CGH profile of chromosome 14. (B) SNP‐arrays profile of chromosome 14 showing proximal and distal isodisomy (only homozygous SNPs) and central heterodisomy (homozygous and heterozygous SNPs). (C) Microsatellites analysis along chromosome 14, with a comparison of the results from the maternal blood sample and from various proband's tissues samples (paternal blood sample unavailable). Profiles 1 and 5 are informative for iso‐upd(14)mat and profile 4 is informative for hetero‐upd(14)mat, consistent with SNP‐arrays profile. In cells from blood, urine, and saliva, T14 mosaicism is suggested by the presence of a paternal allele (black arrows) in addition to maternal alleles. This paternal allele is completely loss in cells from the skin, by trisomic rescue.
Discussion
To our knowledge, we report the first case of concomitant complete T14 mosaicism and upd(14)mat, in a 15‐year‐old girl. These genetic findings seem consistent with the patient's phenotype. Indeed, developmental delay, abnormal skin pigmentation pattern, and dysmorphic features may be attributed to mosaic trisomy 14, while low birth weight, short stature, precocious puberty, joint hyperlaxity, and truncal obesity may be attributed to upd(14)mat 1, 2, 3, 4. Intellectual disability may find its origin in both cytogenetic abnormalities. Clinical features of patients reported in the literature with T14 or upd(14)mat, compared to our patient, are summarized in Table 2 3, 4. Interestingly, skin analysis by electron microscopy showed abnormal collagen and elastic fibers (Fig. 2). These findings were not described so far in patients with mosaic T14 or upd(14)mat, but could be related to the joint hyperlaxity found in upd(14)mat patients.
Table 2.
Phenotype comparison between our patient and previously reported cases
| Reported patients | Our patient | |
|---|---|---|
| Clinical findings | % | Clinical findings |
| UPD(14)mat (Temple et al. 8) | ||
| Intrauterine growth retardation | 75 | − |
| Low birth weight | 87 | + |
| Low birth length | 57 | − |
| Postnatal short stature | 79 | + |
| Obesity | 49 | + |
| Early‐onset puberty | 86 | + |
| Hypotonia | 93 | + |
| Feeding difficulties | n/a | + |
| Motor developmental delay | 83 | + |
| Mental retardation | 39 | + |
| Speech delay | 59 | + |
| Hyperextensible joints | 63 | + |
| Scoliosis | 23 | − |
| Trisomy 14 mosaicism (Salas‐Labadia et al. [4]) | ||
| Growth retardation | 70 | + |
| Developmental/mental delay | 70 | + |
| Hypotonia | 24 | + |
| Microcephaly | 25 | − |
| Hearings problems | 20 | − |
| Eye abnormalities | 75 | − |
| Heart defect | 60 | − |
| Hip problems | 7.5 | − |
| Extremities abnormalities | 58 | + |
| Genitourinary abnormalities | 38 | − |
| Pigmentary skin lesions | 38 | + |
Clinical characteristics of upd(14)mat are caused by dysregulation of the expression of imprinted genes. Human chromosome 14 carries the 14q32 imprinted locus, controlled by paternally intergenic differentially methylated region (IG‐DMR), and containing genes expressed only on the paternal allele (paternally expressed genes, PEGs, such as DLK1, DIO3, and RTL1) and genes expressed only on the maternal allele (maternally expressed genes, MEGs, such as GTL2, RTL1as, and MEG8). In case of upd(14)mat, the overexpression of MEGs and the nonexpression of PEGs explain the patient's phenotype 3, 5.
Several cases 6, 7, 8, 9 of upd(14)mat‐like phenotypes have been published in patients with normal karyotype, supporting the assumption that point mutations or rearrangements at the 14q32 imprinted locus were responsible for clinical features of upd(14)mat. Indeed, in 2007, Temple et al. 8 reported the case of an upd(14)mat‐like patient with isolated mutation in the DLK1/GTL2 locus. In 2008, Kagami et al. 7 reviewed 11 cases of upd(14)mat‐like phenotypes. Among these 11 patients, three harbored a deletion in the 14q32.2 imprinted locus.
In 2014, Zada et al. 9 described another case of upd(14)mat‐like patient in which a 14q32.2q32.31 microdeletion was detected by CGH array analysis.
Three mechanisms may be involved in the occurrence of upd(14)mat: the mitotic loss of one copy in a trisomic cell (called trisomy rescue), the fusion of nullisomic gamete with a disomic gamete, and the mitotic duplication of one copy of a chromosome in a monosomic cell (monosomy rescue) 10, 11. Trisomy rescue is the most frequent mechanism, already described in other imprinting‐related syndromes, as in Prader–Willi syndrome.
In cases of T14 rescue mechanism, the T14 is mainly due to a desequilibrated Robertsonian translocation involving chromosome 14. In a minority of cases, initial T14 results from maternal meiosis nondisjunction or possibly postzygotic mitotic nondisjunction 12. Subsequently, upd(14)mat arises through loss of the paternal homologue chromosome 14.
The association of confined placental complete T14 mosaicism and fetal upd(14)mat was previously reported, providing arguments in favor of this last mechanism 13. In 1999, a case of upd(14)mat detected prenatally in a fetus with mosaic T14 was also described 14. However, T14 cell lines are usually not found with cytogenetic investigations in case of postnatal upd(14)mat. Here, we report on the coexistence of T14 cell lines and euploid cell lines with upd(14)mat, strongly suggesting that upd(14)mat results from loss of the paternal homologue in a T14 zygote. We assume that the origin of T14 is probably due to a maternal meiosis nondisjunction. In 2010, a comparable case of T14 mosaicism coexisting with euploid upd(14) cell lines in a live patient was already described 11, but resulted in a different phenotype as, in upd cell lines, both chromosomes 14 were of paternal origin.
Surprisingly, in our patient, T14 cells were found in blood, saliva, and urine but not significantly in skin. This suggests that rescue mechanism could have occurred at a very early stage of embryogenesis. Moreover, we found, in the skin sample, maternal isodisomy at the telomeric parts of chromosome 14 and maternal heterodisomy at the centromeric part of chromosome 14 (Fig. 4). This may be hypothesized as being a double crossing‐over between homologous chromosomes during maternal meiosis I, followed by the nondisjunction of sister chromatids during maternal meiosis II. After fertilization, some T14 cells will undergo trisomy rescue mechanism with the loss of the paternal copy of chromosome 14, leading to euploid cells showing maternal hetero and isodisomy regions on chromosome 14. This mechanism is illustrated in Figure 5.
Figure 5.
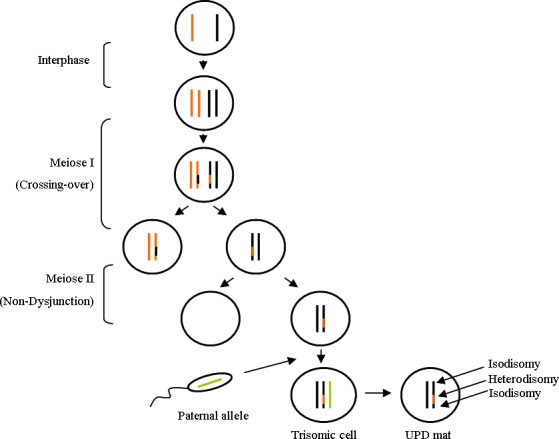
Mechanism of upd(14)mat in our patient: Meiosis I with double crossing‐over between homologous chromosomes 14, followed by Meiosis II with nondisjunction of sister chromatids, leading after fertilization to trisomic cells for chromosome 14. The elimination of the paternal copy of chromosome 14 is responsible for upd(14)mat in nontrisomic cells. Coexistence of proximal/distal isodisomy and central heterodisomy is the consequence of the double crossing‐over in Meiosis I.
In conclusion, this is a unique case of postnatal coexistence of mosaic trisomy 14 and maternal upd(14) cell lines in a patient presenting phenotypic characteristics of both cytogenetic entities.
This confirms that upd(14)mat resulted, in the present case, from loss of the paternal homologue 14 in a maternal meiosis II nondisjunction trisomic zygote.
While no specific treatment exists, making this diagnosis allows us to establish a close and multidisciplinary clinical follow‐up with special attention to symptoms and deficiencies usually seen in this syndrome. Patients with concomitant mosaic trisomy 14 and maternal uniparental disomy 14 should especially benefit from neurological and endocrinological follow‐up. Physiotherapy, ergotherapy, and speech therapy may improve the patients' outcome.
Conflict of Interest
None declared.
Clinical Case Reports 2016; 4(3): 265–271
References
- 1. Hosoki, K. , Kagami M., Tanaka T., Kubota M., Kurosawa K., Kato M., et al. 2009. Maternal uniparental disomy 14 syndrome demonstrates prader‐willi syndrome‐like phenotype. J. Pediatr. 155:900–903. [DOI] [PubMed] [Google Scholar]
- 2. Rocha, C. F. , and Paiva C. L.. 2014. Prader‐Willi‐like phenotypes: a systematic review of their chromosomal abnormalities. Genet. Mol. Res. 13:2290–2298. [DOI] [PubMed] [Google Scholar]
- 3. Ioannides, Y. , Lokulo‐Sodipe K., Mackay D. J., Davies J. H., and Temple I. K.. 2014. Temple syndrome: improving the recognition of an underdiagnosed chromosome 14 imprinting disorder: an analysis of 51 published cases. J. Med. Genet. 51:495–501. [DOI] [PubMed] [Google Scholar]
- 4. Salas‐Labadia, C. , Lieberman E., Cruz‐Alcivar R., Navarrete‐Meneses P., Gòmez S., Cantú‐Reyna C., et al. 2014. Partial and complete trisomy 14 mosaicism: clinical follow‐up, cytogenetic and molecular analysis. Mol. Cytogenet. 7:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ogata, T. , and Kagami M.. 2008. Molecular mechanisms leading to the phenotypic development in paternal and maternal uniparental disomy for chromosome 14. Clin. Pediatr. Endocrinol. 17:103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hordijk, R. , Wierenga H., Scheffer H., Leegte B., Hofstra R. M., and Stolte‐Dijkstra I.. 1999. Maternal uniparental disomy for chromosome 14 in a boy with a normal karyotype. J. Med. Genet. 36:782–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kagami, M. , Sekita Y., Nishimura G., Irie M., Kato F., Okada M., et al. 2008. Deletions and epimutations affecting the human 14q32.2 imprinted region in individuals with paternal and maternal upd(14)‐like phenotypes. Nat. Genet. 40:237–242. [DOI] [PubMed] [Google Scholar]
- 8. Temple, I. K. , Shrubb V., Lever M., Bullman H., and Mackay D. J.. 2007. Isolated imprinting mutation of the DLK1/GTL2 locus associated with a clinical presentation of maternal uniparental disomy of chromosome 14. J. Med. Genet. 44:637–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zada, A. , Mundhofir F. E., Pfundt R., Leijsten N., Nillesen W., Faradz S. M. H., et al. 2014. A rare, recurrent, de novo 14q32.2q32.31 microdeletion of 1.1 Mb in a 20‐year‐old female patient with a maternal UPD(14)‐like phenotype and intellectual disability. Case Rep. Genet. 2014:530134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Antonarakis, S. E. , Blouin J. L., Maher J., Avramopoulos D., Thomas G., and Talbot C. C. Jr. 1993. Maternal uniparental disomy for human chromosome 14, due to loss of a chromosome 14 from somatic cells with t(13;14) trisomy 14. Am. J. Med. Genet. 52:1145–1152. [PMC free article] [PubMed] [Google Scholar]
- 11. Conlin, L. K. , Thiel B. D., Bonnemann C. G., Medne L., Ernst L. M., Zackai E. H., et al. 2010. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum. Mol. Genet. 19:1263–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sanlaville, D. , Aubry M. C., Dumez Y., Nolen M., Amiel J., Pinson M., et al. 2000. Maternal uniparental heterodisomy of chromosome 14: chromosomal mechanism and clinical follow up. J. Med. Genet. 37:525–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Towner, D. R. , Shaffer L. G., Yang S. P., and Walgenbach D. D.. 2001. Confined placental mosaicism for trisomy 14 and maternal uniparental disomy in association with elevated second trimester maternal serum human chorionic gonadotrophin and third trimester fetal growth restriction. Prenat. Diagn. 21:395–398. [DOI] [PubMed] [Google Scholar]
- 14. Ralph, A. , Scott F., Tiernan C., Caubere M., Kollegger S., Junio J., et al. 1999. Maternal uniparental isodisomy for chromosome 14 detected prenatally. Prenat. Diagn. 19:681–684. [DOI] [PubMed] [Google Scholar]