

Key Clinical Message
Patients with Williams–Beuren Syndrome can be recognized clinically, given the characteristic dysmorphism, intellectual disability, and behavior. We report on a Congolese boy with typical WBS facial characteristics. He suffered meningitis and coma at the age of 2 years then subsequently presented with profound intellectual disability and atypical behavior. The WBS was only made at age 8.2 years and confirmed with FISH testing and microarray‐CGH. The present report aims to warn clinicians that infections may associate and/or modify a genetic disease as this may be observed in developing countries given the prevalence of infectious diseases.
Keywords: Central Africa, Democratic Republic of Congo, phenotype, Williams syndrome
Introduction
Williams–Beuren Syndrome (WBS) is one of the most common recurrent microdeletion syndromes, with an estimated incidence varying from 1 in 7500 to 1 in 20,000 births 1, 2. Patients can be recognized clinically, given their characteristic features, encompassing congenital heart defect, typically supravalvular aortic stenosis (SVAS) and/or peripheral pulmonic stenosis, short stature/growth retardation, recognizable facial dysmorphism, developmental delay and intellectual disability, distinct behavioral phenotype; and, in some instances, hypercalcemia. Facial dysmorphism includes a wide mouth, short nose with bulbous or upturned nasal tip, long philtrum, thick everted vermillion of the lips, bitemporal narrowing, periorbital fullness, full cheeks, small jaw, and the characteristic stellate pattern of the iris 3, 4. The cognitive profile in WBS ranges from mild to severe ID 3, 5, 6. Although acquisition of language is often delayed in WBS patients, they later develop good language skills and become talkative 7, 8. Behavioral issues in WBS comprise anxiety, hyperacousis, attention problems, hypersociability or friendly behavior, and strong affinity and interest to music 7, 9, 10. Recognition of the typical dysmorphism and characteristic behavior lead to a clinical diagnosis.
Some cases are more difficult to diagnose, since they present with atypical features 11, or because they have a different ethnicity where the features may be different from those observed in Caucasians. For instance, African patients with WBS from Cameroon have been reported with incomplete WBS facial dysmorphism 8. Finally, cases with a deletion smaller or larger than the recurrent 1.5 Mb 7q11.23 microdeletion may also present with a milder or more severe phenotype 12.
We here report a WBS patient from Central Africa carrying the recurrent WBS microdeletion who presented with a typical facial dysmorphism but more severe intellectual disability and atypical behavioral phenotype due to a confounding factor, that is, meningitis with coma supposedly resulting in brain damage.
Case History and Examination
An 8.2 years old Congolese boy was referred by his pediatrician to the genetic clinic at the University Hospitals of the University of Kinshasa, DR Congo, for evaluation of intellectual disability, allegedly resulting from an episode of meningitis and coma. He was born from unrelated, healthy parents after an uneventful pregnancy and delivery. At the age of 2 years, he wasn't able to stand‐up without support or build simple sentences although he could call names of his relatives. Four months later, he suffered from meningitis and remained in coma for 2 days. After recovery, he presented severe intellectual disability and complete loss of intelligible speech. His behavior changed, including hyperactivity, irritability, hypersensitivity to noise and episodes of aggressiveness against strangers. In contrast, he was friendly toward familiar persons and demonstrated a strong attraction to music and musical sounds. He was diagnosed with sequels from meningitis and was followed by a neurologist until the age of 8 years, when he was referred for a genetic evaluation.
At that time, he had severe global growth delay with microcephaly (OFC 47 cm; −3.82 SD), short stature (length 109 cm; −3.43 SD) and low weight (17 kg; <−3 SD). Family history revealed that two siblings of the patient (Fig. 1, upper panel) died from infectious diseases. There were two other males with ID in the pedigree, related through their mothers. Unfortunately, these were not available for evaluation. Patient's phenotype consisted of a triangular face with low set ears, long philtrum, flat nose bridge, bulbous nose tip, anteverted nostrils, pronounced nasolabial folds, large mouth, thick and everted vermillion of the upper and lower lip, and a small jaw (Fig. 1, lower panel). He also had widely spaced nipples, a small penis, bilateral cryptorchidism, and deep palmar creases. Cardiac ultrasound revealed bicuspid aorta with aortic insufficiency.
Figure 1.
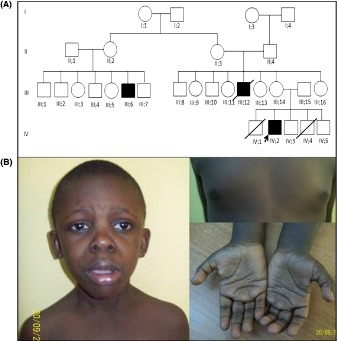
Pedigree and dysmorphic features. (A) Pedigree showing three males with ID, consistent with the X‐linked inheritance; (B) Clinical features. Note triangular face with low set ears, long philtrum, flat nasal bridge, bulbous nose tip, anteverted nostrils, pronounced nasolabial folds, large mouth, thick and everted vermillion of the upper and lower lip and a small jaw, widely spaced nipples, and deep palmar creases.
Differential Diagnosis and Investigations
Although the pedigree was consistent with X‐linked ID, a clinical diagnosis of WBS was retained.
Fluorescent in situ hybridization (FISH) was performed on nuclei from peripheral white blood cells using the commercially available Vysis LSI ELN Kit (Vysis; Abbott Laboratories, Abbott Park, IL). A total of 100 nuclei were examined and a single ELN was observed in all of them, whereas the control probe always showed two signals. This confirmed the diagnosis of WBS (Fig. 2A). Microarray‐CGH was performed using a stripped CytoSure™ ISCA v2 array 8 × 60 k format (OGT, Oxford UK) slide, the CytoSure™ Genomic DNA Labelling Kits (Oxford, UK), and the Oligo aCGH & ChIP‐on‐Chip Hybridization Kits (Agilent, Santa Clara, CA) following manufacturers’ procedures. This revealed the presence of a recurrent 1.57 Mb deletion arr[hg19]7q11.23 (72,634,874–74,203,685)×1 (Fig. 2B).
Figure 2.
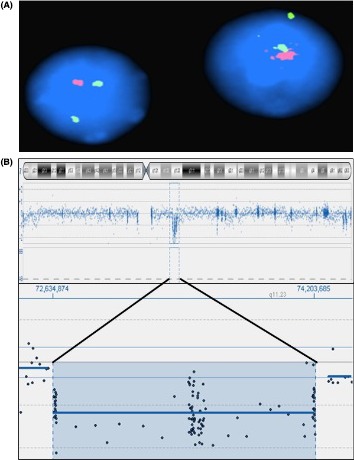
FISH and Array CGH ideograms. (A) Two interphase nuclei showing a single signal for the Elastin probe (red) and two signals for the control probe (green); (B) Chromosome icon and the zoom‐in on the deleted region.
Discussion
We report on a central African patient with typical gestalt for WBS. Retrospectively, we uploaded facial photographs of the patient to Face2Gene (http://www.fdna.com/face2gene/). Basically, this tool captures gestalt signatures from patient's facial photograph and compares these to patients in the database, then reports the 10 most likely matches. Interestingly, WBS was reported as the fifth match. This indicates that our patient's gestalt did not vary from common WBS gestalt.
It is surprising that the patient was only diagnosed at age 8.2 years, despite the typical facial appearance. Mean age of diagnosis for WBS in Western countries is 3.5 years 13, 14. Several factors may have contributed to a delayed diagnosis. First, the facial dysmorphism in WBS evolves over age and may be discrete in infancy and early childhood 4, 13, 14. Second, our patient had no SVAS, one of the most characteristic features often leading to the diagnosis 13, 15. Finally, severe phenotypes have been associated with larger deletions. As the deletion in our patient corresponds to the classical WBS microdeletion, we assume that the severe speech deficit and profound intellectual disability in the patient are more likely the complications of the meningitis and coma. It is thus undeniable that an acquired condition modified the clinical course of the patient toward the more severe and atypical phenotype for WBS causing the delay in diagnosis.
Such overlap of genetic disease with an infectious disease is expected to be frequent in the developing world, where infections represent the major medical problem. As previously reported, infections are major cause for misdiagnosis of genetic diseases in developing countries 16. The present report supports the assumption that infections constitutes a major pitfall for genetic diagnosis and warns for clinicians working in developing world that a genetic disease may be masked by a intercurrent infectious disease.
To date, only one report exists concerning WBS in Central Africa 8. Given the incidence of 1/7500 for WBS 2, this suggests that most cases remain undiagnosed. This is most likely due to a lack of training in the field of dysmorphology and lack of access to genetic facilities in that part of the world.
Conflict of Interest
None declared.
Acknowledgments
AL acknowledge the contribution of the FWO travel grants for research abroad (Ref: V405213N and K210115) to his research. The authors are grateful to Reinhilde Thoelen for the FISH analysis; Bijou Myndyo, Ervie‐Winner, Eben, and Etsa‐Edi Lumaka for their support and contribution to this work.
Clinical Case Reports 2016; 4(3): 294–297
References
- 1. Morris, C. A. , and Mervis C. B.. 2000. Williams syndrome and related disorders. Annu. Rev. Genomics Hum. Genet. 1:461–484. [DOI] [PubMed] [Google Scholar]
- 2. Stromme, P. , Bjornstad P. G., and Ramstad K.. 2002. Prevalence estimation of Williams syndrome. J. Child Neurol. 17:269–271. [DOI] [PubMed] [Google Scholar]
- 3. Merla, G. , Brunetti‐Pierri N., Micale L., and Fusco C.. 2010. Copy number variants at Williams‐Beuren syndrome 7q11.23 region. Hum. Genet. 128:3–26. [DOI] [PubMed] [Google Scholar]
- 4. Patil, S. J. , Madhusudhan B. G., Shah S., and Suresh P. V.. 2012. Facial phenotype at different ages and cardiovascular malformations in children with Williams‐Beuren syndrome: a study from India. Am. J. Med. Genet. Part A 158A:1729–1734. [DOI] [PubMed] [Google Scholar]
- 5. Martens, M. A. , Wilson S. J., and Reutens D. C.. 2008. Research Review: Williams syndrome: a critical review of the cognitive, behavioral, and neuroanatomical phenotype. J. Child Psychol. Psychiatry 49:576–608. [DOI] [PubMed] [Google Scholar]
- 6. Mervis, C. B. , and John A. E.. 2010. Cognitive and behavioral characteristics of children with Williams syndrome: implications for intervention approaches. Am. J. Med. Genet. Part C Semin. Med. Genet. 154C:229–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pegoraro, L. F. , Steiner C. E., Celeri E. H., Banzato C. E., and Dalgalarrondo P.. 2014. Cognitive and behavioral heterogeneity in genetic syndromes. J. Pediatr. 90:155–160. [DOI] [PubMed] [Google Scholar]
- 8. Tekendo‐Ngongang, C. , Dahoun S., Nguefack S., Gimelli S., Sloan‐Bena F., and Wonkam A.. 2014. Challenges in clinical diagnosis of Williams‐Beuren syndrome in sub‐Saharan Africans: case reports from Cameroon. Mol. Syndromol. 5:287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Doyle, T. F. , Bellugi U., Korenberg J. R., and Graham J.. 2004. “Everybody in the world is my friend” hypersociability in young children with Williams syndrome. Am. J. Med. Genet. Part A 124A:263–273. [DOI] [PubMed] [Google Scholar]
- 10. Ng, R. , Lai P., Levitin D. J., and Bellugi U.. 2013. Musicality correlates with sociability and emotionality in Williams syndrome. J. Ment. Health Res. Intellect. Disabil. 6:268–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sakhuja, P. , Whyte H., Kamath B., Martin N., and Chitayat D.. 2015. Williams syndrome presenting with findings consistent with Alagille syndrome. Clin. Case Rep. 3:24–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fusco, C. , Micale L., Augello B., Teresa Pellico M., Menghini D., Alfieri P., et al. 2014. Smaller and larger deletions of the Williams Beuren syndrome region implicate genes involved in mild facial phenotype, epilepsy and autistic traits. Eur. J. Hum. Genet. 22:64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ferrero, G. B. , Biamino E., Sorasio L., Banaudi E., Peruzzi L., Forzano S., et al. 2007. Presenting phenotype and clinical evaluation in a cohort of 22 Williams‐Beuren syndrome patients. Eur. J. Med. Genet. 50:327–337. [DOI] [PubMed] [Google Scholar]
- 14. Huang, L. , Sadler L., O'Riordan M. A., and Robin N. H.. 2002. Delay in diagnosis of Williams syndrome. Clin. Pediatr. 41:257–261. [DOI] [PubMed] [Google Scholar]
- 15. Eronen, M. , Peippo M., Hiippala A., Raatikka M., Arvio M., Johansson R., et al. 2002. Cardiovascular manifestations in 75 patients with Williams syndrome. J. Med. Genet. 39:554–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lumaka, A. , Mubungu G., Nsibu C., Tady B. P., Lukusa T., and Devriendt K.. 2012. X‐linked adrenal hypoplasia congenita: a novel DAX1 missense mutation and challenges for clinical diagnosis in Africa. Eur. J. Pediatr. 171:267–270. [DOI] [PubMed] [Google Scholar]