

Abstract
Exercise training (ExT) is currently being used as a nonpharmacological strategy to improve cardiac function in diabetic patients. However, the molecular mechanism(s) underlying its beneficial effects remains poorly understood. Oxidative stress is known to play a key role in the pathogenesis of diabetic cardiomyopathy and one of the enzyme systems that produce reactive oxygen species is NADH/NADPH oxidase. The goal of this study was to investigate the effect of streptozotocin- (STZ-) induced diabetes on expression of p47phox and p67phox, key regulatory subunits of NADPH oxidase, in cardiac tissues and determine whether ExT can attenuate these changes. Four weeks after STZ treatment, expression of p47phox and p67phox increased 2.3-fold and 1.6-fold, respectively, in left ventricles of diabetic rats and these increases were attenuated with three weeks of ExT, initiated 1 week after onset of diabetes. In atrial tissues, there was increased expression of p47phox (74%), which was decreased by ExT in diabetic rats. Furthermore, increased collagen III levels in diabetic hearts (52%) were significantly reduced by ExT. Taken together, ExT attenuates the increased expression of p47phox and p67phox in the hearts of diabetic rats which could be an underlying mechanism for improving intracardiac matrix and thus cardiac function and prevent cardiac remodeling in diabetic cardiomyopathy.
1. Introduction
Diabetes mellitus (DM) is leading metabolic disease with the highest morbidity and mortality and according to 2014 estimate of International Diabetes Federation the disease now affects more than 387 million people worldwide. DM is associated with an increased risk of cardiovascular complications, including hypertension and coronary artery disease [1]. There is growing recognition of diabetic cardiomyopathy, a primary myocardial disease, defined as either systolic or diastolic left ventricular dysfunction in otherwise healthy diabetic person. This is one of the most serious complications of diabetes; however, the underlying mechanism/s for this dysfunction are not clearly understood [2, 3]. Originally, Rubler et al. in 1972 [4], based on postmortem findings, proposed that diabetic cardiomyopathy is secondary to underlying hyperglycemia resulting in a multitude of adverse downstream effects, including impaired myocyte calcium handling, renin-angiotensin-aldosterone activation, microangiopathy, myocardial fibrosis, and increased oxidative stress [5–7]. Mechanistically, chronic hyperglycemia exerts oxidative stress to cardiomyocytes by the production of reactive oxygen species (ROS) culminating in these pathological abnormalities [8, 9]. Indeed, a number of studies have reported increased ROS formation in cultured cells exposed to high glucose concentrations. Similarly, in animal models of diabetes, ROS are generated as natural byproducts of oxygen metabolism and their moderate levels are thought to function as intracellular signaling molecules; however, high levels of ROS are detrimental to cardiomyocytes and lead to cell death, mitochondrial dysfunction due to mitochondrial fragmentation [10], or impaired insulin signaling [11]. ROS contribute to inducing various cardiovascular complications including cardiac dysfunction accelerated by inflammation, apoptosis, and fibrosis [12–14]. Furthermore, O2 − has been shown to impair nitric oxide (NO) dependent vasodilation [15, 16] and thus enhance endothelium-dependent vasoconstriction [17, 18] and precipitate cardiomyocyte hypertrophy [19].
ROS is generated by all cell types within the heart, including cardiomyocytes, endothelial cells, vascular smooth muscle cells (VSMCs), fibroblasts, and infiltrating inflammatory cells [20]. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs) are membrane-associated enzymes which are the primary physiological producers of O2 − [21] and ROS [20, 22]. NOXs consist of 4 major subunits: a plasma membrane-spanning cytochrome b558 composed of a large gp91phox subunit and a smaller p22phox subunit, whereas 2 cytosolic subunits are p47phox and p67phox. Endothelial cells (ECs) and myocytes express all four components of NADPH oxidase [23, 24], while VSMCs express all of the subunits with the exception of p67phox subunit [25]. The low molecular weight G protein rac2 (in some cells rac1) participates in the assembly of the active complex, and a second G protein, rap1A, is thought to be involved in the deactivation of NADPH oxidase enzyme activity.
DM is associated with increased levels of Ang II [26–30] and numerous studies have shown that Ang II upsurges production of O2 − by activating NADPH oxidases [31–34] in VSMCs [35–37], adventitial fibroblasts [32, 38], ECs [33, 39], and cardiomyocytes [34]. Additionally, hyperglycemia, a key clinical manifestation of diabetes, also produces ROS via NADPH oxidase activation by glycation end products [40], while blocking of ROS formation is known to prevent hyperglycemic damage [41]. In rat ventricular myocytes, high glucose conditions induced cardiac contractile dysfunction and cardiomyopathy occurs via Ang II type 1 receptor (AT1R) mediated activation of NADPH oxidase [27, 28, 30]. High glucose conditions may also increase the activity of NADPH oxidase by increasing the expression of its various subunits resulting in increased production of ROS [8, 42]. Infusion of Ang II has also been shown to increase NADPH oxidase activity and O2 − production by increasing expression of p67phox and gp91phox in aortic adventitial cells [32]. Exposure of ventricular myocytes to high glucose also increases expression of p47phox and production of ROS and these increases were blocked by AT1R antagonist, L-158,809 [42]. Additionally, Hink et al. [43] found increased NADPH oxidase activity and a 7-fold increase in gp91phox mRNA in aortic tissue from rats with streptozotocin- (STZ-) induced diabetes.
Aerobic exercise of moderate intensity and frequency is one of a number of cardiac rehabilitation programs prescribed to patients with chronic heart failure [44]. Exercise training (ExT) reduces blood glucose, body fat, and insulin resistance and improves glycemic control, lipid metabolism, and baroreflex sensitivity in diabetes [45–48]. Additionally, ExT lowers plasma Ang II levels [49–51] and reduces renal sympathetic nerve activity and arterial pressure responses to Ang II [50–53]. Previous studies have also shown that ExT increases expression of superoxide dismutase (SOD), catalase, and glutathione in cardiac tissues [49, 54–56]. The ability of these antioxidant enzymes to reduce ROS has also been well documented [57, 58]. However, the impact of ExT on NADPH oxidase activity and expression of its subunits in diabetes is not entirely clear. These findings have led us to investigate whether diabetes-induced cardiac dysfunction may be due in part to increased expression of p47phox and p67phox and further whether ExT could improve/attenuate the increased expression of these subunits induced by diabetes.
2. Materials and Methods
2.1. Induction and Verification of Experimental Diabetes
Animals used for this study were approved by the Animal Care and Use Committee of the University of Nebraska Medical Center and all experiments were conducted according to the APS's Guiding Principles for Research Involving Animals and Human Beings and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Male Sprague-Dawley rats weighing between 180 and 190 g were purchased from Sasco Breeding Laboratories (Omaha, NE). After 1 week of acclimatization in housing facilities diabetes was induced by a single intraperitoneal injection (65 mg/kg) of streptozotocin (STZ, Sigma) in a 2% solution of cold 0.1 M citrate buffer (pH 4.5). Onset of diabetes was identified by polydipsia, polyuria, and blood glucose levels >250 mg/dL. Control animals were injected with citrate buffer containing no STZ. Throughout the study, animals were housed in pairs (similar weights to minimize dominance) at 22°C with fixed 12 h light/dark cycles and humidity at 30–40%. Laboratory chow (Harlan, Madison, WI) and tap water were available ad libitum. Experiments were performed 4 weeks after the injection of STZ or vehicle.
2.2. Exercise Protocol
After one week, rats in each of control and diabetic groups were randomly divided into two groups. One subgroup of control and one subgroup of diabetic rats remained sedentary, while the other two subgroups were subjected to ExT according to the protocols used by Musch and Terrell [59] with modifications [60] for three weeks after one week after diabetes induction. The resulting four subgroups of animals were referred to as nonexercised controls (Sed-control, n = 16), diabetic nonexercise (Sed-Dia, n = 16), exercise trained controls (ExT-control, n = 20), and exercise trained diabetic (ExT-Dia, n = 20). During the training period, rats were exercised between 5 and 15 min/day at an initial treadmill speed of 10 m/min up a 0% grade for 5 days. In order to ensure a significant endurance ExT regimen, the treadmill grade and speed were gradually increased to 10% and 25 m/min, respectively, and the exercise duration was increased to 60 min/day. Animals demonstrating the ability to run steadily on the treadmill with very little or no prompting (with electrical stimulation) were used in the study. The Sed-control and Sed-Dia rats were treated similarly to the ExT-control and ExT-Dia subgroup and handled daily except for the treadmill running.
2.3. Sample Collection
At the end of the in vivo protocol, animals in all four subgroups (Sed-control, Sed-Dia, ExT-control, and ExT-Dia) were anesthetized using overdose of pentabarbital (65 mg/kg, i.p.). Chest cavities were opened and hearts were removed, quick-frozen by dropping into liquid nitrogen, and stored at −80°C. Soleus muscle from hind legs was also removed, quick-frozen, and stored at −80°C.
2.4. Citrate Synthase Assay
The efficacy of ExT was assessed by measuring citrate synthase activity in whole muscle homogenate as previously described [51, 61]. Citrate synthase activities were normalized to total protein content and reported as micromoles per milligram protein per minute.
2.5. Semiquantitative RT-PCR
Relative levels of p47phox and p67phox in left and right ventricles and atria tissues were determined using semiquantitative RT-PCR. One hundred micron (100 μm) thick coronal sections were cut on a cryostat from the left and right ventricles and a 15-gauge needle stub was used to punch atrial samples. Total RNA from left ventricle, right ventricle, and atrial tissue was isolated by TRI Reagent (MRC) method as per the manufacturer's instructions as previously described [62, 63]. Equivalent amounts of total RNA (1 μg) from each of Sed-control, Sed-Dia, ExT-control, and ExT-Dia rats were then reverse-transcribed for 40 min at 37°C in the presence of 1.5 μM of random hexamers and 100 U of MMLV-Reverse transcriptase. Primers for p47phox and p67phox (200 nM) were used in polymerase chain reactions to determine the amount of transcripts in each sample. β-actin was coamplified as an internal control. After 10 min of denaturing at 94°C, the amplification was performed at 94°C for 1 min, at 56°C for 1 min, and at 72°C for 1 min for 33 cycles with the final extension at 72°C for 10 min. At the end of the reaction, 7 μL from each PCR reaction was mixed with Blue/Orange loading dye (Promega) and electrophoresed for 45 min at 100 V using 1% agarose gels containing ethidium bromide. The gels were visualized with gel doc system (Kodak ID gel Imager). Band intensities were then analyzed with the Kodak analysis software and normalized to that of their respective β-actin bands. Oligo-primers for PCR were synthesized in-house at University of Nebraska Medical Center. Sequences of primers used were as follows: p47phox: 5′-ACCTGTCGGAGAAGGTGGT (forward), 5′-TAGGTCTGAAGGATGATGGG (reverse); p67phox: 5′-AGGACTATCTGGGCAAGGC (forward), 5′-GCTGCGACTGAGGGTGAAT (reverse); β-actin 5′-GGGAAATCGTGCGTGACATT (forward), 5′-CGGATGTCAACGTCACACTT (reverse).
2.6. Western Blotting
Western blot analyses were used to determine p47phox and p67phox in the left ventricle, right ventricle, and atria tissues in the four subgroups of rats. Protein was extracted from the heart after homogenization in RIPA buffer (cat. number BP-115, Boston BioProducts, Worcester, MA, USA) supplemented with 1 mM phenylmethylsulfonyl fluoride and protease inhibitor cocktail. Collagen III levels were measured as an index of myocardial stiffness (diabetic cardiomyopathy) in atrial tissue. 30–40 μg of each protein sample was mixed with an equal volume of 4% SDS sample buffer, fractionated on 7.5% polyacrylamide-sodium dodecyl sulfate gel, and transferred to a PVDF membrane (Millipore). After transfer, the membranes were blocked with 5% nonfat dry milk powder in TBST (20 mM Tris/HCl, pH 7.4, 150 mM NaCl, 0.1% (v/v) Tween 20) at room temperature for 1 h. Subsequently, membranes were probed with a primary anti-p47phox rabbit polyclonal or anti-p67phox rabbit polyclonal or anti-collagen III goat polyclonal antibody from Santa Cruz Biotechnology overnight at 4°C followed by incubation with a corresponding peroxidase-conjugated secondary antibody for one hour. The signals were visualized using an enhanced chemiluminescence (Pierce Chemical, Rockford) and detected by a UVP digital imaging system. ImageJ-NIH program was used to quantify the signal.
2.7. Statistical Analysis
Data was presented as mean ± SEM and subjected to Student-Newman-Keuls test. P values < 0.05 were considered to indicate statistical significance.
3. Results
3.1. General Characteristics of Animals
Table 1 summarizes the characteristics of the Sed-control, Sed-Dia, ExT-control, and ExT-Dia animals used in the present study. Mean body weight of all animals at the start of the study was 186 ± 2 g. As shown in Table 1, after 28 days, the mean body weight of Sed-control animals increased to 323 ± 7 g, whereas the mean body weight of ExT-control animals was 226 ± 2 g. Mean body weight of sedentary and exercised trained diabetic animals increased modestly to 209 ± 7 and 208 ± 12 g, respectively. In the animals injected with citrate buffer only, mean blood glucose levels did not change significantly during the study (72.0 ± 6.0 mg/dL at start and 78.0 ± 7.0 mg/dL at the time of sacrifice). Sed-Dia animals had significant high blood glucose levels at the time of sacrifice, 362 ± 18 mg/dL, whereas ExT had significantly lowered blood glucose levels, 315 ± 11 mg/dL. We have used citrate synthase activity as a marker of increased aerobic metabolism in the soleus muscle. The enzyme citrate synthase catalyzes the formation of citrate from acetic acid and oxaloacetic acid in the first step of Krebs cycle. Regular exercise increases the size and number of skeletal muscle mitochondria to increase respiratory capacity of the muscle. Increased mitochondria translate to increased citrate synthase activity, reflecting the increased activity of muscle associated with ExT [51, 61]. Cardiac muscle does not increase respiratory capacity in response to ExT as skeletal muscle does [64]. Therefore measuring the citrate synthase activity in soleus muscle is extensively used as a metabolic marker in assessing oxidative and respiratory capacity. In the current study, Sed-control and diabetic animals had a lower citrate synthase activity (4.58 ± 0.34 μmol/g/min and 4.01 ± 0.42 μmol/g/min, resp.) than ExT animals (6.93 ± 0.56 μmol/g/min for ExT-control and 6.94 ± 0.56 μmol/g/min for ExT-diabetic). These data also indicate that similar level of ExT was performed in the two groups of animals.
Table 1.
Characteristics of the Sed-control, Sed-Dia, ExT-control, and ExT-Dia groups. n = 10 in each group. Values represent mean ± S.E. ∗ P < 0.01 versus Sed-control. # P < 0.05 versus Sed-Dia group.
| Sed-control | Sed-Dia | ExT-control | ExT-Dia | |
|---|---|---|---|---|
| (n = 10) | (n = 10) | (n = 10) | (n = 10) | |
| Body weight (g) | 323 ± 7 | 209 ± 7∗ | 226 ± 2∗ | 208 ± 12∗ |
| Blood glucose (mg/dL) | 72 ± 6 | 362 ± 18∗ | 78 ± 7 | 315 ± 11# |
| Citrate synthase (mmol/g/min) | 4.58 ± 0.34 | 4.01 ± 0.42 | 6.93 ± 0.56∗ | 6.94 ± 0.56∗ |
3.2. Increased Expression of p47phox and p67phox in Left Ventricle Alleviated with ExT in Diabetic Animals
Expressions of p47phox and p67phox were determined by semiquantitative RT-PCR and Western blot using actin as an internal reference in four groups of rats, namely, Sed-control, Sed-Dia, ExT-control, and ExT-Dia (Figure 1). There were significantly increased transcripts of p47phox I (45.8% increase) and protein (2.3-fold) in the left ventricle of 4-week Sed-Dia versus 4-week Sed-control rats (Figures 1(a) and 1(b)). Sed-Dia animals also demonstrated increased steady-state levels of mRNA encoding p67phox (increased 78.1% of the 4-week Sed-control) (Figure 1(c)) and protein (1.6-fold) (Figure 1(d)) in the left ventricle. Three weeks of ExT, initiated after 1 week of diabetes, reduced mRNA and protein levels of p47phox and p67phox in the left ventricle of the heart to levels similar to those of the ExT controls (≈40–50% less than Sed-Dia).
Figure 1.

Expression of left ventricle p47phox and p67phox in Sed-control, Sed-Dia, ExT-control, and ExT-Dia animals. (a) RT-PCR of p47phox: top: a representative gel; bottom: quantification of p47phox transcript normalized to β-actin as loading control. n = 10 in each group. (b) Western blot analysis of left ventricle p47phox in four groups: top: a representative gel; bottom: bar graph of summary data of densitometry analyses of p47phox protein level normalized to actin for loading variations. n = 6–8 in each group. Values represent mean ± S.E. (c) RT-PCR of left ventricle p67phox in Sed-control, Sed-Dia, ExT-control, and ExT-Dia animals: top: a representative gel; bottom: quantification of p67phox transcript normalized to β-actin as loading control. n = 6 in each group. (d) Protein expression of p67phox in four groups: top: a representative Western blot; bottom: densitometry analysis of p67phox protein normalized to β-actin as loading control. n = 3-4 in each group. Values are represented as mean ± S.E. ∗ P < 0.05 versus Sed-control. + P < 0.05 versus Sed-Dia group.
3.3. Increased Expression of p47phox and p67phox in Right Ventricle Alleviated with ExT in Diabetic Animals
Similar to the left ventricle, 4 weeks of diabetes also increased p47phox mRNA and protein expression in the right ventricle, albeit not reaching statistical significance compared to Sed-control (Figure 2(a)). Steady-state levels of mRNA encoding p67phox in the right ventricle of the heart increased slightly (17.7%). However, there were no significant differences in p67phox protein expression between the two groups (Figures 2(c) and 2(d)). Three weeks of ExT regimen attenuated the increase in steady-state levels of mRNA encoding p47phox to similar levels in ExT-control and ExT-Dia groups.
Figure 2.

Expression of right ventricle p47phox and p67phox in Sed-control, Sed-Dia, ExT-control, and ExT-Dia animals. (a) RT-PCR of p47phox: top: a representative gel; bottom: bar graph shows quantification of densitometry analysis of p47phox normalized to β-actin. n = 10 in each group. Values represent mean ± S.E. (b) Protein expression of p47phox in four groups: top: a representative gel; bottom: densitometry analyses of p47phox protein level normalized to actin. n = 3-4 in each group. Values represent mean ± S.E. (c) RT-PCR of right ventricle p67phox in Sed-control, Sed-Dia, ExT-control, and ExT-Dia animals: top: a representative gel; bottom: quantification of p67phox transcript normalized to β-actin as loading control. n = 6 in each group. (d) Protein expression of p67phox in four groups: top: a representative Western blot; bottom: densitometry analysis of p67phox protein normalized to β-actin as loading control. n = 3-4 in each group. Values are represented as mean ± S.E. ∗ P < 0.05 versus Sed-control. + P < 0.05 versus Sed-Dia group.
3.4. Increased Expression of p47phox and p67phox in the Atria Alleviated with ExT in Diabetic Animals
As shown in Figure 3, steady-state levels of mRNA encoding p47phox and p67phox were increased significantly in atria of the diabetic group. The increase was 30.1% for p47phox and 35.5% for p67phox over sedentary controls (Figures 3(a) and 3(c)). As expected, three weeks of ExT, initiated after 1 week of diabetes, significantly lowered mRNA levels of p47phox and p67phox in the atria of ExT-Dia heart compared to Sed-Dia group. Protein expression of p47phox was increased 74%, while p67phox protein expression increased but did not reach statistical significance (Figures 3(b) and 3(d)). Interestingly, p47phox mRNA and protein expression in ExT-control and ExT-Dia atria was decreased to almost similar levels suggesting that ExT attenuated the increase in steady-state levels of p47phox expression in atria.
Figure 3.

Expression of atrium p47phox and p67phox in Sed-control, Sed-Dia, ExT-control, and ExT-Dia animals. (a) RT-PCR of p47phox: top: a representative gel; bottom: bar graph shows quantification of densitometry analysis of p47phox normalized to β-actin. n = 10 in each group. Values represent mean ± S.E. (b) Protein expression of p47phox in four groups: top: a representative gel; bottom: densitometry analyses of p47phox protein level normalized to actin. n = 6–8 in each group. Values represent mean ± S.E. (c) RT-PCR of atrium p67phox in Sed-control, Sed-Dia, ExT-control, and ExT-Dia animals: top: a representative gel; bottom: quantification of p67phox transcript normalized to β-actin as loading control. n = 6 in each group. (d) Protein expression of p67phox in four groups: top: a representative Western blot; bottom: densitometry analysis of p67phox protein normalized to β-actin as loading control. n = 3-4 in each group. Values are represented as mean ± S.E. ∗ P < 0.05 versus Sed-control. + P < 0.05 versus Sed-Dia group.
3.5. Increased Expression of Arterial Collagen III Is Alleviated with ExT in Diabetic Animals
As shown in Figure 4, expression of the extracellular matrix protein collagen III significantly increased by 53% in 4 weeks Sed-Dia group. This increase in steady-state levels of collagen III suggests increasing ventricular stiffening, a characteristic feature of diabetic cardiomyopathy. Three weeks of ExT initiated after 1 week of STZ-induced diabetes attenuated the increase in collagen III protein expression (38%) in the diabetic group at the same level as control, while three weeks of ExT had no effect on collagen III expression in nondiabetic control animals.
Figure 4.
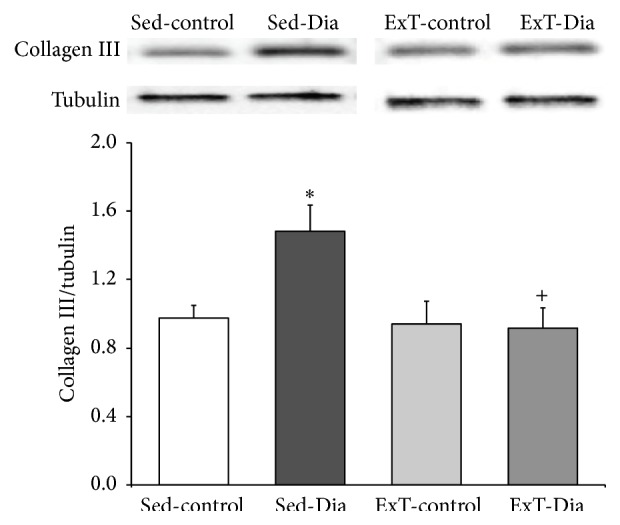
Collagen III expression in atria: top: representative Western blot showing collagen III protein expression in Sed-control, Sed-Dia, ExT-control, and ExT-Dia animals; bottom: densitometry analysis of collagen III expression normalized to β-tubulin as loading control. n = 6 in each group. Values represent mean ± S.E. ∗ P < 0.01 versus Sed-control group. + P < 0.05 versus Sed-Dia group.
4. Discussion
The principal finding of the present study is that expression of the cytosolic subunits of NADPH oxidase, p47phox and p67phox, is upregulated in hearts of STZ-induced diabetic rats and the increased expression was attenuated with ExT. Since ExT was initiated after 1 week of diabetes, it is likely that it is preventing or minimizing rather than reversing the increase in expression induced by diabetes. Previously, we have reported that Sed-Dia animals demonstrated significant reductions in fractional shortening, ejection fraction, stroke volume, and cardiac output compared with Sed-control animals and ExT (for 3 weeks) implemented 4 weeks after the onset of diabetes significantly increased percent ejection fraction, attenuated the increase in left ventricular end-systolic diameter, and improved dP/dt and isoproterenol induced increase in dP/dt in the diabetic group [60]. It is possible that these changes in expression of NADPH oxidase subunits in the heart are associated with improvement in cardiac function in diabetes.
There is increasing information that mitochondria and NADPH oxidase play a fundamental role in ROS production in the diabetic heart [12]. Furthermore, ROS-mediated increase in peroxynitrite formation induces apoptosis in cardiomyocytes in vitro and in the myocardium in vivo as shown by Levrand et al. [65] suggesting that oxidative stress is a common mediator for apoptosis induced cardiac damage in diabetic rats [13]. Our data suggest that the increased oxidative stress levels observed in diabetic hearts may be in part due to the upregulation of NADPH oxidase subunits and this change can be alleviated by ExT. These results suggest that beneficial effects of ExT may involve improvement in oxidative stress commonly observed in diabetic cardiomyopathy.
Diabetic cardiomyopathy, one of the leading causes of increased morbidity and mortality in the diabetic population, is characterized by the prolongation of action potential duration, delayed cytosolic Ca2+ clearance, and impaired systolic and diastolic left ventricular function [42, 60]. Several factors are believed to contribute to the pathogenesis of diabetic cardiomyopathy, including insulin resistance, enhanced renin-angiotensin system activation, hyperglycemia, and damage from ROS [66]. These pathophysiological factors associated with diabetes, including hyperglycemia and elevated Ang II levels, have been shown to increase the expression of NADPH oxidase subunits and the enzyme's activity [32, 43, 67] leading to myocardial oxidative stress. A pivotal role of NADPH oxidase in Ang II-induced cardiac hypertrophy and interstitial fibrosis was demonstrated by using mice with targeted disruption of the NADPH oxidase subunit gp91phox [34]. p47phox subunit of NADPH oxidase has also been demonstrated to play a key role in the enzyme's activation by agonists. Using p47phox knockout mice and p47phox cDNA transfection and antisense techniques, Li et al. [68] demonstrated the necessity of p47phox subunit for the activation of NADPH oxidase when exposed to the agonists Ang II, TNF-α, or phorbol 12-myristate 13-acetate, an activator of protein kinase C. The phosphorylation of p47phox is believed to induce conformational changes that facilitate its binding to the other cytosolic subunits, including p67phox, and their subsequent binding to the membrane-bound cytochrome b558 to allow optimal O2 − production [69]. Precise mechanisms underlying agonist-induced nonphagocytic NADPH oxidase activation are not yet understood, but it is clear that both acute protein modification and chronic changes in expression levels may be involved.
Our findings indicate that the cardiac tissue expressions for p47phox and p67phox are upregulated in the diabetic rats. This upregulation was greatest in the left ventricle and atria, while it was slightly elevated in the right ventricle of the diabetic group. These findings are consistent with clinical data showing an association of DM with an increased risk of systolic, diastolic, and any left ventricular dysfunction and the relative sparing of the right ventricle [1, 2]. In ventricular myocytes, it was reported that hyperglycemic conditions increase the production of ROS via an enhanced expression of p47phox protein, which could be blocked by an AT1R antagonist [42]. Cifuentes et al. [32] have also shown that Ang II infusion, via the AT1R activation, induces an enhanced expression of p67phox and gp91phox proteins in aortic adventitia. Additionally, an increased NADPH oxidase activity and a 7-fold upregulation of gp91phox mRNA in aortic tissue have been reported in STZ-induced diabetic rats [43]. Furthermore, increased NADPH oxidase activity and collagen 3 were demonstrated in pathogenesis of the heart failure as seen in hearts of Duchenne muscular dystrophy rat model [70] and also a significant increase in collagen III was found in endomyocardial biopsies obtained from patients with DM [71]. Ang II has also been shown to stimulate collagen I and collagen III in cardiac fibroblast, and apocynin, an inhibitor of NADPH oxidase, prevents this initiation suggesting a critical role for ROS in cardiac remodelling [72]. Although a medical treatment with different antioxidant has been proposed for diabetic cardiomyopathy for decades, unfortunately, such treatments have failed to attenuate cardiac dysfunction or improve outcomes [73]. This may be due to lack of improving the ROS load in an appropriate measure in the myocardium rather than an overall reduction in ROS.
Physical activity is widely accepted as a key element in the prevention of type 2 diabetes [74] and also has beneficial effects in patients with established heart disease [75], although the physiological mechanisms explaining how physical activity promotes health remain to be fully elucidated. ExT maintains cardiac output by blunting diabetes-induced bradycardia and the reduction in force of myocardial contractility [60]. ExT has been shown to elicit a number of beneficial physiological changes in animals and clinical studies. These include (1) a reduction in blood glucose, body fat, and insulin resistance [45, 47], (2) improved glycemic control and lipid metabolism [45, 76], (3) improved baroreflex sensitivity [46], (4) reduced plasma Ang II levels [49–51, 54], and (5) reduced renal sympathetic nerve activity and arterial pressure responses to Ang II [50, 52, 54, 77]. Additionally, the ability of ExT to upregulate antioxidant enzyme expression and activity in the cardiovascular system has been clearly demonstrated [48, 54, 55, 78, 79]. Previously, our lab showed ExT initiated after the onset of diabetes blunts primarily beta(1)-adrenoceptor expression loss and improves cardiac function in diabetic rats [60].
The current study adds to this literature by demonstrating for the first time that ExT significantly reduces p47phox mRNA expression in the left and right ventricles and atria of diabetic rats. In diabetic rats, ExT significantly reduced p67phox mRNA expression of the left ventricle and attenuated the increased p67phox message in the atria. The increase in NADPH oxidase subunit expression observed in the diabetic condition and its attenuation by ExT may occur via a variety of mechanisms. Hyperglycemia, enhanced renin-angiotensin system activity, and elevated levels of circulating cytokines are all clinical manifestations associated with the diabetic condition, and each is known to promote NADPH-derived O2 − production. TNF-α, a cytokine that activates transcription factors which induce the expression of genes involved in inflammation and cell growth, has been identified as an agonist for NADPH oxidase [68]. Levels of TNF-α are increased in response to both hyperglycemic conditions and elevated Ang II levels, and the increase is mediated via AT1R [68, 80]. Hyperglycemia and elevated Ang II levels have been clearly shown to upregulate the expression of NADPH oxidase subunits and increase NADPH oxidase activity and O2 − production [32, 39, 67, 81]. Based on these findings, it appears that hyperglycemia and elevated Ang II associated with diabetes work via the AT1 receptor to increase TNF-α levels and NADPH oxidase activity, while ExT suppresses expression of TNF-alpha and thus offers a potential protection against TNF-alpha-induced insulin resistance [82] and increased ROS production [83, 84].
Our study provides evidence that increase in oxidative stress, specifically in left ventricle, may be due to an enhanced transcription as well as translation of p47phox and p67phox subunits of NADPH oxidase. Expression of p67phox was increased at transcription level but not at the protein level in atria of diabetic rats suggesting that increased transcription did not contribute to changes in protein expression in this tissue. Translocation of cytosolic subunits (p47phox, p67phox, p40phox, and rac1) and their association with membrane-bound cytochrome b558 (consisting of one p22phox and one gp91phox subunit) follows during acute oxidase activation [85]. In present studies perhaps there was only translocation of p67phox in the atria of diabetic heart rather than overall expression of synthesis, a possibility that remains to be explored.
The numerous physiological benefits of ExT offer several mechanisms via which the expression and activity of NADPH oxidase may be attenuated in diabetes. ExT has been shown to normalize diabetes-related elevations in blood glucose [45–47], plasma Ang II [49, 50, 53], and TNF-α [83, 84]. Normalizing these factors could lower levels of ROS-inducing agonists, thus dampening NADPH oxidase activity. Additionally, ExT has repeatedly been shown to upregulate antioxidant expression and activity, including SOD, catalase, and glutathione, in aortic and cardiac tissues [48, 49, 54, 56, 78, 79]. However, while the literature provides some insight, the specific mechanism by which ExT lowered the mRNA expression of p47phox and p67phox in our current study remains to be elucidated.
5. Conclusions
In summary, our data show that the mRNA expressions of the NADPH oxidase subunits p47phox and p67phox are upregulated in cardiac tissue in the diabetic condition. Furthermore, our data show that ExT attenuates the upregulated expression of these NADPH oxidase subunits and normalizes the increased collagen III levels (Figure 5) and provides support with a potential mechanistic link for exercise training as being an effective nonpharmacological tool in regulating oxidative stress levels in the diabetic heart. Future areas of research will need to focus on understanding some of the mechanisms involved in diabetic cardiomyopathy and the therapeutic strategies such as ExT to halt or slow its progression.
Figure 5.
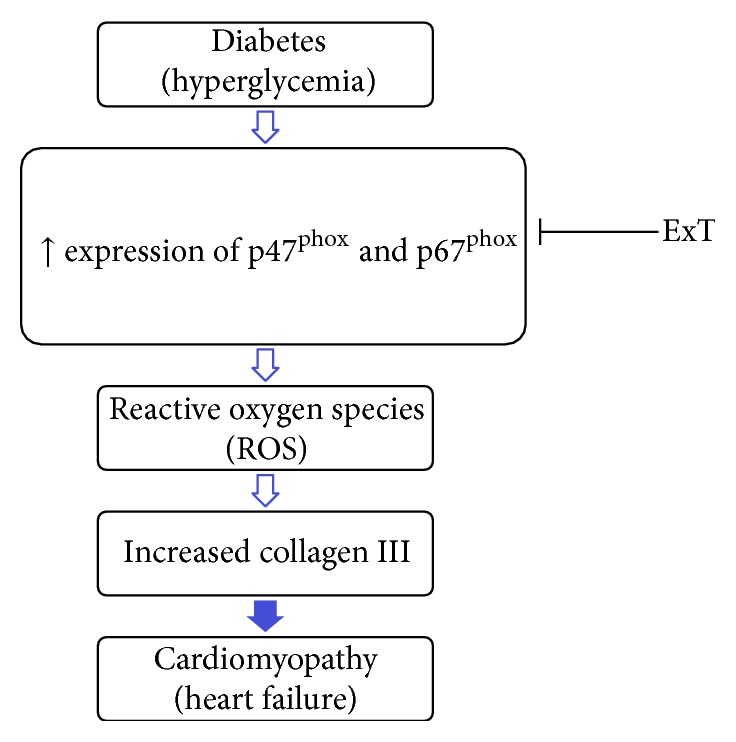
Amelioration of cardiomyopathy by exercise training. Hyperglycemia induces overexpression of cytoplasmic subunits of NADH oxidase (p47phox and p67phox) in left ventricle. Overexpression of these subunits exhibits high reactive oxygen species and leads to increased collagen III and hence cardiomyopathy. Exercise training (ExT) mitigates the expression of p47phox, and p67phox consequently ameliorates heart dysfunctions.
Acknowledgments
Funding from National Institutes of Health, Heart, Lung, and Blood Institute, R56 HL124104, and American Heart Association 14SDG19980007 grant supported the work. The technical assistance of Dr. Xuefei Liu is greatly appreciated.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Dandamudi S., Slusser J., Mahoney D. W., Redfield M. M., Rodeheffer R. J., Chen H. H. The prevalence of diabetic cardiomyopathy: a population-based study in Olmsted County, Minnesota. Journal of Cardiac Failure. 2014;20(5):304–309. doi: 10.1016/j.cardfail.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Devereux R. B., Roman M. J., Paranicas M., et al. Impact of diabetes on cardiac structure and function: the Strong Heart study. Circulation. 2000;101(19):2271–2276. doi: 10.1161/01.cir.101.19.2271. [DOI] [PubMed] [Google Scholar]
- 3.Ilercil A., Devereux R. B., Roman M. J., et al. Relationship of impaired glucose tolerance to left ventricular structure and function: the Strong Heart study. American Heart Journal. 2001;141(6):992–998. doi: 10.1067/mhj.2001.115302. [DOI] [PubMed] [Google Scholar]
- 4.Rubler S., Dlugash J., Yuceoglu Y. Z., Kumral T., Branwood A. W., Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. The American Journal of Cardiology. 1972;30(6):595–602. doi: 10.1016/0002-9149(72)90595-4. [DOI] [PubMed] [Google Scholar]
- 5.Boudina S., Abel E. D. Diabetic cardiomyopathy revisited. Circulation. 2007;115(25):3213–3223. doi: 10.1161/circulationaha.106.679597. [DOI] [PubMed] [Google Scholar]
- 6.Aneja A., Tang W. H. W., Bansilal S., Garcia M. J., Farkouh M. E. Diabetic cardiomyopathy: insights into pathogenesis, diagnostic challenges, and therapeutic options. American Journal of Medicine. 2008;121(9):748–757. doi: 10.1016/j.amjmed.2008.03.046. [DOI] [PubMed] [Google Scholar]
- 7.Falcão-Pires I., Leite-Moreira A. F. Diabetic cardiomyopathy: understanding the molecular and cellular basis to progress in diagnosis and treatment. Heart Failure Reviews. 2012;17(3):325–344. doi: 10.1007/s10741-011-9257-z. [DOI] [PubMed] [Google Scholar]
- 8.Baynes J. W., Thorpe S. R. Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes. 1999;48(1):1–9. doi: 10.2337/diabetes.48.1.1. [DOI] [PubMed] [Google Scholar]
- 9.Giugliano D., Ceriello A., Paolisso G. Oxidative stress and diabetic vascular complications. Diabetes Care. 1996;19(3):257–267. doi: 10.2337/diacare.19.3.257. [DOI] [PubMed] [Google Scholar]
- 10.Yu T., Sheu S.-S., Robotham J. L., Yoon Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovascular Research. 2008;79(2):341–351. doi: 10.1093/cvr/cvn104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boudina S., Bugger H., Sena S., et al. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation. 2009;119(9):1272–1283. doi: 10.1161/circulationaha.108.792101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teshima Y., Takahashi N., Nishio S., et al. Production of reactive oxygen species in the diabetic heart: roles of mitochondria and NADPH oxidase. Circulation Journal. 2014;78(2):300–306. doi: 10.1253/circj.cj-13-1187. [DOI] [PubMed] [Google Scholar]
- 13.Dallak M. M., Mikhailidis D. P., Haidara M. A., et al. Oxidative stress as a common mediator for apoptosis induced-cardiac damage in diabetic rats. Open Cardiovascular Medicine Journal. 2008;2(1):70–78. doi: 10.2174/1874192400802010070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takahashi N., Kume O., Wakisaka O., et al. Novel strategy to prevent atrial fibrosis and fibrillation. Circulation Journal. 2012;76(10):2318–2326. doi: 10.1253/circj.cj-12-1099. [DOI] [PubMed] [Google Scholar]
- 15.Gryglewski R. J., Palmer R. M. J., Moncada S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature. 1986;320(6061):454–456. doi: 10.1038/320454a0. [DOI] [PubMed] [Google Scholar]
- 16.Mugge A., Elwell J. H., Peterson T. E., Harrison D. G. Release of intact endothelium-derived relaxing factor depends on endothelial superoxide dismutase activity. The American Journal of Physiology—Cell Physiology. 1991;260(2, part 1):C219–C225. doi: 10.1152/ajpcell.1991.260.2.C219. [DOI] [PubMed] [Google Scholar]
- 17.Tesfamariam B., Cohen R. A. Enhanced adrenergic neurotransmission in diabetic rabbit carotid artery. Cardiovascular Research. 1995;29(4):549–554. doi: 10.1016/0008-6363(96)88533-6. [DOI] [PubMed] [Google Scholar]
- 18.Katusic Z. S., Vanhoutte P. M. Superoxide anion is an endothelium-derived contracting factor. The American Journal of Physiology—Heart and Circulatory Physiology. 1989;257(1, part 2):H33–H37. doi: 10.1152/ajpheart.1989.257.1.H33. [DOI] [PubMed] [Google Scholar]
- 19.Sugden P. H., Clerk A. Cellular mechanisms of cardiac hypertrophy. Journal of Molecular Medicine. 1998;76(11):725–746. doi: 10.1007/s001090050275. [DOI] [PubMed] [Google Scholar]
- 20.Akki A., Zhang M., Murdoch C., Brewer A., Shah A. M. NADPH oxidase signaling and cardiac myocyte function. Journal of Molecular and Cellular Cardiology. 2009;47(1):15–22. doi: 10.1016/j.yjmcc.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 21.Griendling K. K., Sorescu D., Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circulation Research. 2000;86(5):494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 22.Wang H. D., Xu S., Johns D. G., et al. Role of NADPH oxidase in the vascular hypertrophic and oxidative stress response to angiotensin II in mice. Circulation Research. 2001;88(9):947–953. doi: 10.1161/hh0901.089987. [DOI] [PubMed] [Google Scholar]
- 23.Li J.-M., Shah A. M. Differential NADPH- versus NADH-dependent superoxide production by phagocyte-type endothelial cell NADPH oxidase. Cardiovascular Research. 2001;52(3):477–486. doi: 10.1016/S0008-6363(01)00407-2. [DOI] [PubMed] [Google Scholar]
- 24.Cave A., Grieve D., Johar S., Zhang M., Shah A. M. NADPH oxidase-derived reactive oxygen species in cardiac pathophysiology. Philosophical Transactions of the Royal Society B: Biological Sciences. 2005;360(1464):2327–2334. doi: 10.1098/rstb.2005.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patterson C., Ruef J., Madamanchi N. R., et al. Stimulation of a vascular smooth muscle cell NAD(P)H oxidase by thrombin: evidence that p47phox may participate in forming this oxidase in vitro and in vivo . Journal of Biological Chemistry. 1999;274(28):19814–19822. doi: 10.1074/jbc.274.28.19814. [DOI] [PubMed] [Google Scholar]
- 26.Patel K. P., Mayhan W. G., Bidasee K. R., Zheng H. Enhanced angiotensin II-mediated central sympathoexcitation in streptozotocin-induced diabetes: role of superoxide anion. American Journal of Physiology—Regulatory Integrative and Comparative Physiology. 2011;300(2):R311–R320. doi: 10.1152/ajpregu.00246.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siragy H. M. AT(1) and AT(2) receptors in the kidney: role in disease and treatment. American Journal of Kidney Diseases. 2000;36(3, supplement 1):S4–S9. doi: 10.1053/ajkd.2000.9684. [DOI] [PubMed] [Google Scholar]
- 28.Sowers J. R., Epstein M., Frohlich E. D. Diabetes, hypertension, and cardiovascular disease—an update. Hypertension. 2001;37(4):1053–1059. doi: 10.1161/01.hyp.37.4.1053. [DOI] [PubMed] [Google Scholar]
- 29.Koshkin V., Lotan O., Pick E. The cytosolic component p47(phox) is not a sine qua non participant in the activation of NADPH oxidase but is required for optimal superoxide production. The Journal of Biological Chemistry. 1996;271(48):30326–30329. doi: 10.1074/jbc.271.48.30326. [DOI] [PubMed] [Google Scholar]
- 30.Siragy H. M., Awad A., Abadir P., Webb R. The angiotensin II type 1 receptor mediates renal interstitial content of tumor necrosis factor-alpha in diabetic rats. Endocrinology. 2003;144(6):2229–2233. doi: 10.1210/en.2003-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Griendling K. K., Minieri C. A., Ollerenshaw J. D., Alexander R. W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circulation Research. 1994;74(6):1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 32.Cifuentes M. E., Rey F. E., Carretero O. A., Pagano P. J. Upregulation of p67(phox) and gp91(phox) in aortas from angiotensin II-infused mice. The American Journal of Physiology—Heart and Circulatory Physiology. 2000;279(5):H2234–H2240. doi: 10.1152/ajpheart.2000.279.5.H2234. [DOI] [PubMed] [Google Scholar]
- 33.Lang D., Mosfer S. I., Shakesby A., Donaldson F., Lewis M. J. Coronary microvascular endothelial cell redox state in left ventricular hypertrophy: the role of angiotensin II. Circulation Research. 2000;86(4):463–469. doi: 10.1161/01.res.86.4.463. [DOI] [PubMed] [Google Scholar]
- 34.Bendall J. K., Cave A. C., Heymes C., Gall N., Shah A. M. Pivotal role of a gp91phox-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation. 2002;105(3):293–296. doi: 10.1161/hc0302.103712. [DOI] [PubMed] [Google Scholar]
- 35.Griendling K. K., Murphy T. J., Alexander R. W. Molecular biology of the renin-angiotensin system. Circulation. 1993;87(6):1816–1828. doi: 10.1161/01.CIR.87.6.1816. [DOI] [PubMed] [Google Scholar]
- 36.Griendling K. K., Sorescu D., Lassègue B., Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20(10):2175–2183. doi: 10.1161/01.atv.20.10.2175. [DOI] [PubMed] [Google Scholar]
- 37.Griendling K. K., Ushio-Fukai M. Reactive oxygen species as mediators of angiotensin II signaling. Regulatory Peptides. 2000;91(1–3):21–27. doi: 10.1016/s0167-0115(00)00136-1. [DOI] [PubMed] [Google Scholar]
- 38.Pagano P. J., Clark J. K., Cifuentes-Pagano M. E., Clark S. M., Callis G. M., Quinn M. T. Localization of a constitutively active, phagocyte-like NADPH oxidase in rabbit aortic adventitia: enhancement by angiotensin II. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(26):14483–14488. doi: 10.1073/pnas.94.26.14483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang H., Schmeisser A., Garlichs C. D., et al. Angiotensin II-induced superoxide anion generation in human vascular endothelial cells: role of membrane-bound NADH-/NADPH-oxidases. Cardiovascular Research. 1999;44(1):215–222. doi: 10.1016/S0008-6363(99)00183-2. [DOI] [PubMed] [Google Scholar]
- 40.Zhang M., Kho A. L., Anilkumar N., et al. Glycated proteins stimulate reactive oxygen species production in cardiac myocytes: involvement of Nox2 (gp91phox)-containing NADPH oxidase. Circulation. 2006;113(9):1235–1243. doi: 10.1161/circulationaha.105.581397. [DOI] [PubMed] [Google Scholar]
- 41.Nishio S., Teshima Y., Takahashi N., et al. Activation of CaMKII as a key regulator of reactive oxygen species production in diabetic rat heart. Journal of Molecular and Cellular Cardiology. 2012;52(5):1103–1111. doi: 10.1016/j.yjmcc.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 42.Privratsky J. R., Wold L. E., Sowers J. R., Quinn M. T., Ren J. AT1 blockade prevents glucose-induced cardiac dysfunction in ventricular myocytes: role of the AT1 receptor and NADPH oxidase. Hypertension. 2003;42(2):206–212. doi: 10.1161/01.hyp.0000082814.62655.85. [DOI] [PubMed] [Google Scholar]
- 43.Hink U., Li H., Mollnau H., et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circulation Research. 2001;88(2):E14–E22. doi: 10.1161/01.res.88.2.e14. [DOI] [PubMed] [Google Scholar]
- 44.Thijssen D. H. J., Maiorana A. J., O'Driscoll G., Cable N. T., Hopman M. T. E., Green D. J. Impact of inactivity and exercise on the vasculature in humans. European Journal of Applied Physiology. 2010;108(5):845–875. doi: 10.1007/s00421-009-1260-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li S., Culver B., Ren J. Benefit and risk of exercise on myocardial function in diabetes. Pharmacological Research. 2003;48(2):127–132. doi: 10.1016/S1043-6618(03)00099-9. [DOI] [PubMed] [Google Scholar]
- 46.Loimaala A., Huikuri H. V., Kööbi T., Rinne M., Nenonen A., Vuori I. Exercise training improves baroreflex sensitivity in type 2 diabetes. Diabetes. 2003;52(7):1837–1842. doi: 10.2337/diabetes.52.7.1837. [DOI] [PubMed] [Google Scholar]
- 47.Rousseau-Migneron S., Turcotte L., Tancrede G., Nadeau A. Transient increase in basal insulin levels in severely diabetic rats submitted to physical training. Diabetes Research. 1988;9(2):97–100. [PubMed] [Google Scholar]
- 48.Davis M. E., Cai H., McCann L., Fukai T., Harrison D. G. Role of c-Src in regulation of endothelial nitric oxide synthase expression during exercise training. The American Journal of Physiology—Heart and Circulatory Physiology. 2003;284(4):H1449–H1453. doi: 10.1152/ajpheart.00918.2002. [DOI] [PubMed] [Google Scholar]
- 49.Kohno H., Furukawa S., Naito H., Minamitani K., Ohmori D., Yamakura F. Contribution of nitric oxide, angiotensin II and superoxide dismutase to exercise-induced attenuation of blood pressure elevation in spontaneously hypertensive rats. Japanese Heart Journal. 2002;43(1):25–34. doi: 10.1536/jhj.43.25. [DOI] [PubMed] [Google Scholar]
- 50.Braith R. W., Edwards D. G. Neurohormonal abnormalities in heart failure: impact of exercise training. Congestive Heart Failure. 2003;9(2):70–76. doi: 10.1111/j.1527-5299.2003.00277.x. [DOI] [PubMed] [Google Scholar]
- 51.Llewellyn T. L., Sharma N. M., Zheng H., Patel K. P. Effects of exercise training on SFO-mediated sympathoexcitation during chronic heart failure. The American Journal of Physiology—Heart and Circulatory Physiology. 2014;306(1):H121–H131. doi: 10.1152/ajpheart.00534.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Negrao C. E., Irigoyen M. C., Moreira E. D., Brum P. C., Freire P. M., Krieger E. M. Effect of exercise training on RSNA, baroreflex control, and blood pressure responsiveness. The American Journal of Physiology. 1993;265(2, part 2):R365–R370. doi: 10.1152/ajpregu.1993.265.2.R365. [DOI] [PubMed] [Google Scholar]
- 53.Zucker I. H., Pliquett R. U. Novel mechanisms of sympatho-excitation in chronic heart failure. Heart Failure Monitor. 2002;3(1):2–7. [PubMed] [Google Scholar]
- 54.Husain K. Interaction of physical training and chronic nitroglycerin treatment on blood pressure, nitric oxide, and oxidants/antioxidants in the rat heart. Pharmacological Research. 2003;48(3):253–261. doi: 10.1016/S1043-6618(03)00150-6. [DOI] [PubMed] [Google Scholar]
- 55.Fukai T., Siegfried M. R., Ushio-Fukai M., Cheng Y., Kojda G., Harrison D. G. Regulation of the vascular extracellular superoxide dismutase by nitric oxide and exercise training. Journal of Clinical Investigation. 2000;105(11):1631–1639. doi: 10.1172/JCI9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Powers S. K., Lennon S. L., Quindry J., Mehta J. L. Exercise and cardioprotection. Current Opinion in Cardiology. 2002;17(5):495–502. doi: 10.1097/00001573-200209000-00009. [DOI] [PubMed] [Google Scholar]
- 57.Fridovich I. The biology of oxygen radicals. Science. 1978;201(4359):875–880. doi: 10.1126/science.210504. [DOI] [PubMed] [Google Scholar]
- 58.Balzan R., Agius D. R., Bannister W. H. Cloned prokaryotic iron superoxide dismutase protects yeast cells against oxidative stress depending on mitochondrial location. Biochemical and Biophysical Research Communications. 1999;256(1):63–67. doi: 10.1006/bbrc.1999.0285. [DOI] [PubMed] [Google Scholar]
- 59.Musch T. I., Terrell J. A. Skeletal muscle blood flow abnormalities in rats with a chronic myocardial infarction: rest and exercise. The American Journal of Physiology—Heart and Circulatory Physiology. 1992;262(2):H411–H419. doi: 10.1152/ajpheart.1992.262.2.H411. [DOI] [PubMed] [Google Scholar]
- 60.Bidasee K. R., Zheng H., Shao C.-H., Parbhu S. K., Rozanski G. J., Patel K. P. Exercise training initiated after the onset of diabetes preserves myocardial function: effects on expression of beta-adrenoceptors. Journal of Applied Physiology. 2008;105(3):907–914. doi: 10.1152/japplphysiol.00103.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Srere P. A. [1] Citrate synthase: [EC 4.1.3.7. Citrate oxaloacetate-lyase (CoA-acetylating)] Methods in Enzymology. 1969;13:3–11. doi: 10.1016/0076-6879(69)13005-0. [DOI] [Google Scholar]
- 62.Sharma N. M., Zheng H., Mehta P. P., Li Y.-F., Patel K. P. Decreased nNOS in the PVN leads to increased sympathoexcitation in chronic heart failure: role for CAPON and Ang II. Cardiovascular Research. 2011;92(2):348–357. doi: 10.1093/cvr/cvr217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sharma N. M., Zheng H., Li Y.-F., Patel K. P. Nitric oxide inhibits the expression of AT1 receptors in neurons. American Journal of Physiology: Cell Physiology. 2012;302(8):C1162–C1173. doi: 10.1152/ajpcell.00258.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Siu P. M., Donley D. A., Bryner R. W., Alway S. E. Citrate synthase expression and enzyme activity after endurance training in cardiac and skeletal muscles. Journal of Applied Physiology. 2003;94(2):555–560. doi: 10.1152/japplphysiol.00821.2002. [DOI] [PubMed] [Google Scholar]
- 65.Levrand S., Vannay-Bouchiche C., Pesse B., et al. Peroxynitrite is a major trigger of cardiomyocyte apoptosis in vitro and in vivo. Free Radical Biology and Medicine. 2006;41(6):886–895. doi: 10.1016/j.freeradbiomed.2006.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dzau V. J. Tissue angiotensin and pathobiology of vascular disease: a unifying hypothesis. Hypertension. 2001;37(4):1047–1052. doi: 10.1161/01.hyp.37.4.1047. [DOI] [PubMed] [Google Scholar]
- 67.Fukui T., Ishizaka N., Rajagopalan S., et al. p22phox mRNA expression and NADPH oxidase activity are increased in aortas from hypertensive rats. Circulation Research. 1997;80(1):45–51. doi: 10.1161/01.res.80.1.45. [DOI] [PubMed] [Google Scholar]
- 68.Li J.-M., Mullen A. M., Yun S., et al. Essential role of the NADPH oxidase subunit p47phox in endothelial cell superoxide production in response to phorbol ester and tumor necrosis factor-α . Circulation Research. 2002;90(2):143–150. doi: 10.1161/hh0202.103615. [DOI] [PubMed] [Google Scholar]
- 69.Babior B. M. NADPH oxidase: an update. Blood. 1999;93(5):1464–1476. [PubMed] [Google Scholar]
- 70.Williams I. A., Allen D. G. The role of reactive oxygen species in the hearts of dystrophin-deficient mdx mice. The American Journal of Physiology—Heart and Circulatory Physiology. 2007;293(3):H1969–H1977. doi: 10.1152/ajpheart.00489.2007. [DOI] [PubMed] [Google Scholar]
- 71.Shimizu M., Umeda K., Sugihara N., et al. Collagen remodelling in myocardia of patients with diabetes. Journal of Clinical Pathology. 1993;46(1):32–36. doi: 10.1136/jcp.46.1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lijnen P. J., van Pelt J. F., Fagard R. H. Stimulation of reactive oxygen species and collagen synthesis by angiotensin II in cardiac fibroblasts. Cardiovascular Therapeutics. 2012;30(1):e1–e8. doi: 10.1111/j.1755-5922.2010.00205.x. [DOI] [PubMed] [Google Scholar]
- 73.Kasznicki J., Drzewoski J. Heart failure in the diabetic population—pathophysiology, diagnosis and management. Archives of Medical Science. 2014;10(3):546–556. doi: 10.5114/aoms.2014.43748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.De Feo P., Schwarz P. Is physical exercise a core therapeutical element for most patients with type 2 diabetes? Diabetes Care. 2013;36(supplement 2):S149–S154. doi: 10.2337/dcs13-2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Myers J., Prakash M., Froelicher V., Do D., Partington S., Edwin Atwood J. Exercise capacity and mortality among men referred for exercise testing. The New England Journal of Medicine. 2002;346(11):793–801. doi: 10.1056/nejmoa011858. [DOI] [PubMed] [Google Scholar]
- 76.Goguen J. M., Leiter L. A. Lipids and diabetes mellitus: a review of therapeutic options. Current Medical Research and Opinion. 2002;18(supplement 1):s58–s74. doi: 10.1185/030079902125000246. [DOI] [PubMed] [Google Scholar]
- 77.Zheng H., Sharma N. M., Liu X., Patel K. P. Exercise training normalizes enhanced sympathetic activation from the paraventricular nucleus in chronic heart failure: role of angiotensin II. American Journal of Physiology: Regulatory Integrative and Comparative Physiology. 2012;303(4):R387–R394. doi: 10.1152/ajpregu.00046.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rush J. W. E., Turk J. R., Laughlin M. H. Exercise training regulates SOD-1 and oxidative stress in porcine aortic endothelium. The American Journal of Physiology—Heart and Circulatory Physiology. 2003;284(4):H1378–H1387. doi: 10.1152/ajpheart.00190.2002. [DOI] [PubMed] [Google Scholar]
- 79.Husain K., Hazelrigg S. R. Oxidative injury due to chronic nitric oxide synthase inhibition in rat: effect of regular exercise on the heart. Biochimica et Biophysica Acta (BBA)—Molecular Basis of Disease. 2002;1587(1):75–82. doi: 10.1016/s0925-4439(02)00070-4. [DOI] [PubMed] [Google Scholar]
- 80.Shanmugam N., Reddy M. A., Guha M., Natarajan R. High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes. 2003;52(5):1256–1264. doi: 10.2337/diabetes.52.5.1256. [DOI] [PubMed] [Google Scholar]
- 81.Rajagopalan S., Kurz S., Münzel T., et al. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. The Journal of Clinical Investigation. 1996;97(8):1916–1923. doi: 10.1172/jci118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Petersen A. M. W., Pedersen B. K. The anti-inflammatory effect of exercise. Journal of Applied Physiology. 2005;98(4):1154–1162. doi: 10.1152/japplphysiol.00164.2004. [DOI] [PubMed] [Google Scholar]
- 83.Aldhahi W., Hamdy O. Adipokines, inflammation, and the endothelium in diabetes. Current Diabetes Reports. 2003;3(4):293–298. doi: 10.1007/s11892-003-0020-2. [DOI] [PubMed] [Google Scholar]
- 84.Straczkowski M., Kowalska I., Dzienis-Straczkowska S., et al. Changes in tumor necrosis factor-alpha system and insulin sensitivity during an exercise training program in obese women with normal and impaired glucose tolerance. European Journal of Endocrinology. 2001;145(3):273–280. doi: 10.1530/eje.0.1450273. [DOI] [PubMed] [Google Scholar]
- 85.Guo Z., Xia Z., Jiang J., McNeill J. H. Downregulation of NADPH oxidase, antioxidant enzymes, and inflammatory markers in the heart of streptozotocin-induced diabetic rats by N-acetyl-L-cysteine. American Journal of Physiology: Heart and Circulatory Physiology. 2007;292(4):H1728–H1736. doi: 10.1152/ajpheart.01328.2005. [DOI] [PubMed] [Google Scholar]