

Abstract
Eukaryotic chromosomes initiate DNA synthesis from multiple replication origins in a temporally specific manner during S phase. The replicative helicase Mcm2-7 functions in both initiation and fork progression and thus is an important target of regulation. Mcm4, a helicase subunit, possesses an unstructured regulatory domain that mediates control from multiple kinase signaling pathways, including the Dbf4-dependent Cdc7 kinase (DDK). Following replication stress in S phase, Dbf4 and Sld3, an initiation factor and essential target of Cyclin-Dependent Kinase (CDK), are targets of the checkpoint kinase Rad53 for inhibition of initiation from origins that have yet to be activated, so-called late origins. Here, whole-genome DNA replication profile analysis is used to access under various conditions the effect of mutations that alter the Mcm4 regulatory domain and the Rad53 targets, Sld3 and Dbf4. Late origin firing occurs under genotoxic stress when the controls on Mcm4, Sld3, and Dbf4 are simultaneously eliminated. The regulatory domain of Mcm4 plays an important role in the timing of late origin firing, both in an unperturbed S phase and in dNTP limitation. Furthermore, checkpoint control of Sld3 impacts fork progression under replication stress. This effect is parallel to the role of the Mcm4 regulatory domain in monitoring fork progression. Hypomorph mutations in sld3 are suppressed by a mcm4 regulatory domain mutation. Thus, in response to cellular conditions, the functions executed by Sld3, Dbf4, and the regulatory domain of Mcm4 intersect to control origin firing and replication fork progression, thereby ensuring genome stability.
Eukaryotic cells initiate DNA synthesis from multiple replication origins on each chromosome to ensure efficient duplication of the genome in S phase. Activation of replication origins is achieved through two distinct steps that take place at separate stages of the cell division cycle. The first step, licensing of replication origins, occurs in G1 when CDK activity is low (Diffley 2011). During this process, a double hexameric minichromosome maintenance (MCM) complex, composed of two Mcm2-7 hexamers, is loaded onto each replication origin to form a pre-Replicative Complex (pre-RC) by the Origin Recognition Complex (ORC) and licensing factors, Cdc6 and Cdt1 (encoded by the TAH11 gene) (Diffley 2011). The second step, activation of licensed origins, occurs at each origin in a temporally controlled manner throughout S phase and requires activities of two S phase kinases, the S phase Cyclin-dependent Kinases (CDKs), and the Dbf4-dependent Cdc7 kinase (DDK) (Tanaka and Araki 2013). CDK phosphorylates two key substrates, Sld2 and Sld3, and promotes their binding to Dpb11 (Tanaka et al. 2007; Zegerman and Diffley 2007). DDK phosphorylates several subunits of the Mcm2-7 hexamer and, most importantly, blocks an intrinsic inhibitory activity residing within the amino-terminus of the Mcm4 subunit (Sheu and Stillman 2006, 2010; Randell et al. 2010). The action of these S phase kinases facilitates recruitment of Cdc45 and the GINS complex, composed of protein subunits Sld5, Psf1, Psf2, and Psf3, to the inactive MCM double hexamer and converts it into an active helicase complex, composed of Cdc45, Mcm2-7, and GINS (the CMG complex) (Tanaka and Araki 2013). The two-step process separates the loading and activation of replicative helicases at origins and thereby ensures that initiation from each origin occurs once and only once during each cell division cycle. Once origins are fully activated, the double helix unwinds, and DNA polymerase and other replisome components are recruited to establish replication forks, where new DNA is copied bidirectionally from each origin.
Initiation of DNA synthesis from licensed origins across the genome (origin firing) follows a predetermined temporal pattern (Rhind and Gilbert 2013). In budding yeast, the timing of DNA replication can be traced to the activation of individual origins. Origin activation occurs continuously during S phase, but those that fire first in S phase are referred to as early origins, and those that fire later are late origins. Despite being an essential target of CDK, Sld3, together with Sld7 and Cdc45, binds to the loaded Mcm2-7 hexamer in a manner dependent on DDK but not CDK (Heller et al. 2011; Tanaka et al. 2011). This association is a prerequisite for the subsequent CDK-dependent recruitment of a preloading complex, composed of Sld2, Dpb11, GINS, and pol ε (Muramatsu et al. 2010). It was proposed that DDK-dependent recruitment of the limiting Sld3-Sld7-Cdc45 is a key step for determining the timing of origin firing (Tanaka et al. 2011). Furthermore, simultaneous overexpression of several limiting replication factors advances late origin firing (Mantiero et al. 2011; Tanaka et al. 2011).
Under genotoxic stress during S phase, DNA damage checkpoint pathways inhibit late origin firing (Zegerman and Diffley 2009). In budding yeast, DNA damage activates the mammalian ATM/ATR homolog, Mec1 kinase, which in turn activates the Rad53 effector kinase (the homolog of mammalian Chk2) to phosphorylate and inhibit the activities of Sld3 and Dbf4, thereby preventing late origin firing (Lopez-Mosqueda et al. 2010; Zegerman and Diffley 2010). Some firing of late origins could be detected under DNA damaging conditions in phosphorylation mutants of these two targets rendered refractory to the inhibition by Rad53. An initiation inhibitory activity within the nonstructured, amino-terminal regulatory domain of Mcm4 (Fig. 1) also plays a role in regulating origin firing under genotoxic stress (Sheu et al. 2014). Because this domain is a target of DDK (Masai et al. 2006; Sheu and Stillman 2006, 2010), it is conceivable that Mcm4 could mediate the checkpoint control by Rad53 phosphorylation of Dbf4. However, since DDK has targets other than Mcm4 and Mcm4 is regulated by signals in addition to DDK, a more comprehensive picture of how these factors cooperate to control origin firing under stress conditions remains to be addressed.
Figure 1.
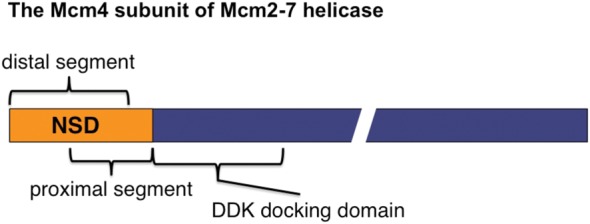
Diagram of the Mcm4 subunit of Mcm2-7 helicase. The two overlapping segments within the Mcm4 structurally disordered N-terminal serine/threonine-rich domain (NSD) are shown.
In addition to origin activation, DNA synthesis can be controlled at the level of replication fork progression. For example, deoxyribonucleoside triphosphate (dNTP) levels influence the rate of replication fork progression (Santocanale and Diffley 1998; Alvino et al. 2007). Hydroxyurea (HU) inhibits the activity of ribonucleotide reductase (RNR) and causes a dramatic slowdown of replication fork progression. In contrast, high dNTP concentration inhibits ORC-dependent initiation of DNA replication (Chabes and Stillman 2007). It has been proposed that dNTP levels are key determinants of replication fork speed, and cells adapt to replication stress by up-regulating dNTP pools (Poli et al. 2012). Methyl methanesulfonate (MMS), a DNA-alkylating agent, also results in slower fork progression while activating the DNA damage checkpoint response (Tercero and Diffley 2001). Although Mec1 and Rad53 are essential for preventing DNA replication fork catastrophe, these checkpoint kinases are not required for fork slowing in MMS (Tercero and Diffley 2001; Tercero et al. 2003). Thus, it is possible that an alternative mechanism might regulate fork progression under stress conditions.
The structurally disordered N-terminal serine/threonine-rich domain (NSD) of Mcm4 participates in both initiation and fork progression (Sheu et al. 2014). It can be subdivided into two overlapping but functionally distinct segments, the proximal segment and the distal segment (Fig. 1). The proximal segment of the NSD (amino acids 74–174) is responsible for the initiation inhibitory activity that is mitigated by DDK through phosphorylation (Sheu and Stillman 2006, 2010). The distal segment (amino acids 2–145) is important for controlling fork progression and checkpoint response under replication stress caused by depletion of dNTP pools and its function is regulated by CDK (Devault et al. 2008; Sheu et al. 2014). Thus, this intrinsic regulatory domain of the replicative helicase may cooperate with additional factors to control origin firing and replication fork progression in response to various environmental conditions. Herein, we examine the contributions of Mcm4, Sld3, and Dbf4 in DNA damage–induced control of both origin activation and DNA replication fork progression.
Results
Eliminating controls on Mcm4, Sld3, and DDK is required for maximal late origin firing in hydroxyurea
To evaluate how deregulating the control on Mcm4, Sld3, and Dbf4 impacts origin firing on a genome-wide scale, the replication profiles of the wild type (WT) and mutant strains with mcm4Δ74-174 (mcm4 mutant lacking the proximal NSD domain, the target of DDK), sld3-38A, and dbf4-19A (alleles of SLD3 and DBF4, respectively, that are resistant to the checkpoint control due to serine/threonine to alanine substitutions at the Rad53 target sites) (Zegerman and Diffley 2010) were analyzed in single, double, and triple mutant combinations. Cells were analyzed by releasing them synchronously from G1 phase into S phase in the presence of HU for 90 min (Fig. 2, data for Chromosome IV). Late origins were inactive at this time in WT (Fig. 2A, profile WT, red arrows), whereas a very low level of late origin firing was detected in each of the single mutants (profiles mcm4Δ74-174, sld3-38A, or dbf4-19A) (Fig. 2A), consistent with previous findings (Lopez-Mosqueda et al. 2010; Zegerman and Diffley 2010; Sheu et al. 2014). Late origin firing appeared more prominent in all of the double mutant combinations (profiles mcm4Δ74-174 sld3-38A, sld3-38A dbf4-19A, and mcm4Δ74-174 dbf4-19A), suggesting that these three factors function in pathways that are not completely overlapping. Among the double mutants, the mcm4Δ74-174 sld3-38A combination showed the most robust late origin firing. Thus, it is likely that Mcm4 and Sld3 function in separate control pathways to regulate origin firing. The mcm4Δ74-174 dbf4-19A combination only increased late origin firing slightly compared with each single mutant, consistent with the finding that Dbf4 and Mcm4 act in pathways that overlap extensively as Mcm4 is the essential target of DDK (Sheu and Stillman 2010). The detectable, but limited increase in late origin firing in sld3-38A dbf4-19A cells in comparison with their single mutants also suggests overlapping of the pathways involving Sld3 and Dbf4. In the triple mutant (Fig. 2A, profile mcm4Δ74-174 sld3-38A dbf4-19A), many late origins fire very robustly, more than any of the double and single mutants, and the efficiency of late origin firing approached the level of the early origins. The massive firing of late origins in the triple mutant further suggests that each of the three factors contribute independently to control of origin firing through overlapping but nonidentical pathways.
Figure 2.

Simultaneously alleviating controls on Mcm4, Sld3, and Dbf4 allows efficient late origin firing in HU despite an elevated checkpoint response. (A,B) Yeast cells were synchronized in G1 phase and released into YPD containing 0.2 M HU and 0.5 mM EdU for 90 min. (A) Replication profiles of Chromosome IV for the wild type (WT), single, double, and triple mutants with mcm4Δ74-174, sld3 38A, and/or dbf4 19A alleles as indicated on the top left of each profile. Gray bars under each profile indicate annotated replication origins in OriDB v2.1.0 (Siow et al. 2012). Orange bars indicate the position of the centromere. Red arrows point out some late origins that are inactive in the wild-type cells but fire in the mutants, in particular the triple mutant. (B) Distribution of fork progression from activated origins. Top panels show the peak width–height plots of all the recorded origins across the entire genome for each strain in A. Box plot shows the fork progression, excluding peaks, with heights <10% of the maximal height (also shown as black dashed line in individual width–height plots). Box and whiskers indicate 25–75 and 10–90 percentiles, respectively. (C) Cells of the indicated yeast strains were synchronized in G1, released into 0.2 M HU, and collected at the indicated time points. Protein samples were prepared using TCA extraction and analyzed by immunoblot. (D) Analysis of chromatin-bound proteins. Cells synchronized in G1 were released into 0.2 M HU and collected at the indicated time points. Chromatin-bound proteins were extracted from cells and analyzed by immunoblot.
Late origin firing in these mutants was not due to defects in the HU induced checkpoint response. Judging from levels of Rad53 hyperphosphorylation and phosphorylation of S129 in histone H2A (γH2A), the checkpoint signaling in mcm4Δ74-174, sld3-38A, and all the double and triple mutants was stronger than WT (Fig. 2C, Rad53 and γH2A). The elevated checkpoint signaling could also reflect more origin firing in these cells and thus more stalled forks (Tercero et al. 2003). Consistent with this idea, higher levels of Cdc45 loading, but not Mcm3 levels, were also detected on S phase chromatin in these mutants (Fig. 2D).
Proximal segment of Mcm4 NSD delays late origin firing in HU when control of Sld3 and Dbf4 by checkpoint is abrogated
To determine whether the triple mutant activated late origins with the same kinetics as early origins, the replication profiles of WT and the triple mutant were compared at different time points after release from G1 phase into S phase in the presence of HU (Fig. 3). At 25 min after release into HU, early origins fired in WT cells, whereas late origins remained inactive (Fig. 3A). At 50 min, the profile of activated origins in WT remained similar to that at 25 min, with a small increase of peak width, indicating progression of replication forks. At 75 min, replication forks progressed further, but the pattern of origin firing remained unchanged. In contrast, the triple mutant activated some late origins by 25 min, and the peak height of late origins continued to increase relative to that of early origins as time progressed to 50 and 75 min, suggesting that late origins continued to fire in this cell population. These data show that removing the proximal NSD segment of Mcm4, together with abolishing Rad53 phosphorylation of Sld3 and Dbf4, allowed late origins to fire efficiently in the presence of HU, but still in a temporally specific manner. In the presence of the proximal NSD segment, however, only low levels of late origin firing were detected in the sld3-38A dbf4-19A mutant at 50 min after release from G1 and only slightly increased at 75 min, but did not reach the same level as the mcm4Δ74-174 sld3-38A dbf4-19A mutant (Fig. 3B). These observations suggest that the proximal segment of the Mcm4 NSD prevents late origin firing at the earlier time in S phase despite the absence of active checkpoint inhibition of Sld3 and DDK function.
Figure 3.

Proximal segment of the Mcm4 NSD delays late origin firing in HU when checkpoint control on Sld3 and Dbf4 is relieved. Yeast cells were synchronized in G1 phase and released into YPD containing 0.2 M HU and 0.5 mM EdU. (A) Replication profiles of Chromosome IV for WT and the mcm4Δ74-174 sld3-38A dbf4-19A triple mutant at 25, 50, and 75 min after release into HU from G1 arrest. (B) Replication profiles of Chromosome IV for the sld3-38A dbf4-19A and mcm4Δ74-174 sld3-38A dbf4-19A mutants at 25, 50, and 75 min after release into HU from G1. (C) Distribution of fork progression from activated origins at the indicated time point for the wild type (blue, data from A), sld3-38A dbf4-19A (orange, data from B), and mcm4Δ74-174 sld3-38A dbf4-19A (red; left panels, data from A; right panels, data from B) mutants. Box graph shows the fork progression, excluding peaks with heights <10% of the maximal height scale.
The distal segment of Mcm4 NSD and Rad53 phosphorylation of Sld3 affect replication fork progression
In addition to revealing patterns of origin activation, whole-genome replication profile analysis also provides information on the average replication fork progression from each origin in the population of cells. For computational analysis, we defined fork progression as the observed peak width at the half maximum of the peak height for each origin in the profile (Sheu et al. 2014). Analysis of replication profiles in HU showed that replication fork progression was much less in all the mutants containing the sld3-38A allele than in the wild type (Figs. 2A,B, 3C, 4A,B; Supplemental Table 1A), suggesting that phosphorylation of Sld3 by the checkpoint kinase Rad53 is needed to allow replication fork progression in HU. The mcm4Δ74-174 mutant lacking the Mcm4 proximal NSD domain also had less fork progression than the wild type, but the difference was subtle, yet reproducible (Figs. 2B, 4B; Supplemental Table 1A). Fork progression in the other Rad53 target mutant dbf4-19A appeared heterogeneous (Fig. 2B). Double mutants sld3-38A dbf4-19A and mcm4Δ74-174 dbf4-19A exhibited fork progression patterns resembling single mutants sld3-38A and mcm4Δ74-174, respectively, rather than that of the dbf4-19A mutant, suggesting that the functions of sld3-38A and mcm4Δ74-174 in fork progression are epistatic to that of dbf4-19A. Thus, Sld3 and the proximal NSD of Mcm4 are likely to function downstream from Dbf4 in regulation fork progression in HU.
Figure 4.

Effects of sld3-38A and the Mcm4 NSD distal segment on fork progression in HU. (A,B) Yeast cells were synchronized in G1 phase and released into YPD containing 0.2 M HU and 0.5 mM EdU. (A) Replication profiles of Chromosome IV for the indicated yeast strains. (B) Distribution of fork progression from origins shown as individual width–height plots and box graph, excluding peaks with heights <30% of the maximal height scale. (C) Cells from the indicated strains were synchronized in G1, released into 0.2 M HU, and collected at the indicated time points. Protein samples were analyzed as in Figure 2C.
Analysis of fork progression of samples from the time course experiment showed that the extent of progression at 25 min after release to HU was similar among WT, sld3-38A dbf4-19A, and mcm4Δ74-174 sld3-38A dbf4-19A mutants (Fig. 3C; Supplemental Table 1A). At 50 min, replication forks advanced further in the wild type. However, in the sld3-38A dbf4-19A and mcm4Δ74-174 sld3-38A dbf4-19A mutants, the progression remained at the same level as those at 25 min. Replication fork progression continued in the wild type at 75 min, whereas the forks in the mutant progressed very little.
In contrast to sld3-38A and mcm4Δ74-174, mutations affecting the function of the distal NSD of Mcm4, such as mcm4Δ2-145 and mcm4Δ74-174, 4(SP→AP), exhibited extensive fork progression (see Fig. 4B; Sheu et al. 2014). To establish the genetic relationship between the function of Sld3 and the distal segment of the NSD of Mcm4 in controlling replication fork progression, replication profiles in HU were analyzed in single and double mutants (Fig. 4). In the mcm4Δ2-145 sld3-38A double mutant, the opposing effects of the individual mutations (mcm4Δ2-145 and sld3-38A) appeared to cancel one another (Fig. 4A,B; Supplemental Table 1A), suggesting their controls operate through separate pathways. The mcm4Δ74-174, 4(SP→AP) has mutations in the Mcm4 distal NSD segment and inactivates CDK phosphorylation sites (Sheu et al. 2014). The mcm4Δ74-174, 4(SP→AP) sld3-38A double mutant and the mcm4Δ2-145 sld3-38A mutant showed similar fork progression phenotypes (Fig. 4B).
Hyperactivation of checkpoint signaling as measured by the degree of Rad53 and H2A phosphorylation was observed in the sld3-38A single mutant and the mcm4Δ74-174 sld3-38A double mutant, but was not observed in the mcm4Δ2-145 sld3-38A and mcm4Δ74-174, 4(SP→AP) sld3-38A double mutants. However, the phosphorylation levels of the latter two were slightly more elevated than that of the mcm4Δ2-145 single mutant (Fig. 4C). Despite the variation in Rad53 and H2A phosphorylation, the downstream response to checkpoint activation, as manifest in degradation of Sml1 and up-regulation of Rnr4, appeared similar among all strains.
Late origin firing in HU in the absence of checkpoint signaling kinases
To gain insight into the extent of late origin firing in the cells lacking the checkpoint kinases Mec1 and Rad53, the main regulators of late origin firing under genotoxic stress, we examined the replication profiles in HU in the absence of these kinases. Mec1 and Rad53 are essential for cell growth; however, their essential role in growth can be bypassed by deleting the SML1 gene, a negative regulator of the dNTP pool (Zhao et al. 1998). Thus, we analyzed sml1Δ, mec1Δ sml1Δ, and rad53Δ sml1Δ cells synchronously replicating in the presence of HU for 90 min, along with the WT and the mcm4Δ74-174 sld3-38A dbf4-19A mutant (Fig. 5). In the sml1Δ cells, a significant amount of late origins were activated in HU while they were inactive in WT (Fig. 5A, profiles WT and sml1Δ), consistent with a previous finding (Poli et al. 2012). In mec1Δ sml1Δ and rad53Δ sml1Δ cells, more late origins were activated in HU than in the sml1Δ cells (Fig. 5A). However, the level of late origin firing relative to the early origins in the absence of Mec1 or Rad53 did not appear to surpass that in the mcm4Δ74-174 sld3-38A dbf4-19A triple mutant cells.
Figure 5.

Late origin firing in HU in the absence of checkpoint signaling kinase. Yeast cells were synchronized in G1 phase and released into YPD containing 0.2 M HU and 0.5 mM EdU for 90 min. (A) Replication profiles of Chromosome IV for WT, sml1Δ, mec1Δ sml1Δ, rad53Δ sml1Δ, and the mcm4Δ74-174 sld3-38A dbf4-19A triple mutant cells. (B) Distribution of fork progression from origins shown as individual width–height plots and box graph, excluding peaks with heights <30% of the maximal height scale.
Replication forks progressed further in sml1Δ cells than in the WT in HU (Fig. 5B; Poli et al. 2012) because dNTP levels increased in the absence of Sml1. In mec1Δ sml1Δ and rad53Δ sml1Δ cells, replication forks progressed less than in sml1Δ cells, although the effect of RAD53 deletion was moderate (Fig. 5B; Supplemental Table 1A). Nevertheless, it suggests that these kinases contribute to modulating fork progression. Alternatively, the lesser fork progression may reflect more aborted replication forks in the population in the absence of these kinases because they are essential for preventing DNA replication fork catastrophe under replication stress (Tercero and Diffley 2001). Importantly, fork progression in the absence of the checkpoint kinases was much greater than fork progression in the mcm4Δ74-174 sld3-38A dbf4-19A triple mutant cells.
High levels of DNA synthesis near the chromosome ends were detected in both mec1Δ sml1Δ and rad53Δ sml1Δ cells replicating in HU, but not in sml1Δ cells (Fig. 5A). This phenomenon is prevalent at most of the chromosome ends in these mutants. Such a high level of DNA synthesis near telomeres does not occur in the unperturbed S phase (data not shown). It was not clear whether these DNA syntheses initiate from newly activated origins near telomeres. Replication profile analysis was also done with the 30 kb at each end of each chromosome masked (Supplemental Fig. S1; Supplemental Table 1A). This had an effect on the scaling of the profiles for the mec1 and rad53 mutants but barely affected the identification of peaks and calculation of fork progression.
The proximal segment of the Mcm4 NSD controls firing of late origins in an unperturbed S phase
Since the proximal segment of the NSD imposes a barrier to refrain late origin firing in HU (Fig. 3B), it is possible that this domain also controls late origin firing in a normal, unperturbed S phase. To test this idea, the replication profiles of WT and various mcm4 mutants containing mutations within the NSD were examined and compared at 25 min after release from G1 arrest (Fig. 6). This time point was selected because peaks representing DNA synthesis from individual origins could be clearly detected without excessive overlap and thus were more suitable for analysis of origin firing and fork progression. At this time, DNA synthesis from early origins was readily detected in WT, whereas very small amounts of DNA synthesis occurred from late origins (Fig. 6A, profile WT, red arrows). Thus, this time represents a point in the early S phase. Because the peak heights of the normally late firing origins were small, this experiment was repeated with biologically duplicate samples (Supplemental Fig. S2A,B), yielding almost identical results. Plots of peak heights for all origins in the genome for duplicate 1 versus duplicate 2 (WT versus WT, etc.) (Supplemental Fig. S2C) show a 100-fold dynamic range of peak height and very little scatter (Supplemental Fig. S2C). Peak heights of duplicate samples were also plotted relative to a WT reference profile (blue for the duplicate sample of WT and orange and red for biological duplicates of mutants) (Fig. 6B). The mutant lacking the distal segment of the NSD (profile mcm4Δ2-145) (Fig. 6A,B) had a similar profile as the WT. In mcm4Δ2-174 and mcm4Δ74-174 mutants that lacked the proximal segment of the NSD, however, peaks corresponding to firing from late origins were clearly observed, although still less than early origins (Fig. 6A; Supplemental Fig. S2A,B), and the peak height distribution of both duplicates diverged from the WT profile (Fig. 6B), suggesting that the proximal NSD segment contributes to the temporal pattern of late origin firing during an unperturbed S phase.
Figure 6.

Removing the Mcm4 proximal NSD allows advanced firing of late origins in an unperturbed S phase. Yeast cells were synchronized in G1 phase and released into YPD containing 0.5 mM EdU for 25 min at 30°C. (A) Replication profiles of Chromosome IV for WT and Mcm4 NSD mutants. Red arrows indicate late origins that are inactive in the wild-type cells but fire in the triple mutant in HU as in Figure 2. (B) Plots comparing the peak height between duplicate samples of mutant strains (red and orange) to a wild-type reference (blue). Data also presented in Supplemental Figure S2. Peak heights for each sample were scaled so that the 90% quantile is equal to 1.0.
Advanced firing of late origins did not occur when, in the context of an Mcm4 lacking the proximal segment of the NSD, the phospho-acceptors for S phase-CDK phosphorylation within the distal segment of the NSD were mutated to alanine (Fig. 6, profile mcm4Δ74-174, 4(SP→AP)). In contrast, mutation of these same residues to the phospho-mimetic aspartic acid (profile mcm4Δ74-174, 4(SP→DP)) allowed earlier firing of late origins, similar to the mcm4Δ74-174 mutant. Note that one mcm4Δ74-174, 4(SP→AP) duplicate sample had a high background due to contamination with non-EdU-labeled DNA of >14% (see profile in Supplemental Fig. S2A; Supplemental Table S1B), thereby skewing the plot in Fig. 6B (mutant 1 in plot for mcm4Δ74-174, 4(SP→AP)), but the same sample from the other two experiments (see profiles in Fig. 6A; Supplemental Fig. S2B) showed little late origin firing, similar to the WT. Thus, phosphorylation of the CDK sites within the distal segment of the NSD is important for efficient firing of late origins during an unperturbed S phase.
The replication profiles of sld3-38A and dbf4-19A mutants were examined in an unperturbed S phase (Fig. 7). Again, the experiment was repeated with biological duplicate samples (Supplemental Fig. S3A–C). None of the sld3-38A, dbf4-19A, and sld3-38A dbf4-19A mutants showed advanced firing of late origins compared to the replication profile of wild-type cells (Fig. 7A; Supplemental Fig. S3A,B). The peak height distributions for these strains were similar to the WT (Fig. 7B). However, the mcm4Δ74-174 single mutant did show late origin firing (Fig. 7A) and a peak height deviation from the WT distribution (Fig. 7B). Combining mcm4Δ74-174 with either sld3-38A or dbf4-19A mutations, or even the triple mutant mcm4Δ74-174 sld3-38A dbf4-19A did not lead to more firing of late origins than the mcm4Δ74-174 single mutant (Fig. 7A,B; Supplemental Fig. S3A,B). Late origin firing at 25 min into S phase was only observed in the four strains that had the mcm4Δ74-174 mutation. Thus, unlike the NSD proximal segment mutant, checkpoint kinase Rad53 phosphorylation of Sld3 or Dbf4 does not control late origin firing during a normal S phase.
Figure 7.

Checkpoint-resistant mutations of SLD3 and DBF4 do not affect timing of late origin firing in an unperturbed S phase. Yeast cells were synchronized in G1 phase and released into YPD containing 0.5 mM EdU for 25 min. (A) Replication profiles of Chromosome IV for WT, single, double, and triple mutants with mcm4Δ74-174, sld3-38A, and/or dbf4-19A alleles as indicated on the top left of each profile. (B) Plots comparing the peak height between duplicate samples of mutant strains (red and orange) to a wild-type reference (blue). Data also presented in Supplemental Figure S3. Peak heights for each sample were scaled so that the 90% quantile is equal to 1.0.
From the replication profile analyses, it appeared that mutations in the Mcm4 NSD did not have a dramatic effect on fork progression in an unperturbed S phase (Supplemental Fig. S4; Supplemental Table 1A). However, subtle differences would be more difficult to detect in such an experiment because DNA synthesis occurred much faster in the absence of HU. Similarly, the sld3-38A mutation did not restrict fork progression in contrast to its effect in HU (Supplemental Fig. S5; Supplemental Table 1A). The distribution of fork progression in the dbf4-19A mutant appeared more heterogeneous, similar to the pattern observed in HU (Fig. 2B). Double mutants of dbf4-19A with either mcm4Δ74-174 or sld3-38A yielded phenotypes resembling the single mutant of mcm4Δ74-174 or sld3-38A with respect to replication fork progression.
Checkpoint response and replication profiles for Mcm4 NSD mutants entering S phase in the presence of MMS
Although the NSD proximal segment controls late origin firing in both a normal S phase and S phase with a depleted pool of dNTP, the effect of NSD mutations on fork progression was only observed in the presence of HU. Furthermore, sld3-38A showed a strong phenotype in restricting fork progression in HU, but no obvious effect in a normal S phase. The differential influence of these mutations on origin firing and replication fork progression in an unperturbed versus a HU-treated S phase raised the question of whether these factors would have the same effect on other types of genotoxic stress than dNTP depletion. Thus, the DNA damage checkpoint response and DNA replication profiles in cells replicating in the presence of the DNA alkylating agent methyl methanesulfonate (MMS) were studied.
For replication profile analyses, cells were synchronized in G1 phase and allowed to enter S phase in the presence of MMS for 50 min. We did not use the 90-min time point that we typically use for analysis of replication profile in HU because replication in MMS is faster than in HU, and 90 min in MMS would have produced profiles difficult to analyze due to numerous merged peaks and passive replication at late/unfired origin loci by replication forks moving from early firing origins. At 50 min in MMS after release from G1 arrest, few late origins fire in WT and in the mutant lacking the distal NSD segment (Fig. 8A, profiles WT and mcm4Δ2-145). In contrast, late origin firing was more evident in the mutants lacking the proximal NSD segment (Fig. 8A, profiles mcm4Δ2-174 and mcm4Δ74-174). In the same context, mutation of the CDK sites to alanines within the distal NSD segment (mcm4Δ74-174,4(SP→AP)) suppressed late origin firing, whereas mutating the same sites to phosphomimetic aspartic acids (mcm4Δ74-174,4(SP→DP)) restored the level of late origin firing. Thus, the proximal NSD segment also mediates control of late origin firing in MMS.
Figure 8.

Replication profiles and checkpoint response in Mcm4 NSD mutants. (A,B) Yeast cells were synchronized in G1 phase and released into YPD containing 0.05% MMS and 0.5 mM EdU for 50 min. (A) Replication profiles of Chromosome IV for the wild type and the indicated Mcm4 NSD mutants. (B) Distribution of fork progression from origins shown as individual width–height plots and box graph, which excludes peaks with heights <30% of the maximal height scale. (C) Cells from the indicated strains were synchronized in G1, released into YPD containing 0.05% MMS, and collected at indicated time points. Protein samples were prepared using TCA extraction and analyzed by immunoblot.
Replication fork progression was also affected in the Mcm4 NSD mutants replicating in the presence of MMS (Fig. 8). Fork progression was more restricted in the mcm4Δ74-174 mutant lacking the NSD proximal segment compared to WT, whereas more expansive fork progression was observed in mutants lacking the distal NSD segment (cf. mcm4Δ2-145 with WT and mcm4Δ2-174 with mcm4Δ74-174) (Fig. 8A,B). Fork progression in MMS was also regulated by phosphorylation at the CDK target sites within the distal segment of the NSD because forks progressed further in mcm4Δ74-174,4(SP→AP) compared to progression in mcm4Δ74-174; whereas in the mcm4Δ74-174,4(SP→DP) mutant, fork progression was more restricted, similar to that in mcm4Δ74-174 (Fig. 8B). Therefore, the distal and proximal segments of the Mcm4 NSD play important roles in mediating control of fork progression in diverse types of genotoxic agents.
In HU, the NSD distal segment was important for checkpoint signaling at the level of Mec1 signaling (Sheu et al. 2014), although Mec1 phosphorylation of Mcm4 is independent of checkpoint activation (Randell et al. 2010). Removing the distal segment of the NSD or mutation of the phospho-acceptor amino acids at CDK sites to alanine within this domain resulted in reduced levels of Rad53 hyperphosphorylation and γH2A. In contrast, other aspects of checkpoint signaling further downstream, such as Sml1 degradation and Rnr4 induction, appeared normal in cells treated with HU. In MMS, however, hyperphosphorylation of Rad53 and S129 phosphorylation in H2A, as well as further downstream events, such as degradation of Sml1 and up-regulation of Rnr4 levels, appeared very similar among wild type and various Mcm4 NSD mutants (Fig. 8C). Thus, the Mcm4 NSD did not play a prominent role in checkpoint signaling in response to DNA damage caused by MMS.
Cooperation between the proximal segment of the Mcm4 NSD and Rad53 in regulating late origin firing in MMS
The Rad53-resistant sld3-38A and dbf4-19A strains alone also showed low levels of late origin firing in cells treated with MMS (Fig. 9A). The difference between the wild type and the dbf4-19A mutant was subtle. Nevertheless, the double mutants sld3-38A dbf4-19A, mcm4Δ74-174 sld3-38A, and mcm4Δ74-174 sld3-38A activated more late origins than the wild type and any of the single mutants. The mcm4Δ74-174 dbf4-19A showed only a slight increase in late origin firing, compared with the mcm4Δ74-174 single mutant, consistent with Mcm4 functioning downstream from Dbf4 in controlling late origin firing in MMS. Furthermore, the triple mutant activated late origins the most efficiently (Fig. 9A, profile mcm4Δ74-174 sld3-38A dbf4-19A). Thus, all three factors contribute to control of late origin firing through overlapping but nonidentical pathways in MMS, as was found for cells treated with HU.
Figure 9.

Analysis of replication profiles and checkpoint response in the wild type, single, double, and triple mutants with mcm4Δ74-174, sld3-38A, and/or dbf4-19A alleles. (A,B) Yeast cells were synchronized in G1 phase and released into YPD containing 0.05% MMS and 0.5 mM EdU for 50 min. (A) Replication profiles of Chromosome IV. (B) Distribution of fork progression constructed as in Figure 4B. (C) Cells from the indicated strains were synchronized in G1, released into YPD containing 0.05% MMS, and analyzed as in Figure 8C.
Like in HU, the sld3-38A mutant had an effect on restricting replication fork progression in MMS (Fig. 9B). The effect of sld3-38A and mcm4Δ74-174 on restricting fork progression was additive under this condition, suggesting that they control fork progression separately. Fork progression in dbf4-19A was more extensive, but became more restricted when sld3-38A and mcm4Δ74-174 were also present. Thus, Mcm4, Sld3, and Dbf4 cooperate to regulate fork progression in MMS.
The DNA damage checkpoint signaling was active in the wild type and all of the single, double, and triple mutant combinations of mcm4Δ74-174, sld3-38A, and dbf4-19A (Fig. 9C). Although we detected elevated H2A S129 phosphorylation in the double mutants of mcm4Δ74-174 sld3-38A and sld3-38A dbf4-19A, as well as the triple mutant, the differences in the signaling at this level among strains did not appear as dramatic as those observed in HU (Fig. 2C).
Suppression of the temperature-sensitive (ts) phenotype of multiple sld3-ts mutants by deletion of the Mcm4 NSD proximal segment
Although replication profile analyses suggest that Sld3 and the Mcm4 NSD mediate controls from separate pathways, the fact that they affect similar processes raises the possibility that the tasks executed by these two factors may converge on a common target. For example, the proximal segment of the Mcm4 NSD may be inhibiting the same molecular process that Sld3 is facilitating. If this is the case, it is likely that removing the proximal NSD segment would compensate for the weakened Sld3 function in hypomorph sld3 mutants. The idea was tested by introducing the mcm4Δ74-174 mutation in sld3-ts mutants sld3-5, sld3-6, and sld3-7 (Kamimura et al. 2001), all of which fail to grow on YPD plates at a nonpermissive temperature >30°C, 37°C, and 34°C, respectively. At 30°C, the sld3-5 mutant grew extremely poorly compared with the wild type and the mcm4Δ74-174 mutant, whereas the mcm4Δ74-174 sld3-5 double mutant grew much better than the sld3-5 mutant (Fig. 10A). At 23°C, the sld3-5 mutant also grew slower than the wild type, but the mcm4Δ74-174 sld3-5 grew similarly to the wild type. Thus, removing the proximal segment of the NSD improved the growth of the sld3-5 mutant. Likewise, removing the proximal segment of the NSD improved the growth of the sld3-6 and sld3-7 mutants at 37°C, but no growth occurred in sld3-5 and mcm4Δ74-174 sld3-5 at this temperature. Thus, removing the Mcm4 proximal segment of the NSD suppresses the defect of multiple hypomorph sld3-ts mutants.
Figure 10.

Partial suppression of sld3-ts by the mcm4Δ74-174 mutant. (A,B) Serial 10-fold dilution of 105 yeast cells on YPD at the indicated temperatures. (C) A model for control of Mcm2-7 helicase.
We also tested if removing the proximal segment of the Mcm4 NSD would also suppress the ts phenotype of mutants affecting other factors that function with Sld3, such as Dbp11 and components of the GINS complex (Kamimura et al. 1998; Takayama et al. 2003). The mcm4Δ74-174 mutation failed to suppress the ts phenotype of sld5-12 or psf1-1, mutants in GINS subunits (30°C, 34°C, and 37°C) (Fig. 10B). A very slight improvement of growth in the mcm4Δ74-174 dpb11-1, compared with the dpb11-1 mutant, at 34°C was observed. However, the suppression was much less effective than the suppression of the sld3-6 defect by mcm4Δ74-174 (34°C and 37°C) (Fig. 10B). The specific and strong suppression of sld3-ts by mcm4Δ74-174 is consistent with the idea that Sld3 and the Mcm4 NSD regulate the same process at the molecular level to control origin firing and influence replication fork progression under genotoxic stress.
Discussion
The inhibition of DNA replication under genotoxic stress requires both Rad53 and Mec1 kinases (Sanchez et al. 1996; Santocanale and Diffley 1998; Zegerman and Diffley 2009). In previous studies, we demonstrated that even in the presence of an active S-phase checkpoint response, late origins fire in the presence of HU when the Mcm4 NSD proximal segment was removed (Sheu et al. 2014). The observation suggested that under replication stress, the checkpoint kinase Rad53 inhibited Dbf4 by phosphorylation (Lopez-Mosqueda et al. 2010; Zegerman and Diffley 2010), rendering DDK incapable of relieving the initiation inhibitory activity of the Mcm4 NSD proximal segment (Fig. 10C). Since the Mcm4 NSD proximal segment is targeted by DDK (Sheu and Stillman 2010), inhibition of DDK by active Rad53 would not prevent initiation in the absence of this initiation inhibitory domain. However, late origin firing in the absence of this domain was still rather inefficient, presumably because checkpoint activation of Rad53 still allowed phosphorylation of the other target, Sld3, thereby inactivating Sld3 activity and preventing robust firing of late origins (Fig. 10C). By using specific probes for analysis by alkaline gel electrophoresis or two-dimensional gel electrophoresis, some firing of certain late origins was detected in sld3 and dbf4 mutants that are refractory to the inhibition by the checkpoint kinase Rad53 (Lopez-Mosqueda et al. 2010; Zegerman and Diffley 2010). In the current study, whole-genome replication profile analysis was used to investigate the individual roles, as well as the combined effect of the two Mcm4 NSD segments and the Rad53 targets, Sld3 and Dbf4, on origin activation and replication fork progression in order to delineate the relationship among these factors in control of replication in response to replication stress.
In both HU and MMS, late origins fire in each of the mcm4Δ74-174, sld3-38A, and dbf4-19A single mutants across the entire genome, albeit very inefficiently (Figs. 2, 8). The fact that all the double mutant combinations among these three mutations activated late origins more efficiently than the respective single mutants and that the triple mutant exhibited the most efficient firing of late origins suggests that each factor contributes a unique function in control of origin activation. Yet, their functions may not be completely independent (Fig. 10C). For example, late origin firing in the mcm4Δ74-174 dbf4-19A double mutant appeared only marginally more efficient than the mcm4Δ74-174 single mutant. This was not surprising given that Mcm4 is the essential target of DDK and mcm4Δ74-174 can bypass the regulation of the kinase (Sheu and Stillman 2010). Nevertheless, because the triple mutant promotes robust firing of late origins, more than any of the single and double mutants, both the Mcm4 NSD proximal segment and Dbf4 must also independently contribute to regulate late origin firing (Fig. 10C). This can be anticipated for Dbf4 because DDK also phosphorylates other factors in addition to Mcm4 NSD. For Mcm4, it raises the possibility that, in addition to DDK, other factors might participate in the regulation of the function of the proximal NSD in controlling late origin firing under replication stress. Identification of factors that interact with the proximal NSD may shed light on this aspect of the control mechanism. Alternatively, Dbf4 may affect Mcm2-7 helicase activity independent of the Mcm4 NSD or participate in feedback regulation of Rad53 kinase activity or specificity (Fig. 10C, dashed lines). One possibility is that they antagonize one another's activity, essentially creating a feedback loop for inactivating the checkpoint once the replication stress has subsided.
The partial overlap of functional pathways involving Sld3 and Dbf4, as revealed by the minimal combined effect of dbf4-19A and sld3-38A on late origin firing in HU compared to each single mutant, can be explained by the fact that the early association of Sld3 to the pre-RC depends on DDK activity (Fig. 10C; Heller et al. 2011; Tanaka et al. 2011). In contrast, the mcm4Δ74-174 sld3-38A exhibited the strongest additive effect among the double mutant combinations, suggesting that these two factors mediate regulation via separate pathways (Fig. 10C). However, the suppression of the hypomorph sld3-ts defect by mcm4Δ74-174 suggests that these two pathways intersect on a common process, and they are likely regulating the same factors. It is possible that this common pathway merges on the direct activation of the Mcm2-7 helicase by recruiting other helicase components, Cdc45 and GINS (Fig. 10C).
In the sld3-38A dbf4-19A double mutant that was expected to be completely refractory to the control by the Rad53-dependent S-phase checkpoint, late origins did not fire until 50 min into S phase in the presence of HU (Fig. 3B). Given that late origin firing was readily detected in a normal, unperturbed S phase at this time, this suggests that a mechanism is functioning to withhold late origins from firing in this double mutant condition. Removing the proximal segment of the Mcm4 NSD in the same genetic background allowed late origins to fire by 25 min after release, similar to what we have observed in unperturbed S phase (Fig. 6), strongly suggesting that activating DDK alone is not sufficient to efficiently block the function of the proximal NSD segment in withholding late origin from firing earlier in HU. Thus, besides DDK, additional factors might participate in relieving the block imposed by proximal NSD to delay late origin firing.
The proximal segment of the Mcm4 NSD also controls late origin firing in an unperturbed S phase (Fig. 10C). In mutants lacking this domain, more late origin firing was detected in early S phase (Fig. 6). In contrast, neither of the sld3-38A, dbf4-19A, or sld3-38A dbf4-19A mutants showed advanced firing of late origins in an unperturbed S phase (Fig. 7). Thus, checkpoint kinase Rad53 does not appear to control late origin firing through Sld3 and Dbf4 in a normal S phase. Phosphorylation of the CDK sites within the distal segment of the Mcm4 NSD (Devault et al. 2008) was also important for advanced firing of late origins in the unperturbed S phase when the proximal segment of the NSD was removed (Fig. 6). Previous studies in budding yeast have shown that, in the absence of the main S phase CDK cyclin, Clb5, only early origins fire, but not late origins (Donaldson et al. 1998). Thus, activation of late origins requires activity of S phase CDK. Together, these results suggest that phosphorylation of the distal segment of the Mcm4 NSD by CDK is an important step for activation of late origins (Fig. 10C). The accumulation of CDK activity as cells progress through S phase may eventually allow late origins to fire.
The role of the Mcm4 NSD in regulating late origin firing and fork progression previously discovered in HU was largely recapitulated in cells replicating in MMS (Fig. 8). Specifically, in MMS, the proximal segment of the NSD mediates control of late origin firing, and the distal segment NSD mediates control of fork progression in a manner that is regulated by phosphorylation at CDK target sites. However, unlike the response in HU, the Mcm4 NSD mutations exhibited little effect on checkpoint signaling in response to DNA damage caused by MMS (Fig. 8C). In our previous study, we noticed an inverse correlation between checkpoint signaling and DNA replication fork progression in HU (Sheu et al. 2014), raising the possibility that one process controls the other. The study here in MMS, in contrast, provides evidence that these two processes might not always influence one another. At least in MMS, the Mcm4 NSD is likely to regulate fork progression through a mechanism independent of the canonical DNA damage checkpoint pathway. One possibility is that the DNA damaging signal would somehow control the activity of CDK or other SP site kinases, which in turn regulate the function of the distal segment of the NSD.
Sld3 also mediates control of DNA replication fork progression in both HU and MMS (Figs. 2B, 8B). The sld3-38A mutant exhibits a dramatic slowdown in replication fork progression. This is a somewhat surprising observation because Sld3 is not considered a replication fork component since it is required for initiation but not for elongation in normal S phase progression (Kanemaki and Labib 2006). It is not clear why DNA synthesis is so limited in this mutant, but the mutation does not seem to result in a defective replication factor because the replication profile of this mutant is similar to the wild type in an unperturbed S phase, and this mutant grows at a rate comparable to wild-type cells. Furthermore, a previous study reported that yeast strains expressing sld3-38A as the sole copy of Sld3 displayed no increase in sensitivity to hydroxyurea or DNA damaging agents and did not exhibit synthetic growth defects with several conditional alleles of essential replication proteins (Zegerman and Diffley 2010). Therefore, this phenotype is likely due to regulation of fork function. Replication profile analysis of the double mutant combining sld3-38A and NSD mutants in the distal segment showed that the effect of the sld3-38A mutant on fork progression in HU and distal NSD mutant are not epistatic to each other (Fig. 4), consistent with the idea that these two factors operate in separate pathways to regulate fork progression in HU. In contrast to its role in HU and MMS, the checkpoint-resistant sld3-38A mutant did not affect fork progression in an unperturbed S phase (Supplemental Fig. S5). Thus, the control of fork progression through Rad53 target sites on Sld3 is a specific feature in the genotoxic-stressed condition.
It has been reported that the level of origin firing inversely affected the rate of fork progression (Zhong et al. 2013) presumably because active forks compete for limiting dNTP pools, which is an important determinant of fork progression (Poli et al. 2012). Since Mcm4 NSD, Sld3, and Dbf4 mutation increased the number of origins that were activated, it was possible that their effects on replication fork progression in HU were in part the consequence of competition for limiting dNTP pools or limiting DNA replication proteins. We suggest, however, that the limited replication fork progression in the sld3-38A and mcm4Δ74-174 mutants was a result of these proteins directly involved in controlling replication fork progression and not only due to more active origins competing for limiting dNTP or replication proteins, since in the rad53 and mec1 mutants, late origins were equally active but replication fork progression was much greater than fork progression in the sld3-38A and mcm4Δ74-174 mutants. Furthermore, MMS treatment should not affect dNTP levels like addition of HU, but the effects of sld3-38A and mcm4Δ74-174 mutations on replication fork progression in MMS paralleled the effects observed in HU. Thus, although increased origin firing may correlate with slowing of DNA replication fork progression, the level of origin firing may not be the sole explanation for changes we observe in replication fork progression.
Since DDK binds directly to the Mcm2-7 helicase subunits Mcm4 and Mcm2 (Varrin et al. 2005; Sheu and Stillman 2006; Jones et al. 2010) and DDK binds directly to Rad53 (Dohrmann et al. 1999; Weinreich and Stillman 1999; Kihara et al. 2000), it is possible that the regulation of the response to DNA replication stress such as limiting dNTP levels involves a local response at the DNA replication fork, essentially a solid-state regulatory complex. How other Mcm2-7–associated replication checkpoint proteins such as Mrc1, Dpb11, Sld2, and the large subunit of DNA polymerase ε control initiation of replication and fork progression remains to be investigated, but we suspect that they integrate with the regulatory system involving Dbf4, Sld3, and Mcm4.
Methods
Yeast strains and methods
Yeast strains generated in this study were derived from W303-1a (MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1) and are described in Supplemental Table 2. A two-step gene replacement method was used to replace the endogenous MCM4 with mcm4 mutants as described (Sheu et al. 2014). All the yeast strains used for the whole-genome DNA replication profile analyses have a copy of the BrdU-Inc cassette inserted into the URA3 locus (Viggiani and Aparicio 2006). For G1 arrest of bar1Δ strains, exponentially growing yeast cells (∼107 cell/mL) in YPD were synchronized in G1 with 25 ng/mL of α-factor for 150 min at 30°C. For G1 arrest of BAR1 strains, exponentially growing cells were grown in normal YPD, then transferred into YPD (pH3.9), grown to ∼107 cell/mL, and then synchronized in G1 with three doses of α-factor at 2 µg/mL at 0-, 50-, and 100-min time point at 30°C. Cells were collected at 150 min for release. To release from G1 arrest, cells were collected by filtration and promptly washed twice on the filter using one culture volume of H2O and then resuspended into YPD medium containing 0.2 mg/mL pronase E (Sigma).
Protein sample preparation and immunoblot analysis
TCA extraction of yeast proteins was as described previously (Sheu et al. 2014). For chromatin fractionation, chromatin pellets were prepared from ∼5 × 108 yeast cells, and chromatin-bound proteins were released using DNase I using a procedure described previously (Sheu et al. 2014). For immunoblot analysis, protein samples were fractionated by SDS-PAGE and transferred to a nitrocellulose membrane. Immunoblot analyses for Mcm3, Cdc45, Orc6, Mcm4, Rad53, γ-H2A, Rnr4, and Sml1 were performed as described (Sheu et al. 2014).
Isolation and preparation of DNA for whole-genome replication profile analysis
Detailed protocol was described previously (Sheu et al. 2014). Briefly, yeast cells were synchronized in G1 with α-factor and released into medium containing 0.2 mg/mL pronase E, 0.5 mM 5-ethynyl-2′-deoxyuridine (EdU) with or without addition of 0.2 M HU or 0.05% MMS as described in the main text. At the indicated time point, cells were collected for preparation of genomic DNA. The genomic DNA were fragmented and then ligated to adaptors containing custom barcodes and then biotinylated, purified, PCR-amplified, quantified, pooled, and submitted for sequencing.
Computational analyses of sequencing data
Read mapping, replication profile analysis, and peak width analysis were performed as previously described (Sheu et al. 2014) with minor modifications. Supplemental Table 1 lists the specific analysis parameters used for each sample. Briefly, each genome-wide replication profile was generated from between 1.0 and 49.2 million reads mapped to the 16 Saccharomyces cerevisiae S288C chromosomes (NC_001133 through NC_001148). As in Sheu et al. (2014), only the first 31 bp of each read were used for mapping. Read counts were then averaged across the genome using a sliding window of 500, 1000, or 2000 bp. Genomic positions that could not be reliably mapped to themselves in a way that substantially affected these smoothed profiles were masked. Positions 450000:500000 of Chromosome XII, which encompass the rDNA locus, were also masked. In the specific analyses performed for Supplemental Figure S1, 30 kb at each end of each chromosome was masked as well. The replication profiles for each data set were then rescaled so that profile height was less than or equal to one “unit” at 99.5% of genomic positions. This rescaling was solely to facilitate the visual comparison of profiles and had no effect on subsequent peak width quantification. Peak widths were quantified using the full width at half maximum of each replication profile peak that encompassed a single annotated origin in oriDB v2.1.0 (Siow et al. 2012). Peaks that encompassed multiple origins or masked genomic regions were discarded. Peaks having a height below either 10% or 30% of the “unit” value were also discarded in width analysis in order to avoid the analysis of widths from insufficiently well-defined peaks. This height cutoff is indicated by a line in each of the peak-width plots in the figures. For peak height plots in Figures 6 and 7 and Supplemental Figures S2C and S3C, peak heights for each sample were scaled so that the 90% quantile is equal to 1.0.
In Supplemental Figures S2 and S3, plots of peak heights for duplicate 1 versus duplicate 2 (e.g., WT versus WT) show a 100-fold dynamic range of peak height and very little scatter. Thus, the reproducibility of these data is such that we can assess with high accuracy the timing of DNA replication of every single origin in the genome. We have also included in Supplemental Table 1, A and B, the median replication profile height for all genomic positions for each strain. Since most DNA does not replicate under our experimental conditions of up to 90 min in HU or early S phase (25 min after release from G1 arrest at 30°C), we expect that this median profile height represents the level of contamination of nonreplicated DNA in the EdU pull down samples. We find that this median profile height is ∼2%–5% of the level for the earliest firing origins. For example, in the plots of the duplicate sample of WT versus WT, we can see deviation between duplicates only for peak heights of 2%–5%.
Data access
All DNA sequencing data from this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra/) under accession number SRA279689. The data analysis scripts are publicly available at GitHub (https://github.com/jbkinney/14_sheu) and in the Supplemental Material.
Acknowledgments
This work was supported by a grant from the National Institute of General Medical Sciences of the National Institutes of Health (GM45436) and core facilities by a National Cancer Institute (NIH) Core grant (CA045508). We thank A. Chabes for the antibodies against Rnr4 and Sml1; H. Araki and J. Diffley for yeast strains; Cold Spring Harbor Laboratory DNA Sequencing Next Gen Shared Resource for high throughput sequencing; Microscopy, Flow Cytometry shared resources, and Bioinformatics core for initial data analysis; and Patty Wendel and the James Building Staff for general assistance.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.195248.115.
References
- Alvino GM, Collingwood D, Murphy JM, Delrow J, Brewer BJ, Raghuraman MK. 2007. Replication in hydroxyurea: It's a matter of time. Mol Cell Biol 27: 6396–6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabes A, Stillman B. 2007. Constitutively high dNTP concentration inhibits cell cycle progression and the DNA damage checkpoint in yeast Saccharomyces cerevisiae. Proc Natl Acad Sci 104: 1183–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devault A, Gueydon E, Schwob E. 2008. Interplay between S-cyclin-dependent kinase and Dbf4-dependent kinase in controlling DNA replication through phosphorylation of yeast Mcm4 N-terminal domain. Mol Biol Cell 19: 2267–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diffley JF. 2011. Quality control in the initiation of eukaryotic DNA replication. Philos Trans R Soc Lond B Biol Sci 366: 3545–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohrmann PR, Oshiro G, Tecklenburg M, Sclafani RA. 1999. RAD53 regulates DBF4 independently of checkpoint function in Saccharomyces cerevisiae. Genetics 151: 965–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson AD, Raghuraman MK, Friedman KL, Cross FR, Brewer BJ, Fangman WL. 1998. CLB5-dependent activation of late replication origins in S. cerevisiae. Mol Cell 2: 173–182. [DOI] [PubMed] [Google Scholar]
- Heller RC, Kang S, Lam WM, Chen S, Chan CS, Bell SP. 2011. Eukaryotic origin-dependent DNA replication in vitro reveals sequential action of DDK and S-CDK kinases. Cell 146: 80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DR, Prasad AA, Chan PK, Duncker BP. 2010. The Dbf4 motif C zinc finger promotes DNA replication and mediates resistance to genotoxic stress. Cell Cycle 9: 2018–2026. [DOI] [PubMed] [Google Scholar]
- Kamimura Y, Masumoto H, Sugino A, Araki H. 1998. Sld2, which interacts with Dpb11 in Saccharomyces cerevisiae, is required for chromosomal DNA replication. Mol Cell Biol 18: 6102–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimura Y, Tak YS, Sugino A, Araki H. 2001. Sld3, which interacts with Cdc45 (Sld4), functions for chromosomal DNA replication in Saccharomyces cerevisiae. EMBO J 20: 2097–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanemaki M, Labib K. 2006. Distinct roles for Sld3 and GINS during establishment and progression of eukaryotic DNA replication forks. EMBO J 25: 1753–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara M, Nakai W, Asano S, Suzuki A, Kitada K, Kawasaki Y, Johnston LH, Sugino A. 2000. Characterization of the yeast Cdc7p/Dbf4p complex purified from insect cells. Its protein kinase activity is regulated by Rad53p. J Biol Chem 275: 35051–35062. [DOI] [PubMed] [Google Scholar]
- Lopez-Mosqueda J, Maas NL, Jonsson ZO, Defazio-Eli LG, Wohlschlegel J, Toczyski DP. 2010. Damage-induced phosphorylation of Sld3 is important to block late origin firing. Nature 467: 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantiero D, Mackenzie A, Donaldson A, Zegerman P. 2011. Limiting replication initiation factors execute the temporal programme of origin firing in budding yeast. EMBO J 30: 4805–4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masai H, Taniyama C, Ogino K, Matsui E, Kakusho N, Matsumoto S, Kim JM, Ishii A, Tanaka T, Kobayashi T, et al. 2006. Phosphorylation of MCM4 by Cdc7 kinase facilitates its interaction with Cdc45 on the chromatin. J Biol Chem 281: 39249–39261. [DOI] [PubMed] [Google Scholar]
- Muramatsu S, Hirai K, Tak YS, Kamimura Y, Araki H. 2010. CDK-dependent complex formation between replication proteins Dpb11, Sld2, Pol ε, and GINS in budding yeast. Genes Dev 24: 602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli J, Tsaponina O, Crabbé L, Keszthelyi A, Pantesco V, Chabes A, Lengronne A, Pasero P. 2012. dNTP pools determine fork progression and origin usage under replication stress. EMBO J 31: 883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randell JC, Fan A, Chan C, Francis LI, Heller RC, Galani K, Bell SP. 2010. Mec1 is one of multiple kinases that prime the Mcm2–7 helicase for phosphorylation by Cdc7. Mol Cell 40: 353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind N, Gilbert DM. 2013. DNA replication timing. Cold Spring Harb Perspect Biol 5: a010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez Y, Desany BA, Jones WJ, Liu Q, Wang B, Elledge SJ. 1996. Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science 271: 357–360. [DOI] [PubMed] [Google Scholar]
- Santocanale C, Diffley JF. 1998. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature 395: 615–618. [DOI] [PubMed] [Google Scholar]
- Sheu YJ, Stillman B. 2006. Cdc7-Dbf4 phosphorylates MCM proteins via a docking site-mediated mechanism to promote S phase progression. Mol Cell 24: 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu YJ, Stillman B. 2010. The Dbf4–Cdc7 kinase promotes S phase by alleviating an inhibitory activity in Mcm4. Nature 463: 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu YJ, Kinney JB, Lengronne A, Pasero P, Stillman B. 2014. Domain within the helicase subunit Mcm4 integrates multiple kinase signals to control DNA replication initiation and fork progression. Proc Natl Acad Sci 111: E1899–E1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siow CC, Nieduszynska SR, Müller CA, Nieduszynski CA. 2012. OriDB, the DNA replication origin database updated and extended. Nucleic Acids Res 40: D682–D686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama Y, Kamimura Y, Okawa M, Muramatsu S, Sugino A, Araki H. 2003. GINS, a novel multiprotein complex required for chromosomal DNA replication in budding yeast. Genes Dev 17: 1153–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Araki H. 2013. Helicase activation and establishment of replication forks at chromosomal origins of replication. Cold Spring Harb Perspect Biol 5: a010371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Umemori T, Hirai K, Muramatsu S, Kamimura Y, Araki H. 2007. CDK-dependent phosphorylation of Sld2 and Sld3 initiates DNA replication in budding yeast. Nature 445: 328–332. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Nakato R, Katou Y, Shirahige K, Araki H. 2011. Origin association of Sld3, Sld7, and Cdc45 proteins is a key step for determination of origin-firing timing. Curr Biol 21: 2055–2063. [DOI] [PubMed] [Google Scholar]
- Tercero JA, Diffley JF. 2001. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 412: 553–557. [DOI] [PubMed] [Google Scholar]
- Tercero JA, Longhese MP, Diffley JF. 2003. A central role for DNA replication forks in checkpoint activation and response. Mol Cell 11: 1323–1336. [DOI] [PubMed] [Google Scholar]
- Varrin AE, Prasad AA, Scholz RP, Ramer MD, Duncker BP. 2005. A mutation in Dbf4 motif M impairs interactions with DNA replication factors and confers increased resistance to genotoxic agents. Mol Cell Biol 25: 7494–7504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viggiani CJ, Aparicio OM. 2006. New vectors for simplified construction of BrdU-Incorporating strains of Saccharomyces cerevisiae. Yeast 23: 1045–1051. [DOI] [PubMed] [Google Scholar]
- Weinreich M, Stillman B. 1999. Cdc7p–Dbf4p kinase binds to chromatin during S phase and is regulated by both the APC and the RAD53 checkpoint pathway. EMBO J 18: 5334–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zegerman P, Diffley JF. 2007. Phosphorylation of Sld2 and Sld3 by cyclin-dependent kinases promotes DNA replication in budding yeast. Nature 445: 281–285. [DOI] [PubMed] [Google Scholar]
- Zegerman P, Diffley JF. 2009. DNA replication as a target of the DNA damage checkpoint. DNA Repair (Amst) 8: 1077–1088. [DOI] [PubMed] [Google Scholar]
- Zegerman P, Diffley JF. 2010. Checkpoint-dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature 467: 474–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Muller EG, Rothstein R. 1998. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell 2: 329–340. [DOI] [PubMed] [Google Scholar]
- Zhong Y, Nellimoottil T, Peace JM, Knott SR, Villwock SK, Yee JM, Jancuska JM, Rege S, Tecklenburg M, Sclafani RA, et al. 2013. The level of origin firing inversely affects the rate of replication fork progression. J Cell Biol 201: 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]