

Abstract
Background
Although adolescent and young adult (AYA) cancers are characterized by biological features and clinical outcomes distinct from those of other age groups, the molecular profile of AYA cancers has not been well defined. In this study, we analyzed cancer genomes from rare types of metastatic AYA cancers to identify driving and/or druggable genetic alterations.
Methods
Prospectively collected AYA tumor samples from seven different patients were analyzed using three different genomics platforms (whole-exome sequencing, whole-transcriptome sequencing or OncoScan™). Using well-known bioinformatics tools (bwa, Picard, GATK, MuTect, and Somatic Indel Detector) and our annotation approach with open access databases (DAVID and DGIdb), we processed sequencing data and identified driving genetic alterations and their druggability.
Results
The mutation frequencies of AYA cancers were lower than those of other adult cancers (median = 0.56), except for a germ cell tumor with hypermutation. We identified patient-specific genetic alterations in candidate driving genes: RASA2 and NF1 (prostate cancer), TP53 and CDKN2C (olfactory neuroblastoma), FAT1, NOTCH1, and SMAD4 (head and neck cancer), KRAS (urachal carcinoma), EML4-ALK (lung cancer), and MDM2 and PTEN (liposarcoma). We then suggested potential drugs for each patient according to his or her altered genes and related pathways. By comparing candidate driving genes between AYA cancers and those from all age groups for the same type of cancer, we identified different driving genes in prostate cancer and a germ cell tumor in AYAs compared with all age groups, whereas three common alterations (TP53, FAT1, and NOTCH1) in head and neck cancer were identified in both groups.
Conclusion
We identified the patient-specific genetic alterations and druggability of seven rare types of AYA cancers using three genomics platforms. Additionally, genetic alterations in cancers from AYA and those from all age groups varied by cancer type.
Electronic supplementary material
The online version of this article (doi:10.1186/s12885-016-2209-1) contains supplementary material, which is available to authorized users.
Keywords: Adolescent and young adult (AYA) cancer, Next-generation sequencing (NGS), Whole exome sequencing, Precision medicine, Genomics
Background
Cancer is one of the leading causes of death worldwide. Abnormal genetic alterations followed by the uncontrolled growth of somatic cells initiate cancer. Although most genetic alterations are passenger mutations that do not contribute to tumorigenesis, an individual cell can proliferate and become a tumor if it acquires a sufficient set of driving mutations. Therefore, finding cancer-driving mutations and targeting the encoded abnormal proteins and related pathways via cancer therapeutics are important strategies to delay cancer progression and prevent metastasis [1].
Previous studies, led by The Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium (ICGC), have identified cancer-driving mutations via large-scale analyses [2]. Although large-scale analyses unveiled frequently altered driving mutations in many cancer types, such as BRAF (V600E) in melanoma and colorectal cancer, finding less frequently altered mutations is a challenge using large-scale analyses, especially in uncommon cancer types [2–4].
Adolescent and young adult (AYA) cancer is a rare type of malignant disease that arises in patients aged 15 to 39 years and is characterized by biological features, therapeutic outcomes, and survival rates that are distinct from those observed in other age groups. Although determining the genomic profiles of AYA cancer is important to investigate the causes of these distinct characteristics, large-scale genomic studies or molecular data for AYA cancer are not available due to the rarity of the disease and the difficulty of collecting tumor samples [5, 6].
In this study, we analyzed seven different AYA cancers from patients with metastatic tumors using three different genomics platforms (whole-exome sequencing, whole-transcriptome sequencing, and OncoScan™). We identified single nucleotide variations (SNVs) and insertion and deletions (indels) by using whole-exome sequencing (WES) and detected fusions by using whole transcriptome sequencing (WTS). For copy number variations (CNVs), we used OncoScan™ that is the genomics platform for analysis of copy number variations which had high performance with samples from FFPE, especially [7]. We processed the WES data with well-known bioinformatics tools (bwa, Picard, GATK, MuTect, and Somatic Indel Detector), as other studies described and processed WTS data with fusion detection tools [8–10]. We then identified candidate genes and suggested potential drugs that are specific to the genetic alterations of each patient. We also compared candidate genes for AYA cancers with the same types of cancers from all age groups using published data.
Methods
Ethics and consent statement
This study was approved by the Institutional Review Board (IRB) of Seoul National University Hospital (1206-086-414). We obtained written informed consent from the patients who participated to this study. All participants in this study gave us written informed consent for publication of their details. Written informed consent for publication of their clinical details and/or clinical images was obtained from the patients. A copy of the consent form is available for review by the Editor of this journal.
Study design and sample information
Samples from seven different tumors, prostate cancer, olfactory neuroblastoma, head and neck squamous cell carcinoma (HNSCC), urachal carcinoma, germ cell tumor, lung cancer, and liposarcoma, were prospectively obtained in three different forms (fresh-frozen tissue, formalin-fixed paraffin-embedded (FFPE), and pleurisy). The samples were analyzed using three different genomics platforms (whole-exome sequencing (WES), whole transcriptome sequencing (WTS), and OncoScan™) as the tumor sources permitted. We first intended to analyze samples from ten patients, but three samples were excluded because the amount of provided tumor sample was insufficient (AYA03) or sufficient DNA/RNA for a genome-scale analysis was not obtained (AYA05, and 08). For sample AYA04 (HNSCC), the HPV infection status was identified by IHC staining (data not shown).
Whole exome sequencing (WES)
A minimum of 3 μg of genomic DNA was randomly fragmented by Covaris, and the sizes of the library fragments were mainly distributed between 250 and 300 bp. adapters were then ligated to both ends of the fragments. Extracted DNA was amplified by ligation-mediated PCR (LM-PCR) and then purified and hybridized to the SureSelect XT Human All Exon v4 + UTR 71 Mb (Agilent Technologies, Santa Clara, CA, USA) for enrichment according to the manufacturer’s recommended protocol. After loading each captured library on the Hiseq2000 platform (Illumina, San Diego, CA, USA), we performed high-throughput sequencing for each captured library. Raw image files were processed by Illumina CASAVA v1.8.2 for base-calling with default parameters, and the sequences from each individual were generated as 101-bp pair-end reads.
Processing WES data to analyze SNVs and indels
WES data were processed using a series of steps. We aligned the sequenced files (Fastq file) to the reference genome (human reference genome g1k v37) using the Burrows-Wheeler Aligner (BWA v0.7.5a) [11] and then sorted the output and removed PCR duplicates using PICARD v1.95 [12]. Using the typical GATK workflow (The Genome Analysis Toolkit v2.6-5), we processed the data for local indel realignment and base quality recalibration [13]. For variant calling, we used MuTect v1.1.6 for single nucleotide variants (SNVs) and Somatic Indel Detector (from GATK v2.2-8) for indels [14]. Whereas we called the SNVs with the default setting value, we altered the tumor indel fraction from 0.3 to 0.05 (T_INDEL_F <0.05) for indel calling after considering false-negatives. The called variants interpreted as somatic mutations were tagged with “KEEP” or “SOMATIC” with MuTect and Somatic Indel Detector, respectively, and used for further study. To avoid false-positive indel variants, we filtered out variants with tumor alterative reads less than 6. All somatic variants were annotated by ANNOVAR [15]. The variants that passed through the steps were called ‘processed WES data’ (Additional file 1: Figure S1).
Analysis of copy number variations (CNVs) by OncoScan™
We used the 330-k OncoScan™ FFPE platform (Affymetrix, Santa Clara, CA, USA) to identify candidate CNVs (amplification/deletion and loss-of-heterozygosity (LOH)). AYA02 was excluded because the amount of DNA was insufficient for OncoScan™. A minimum of 80 ng of DNA from each sample was used for the OncoScan™ platform. The Nexus Express (Affymetrix) software was used to analyze the data and find CNVs. We filtered out CNVs with a CN ≤ 2.5, which were considered insignificant amplification, and analyzed chromosome-level CNVs and focal level CNVs.
Analysis of CNVs by VarScan2 for AYA02
To analyze CNVs in AYA02, we used VarScan2 as an alternative method to OncoScan™. After processing data up to the ‘Realignment/Recalibration’ step in WES processing (Additional file 1: Figure S1), we processed data based on the manufacturer’s recommendations. We generated mpileup data from recalibrated BAM files of both tumor and normal using SAMtools and used the ‘Copy caller’ module of VarScan2 to generate the relative copy number change (C), which was determined as follows: C = log2((DT/DN)*(IN/IT)), where ‘D’ stands for the average depth, ‘I’ for the number of uniquely mapped bases, ‘N’ for normal, and ‘T’ for tumor [16, 17]. After filtering out mapping quality values <15, we adjusted the relative copy number using the re-centering option in ‘Copy caller’ and segmented copy number regions based on the circular binary segmentation algorithm. After merging, the results were represented by IGV (Integrative Genomics Viewer) [18]. Because the VarScan2 results covered only exon regions, not all genome regions, we analyzed only chromosome-level CNVs that were similar to those obtained with OncoScan™ and did not analyze focal-level CNVs. To create graphs of relative copy number changes, we used data from re-centered relative copy number changes displayed on a log2 scale.
Analysis of mutation frequency and mutation spectrum
The mutation frequency was analyzed by counting the number of variants annotated by ANNOVAR from WES data as nonsynonymous SNVs, synonymous SNVs, nonsense mutations, stop-loss mutations, splicing mutations, frameshift insertions/deletions (indels), in-frame indels, and noncoding RNA in exonic regions. These mutations had previously been described in published data from 12 major cancer studies [19]. To analyze the mutation spectrum, we used SNVs processed with MuTect in all sequenced regions not limited to coding regions.
Pathway-drug analysis
After assigning the levels to the variants by pattern-based heuristic annotation, we investigated the biologic pathways of variants using DAVID or the literature [20]. To analyze the druggability of the variants, we concentrated mainly on level-1 (strong) variants using DGIdb [21].
Fusion analysis
We analyzed WTS data from four samples (AYA01, 02, 09, and 10) to identify cancer driving fusions using three different fusion tools, FusionMap, deFuse and ChimeraScan [22–24]. From the results, we selected candidate cancer-driving fusions using a fusion gene list archived in COSMIC (download date: 2015-03-03).
Results
AYA cancer samples, platforms and generation of data from WES
Samples analyzed in this study were collected from AYA patients (median age = 32) who had metastatic tumors. Seven different tumor samples were obtained in three different forms (fresh-frozen tissues, FFPE and pleurisy) and analyzed using three different genomics platforms (WES, WTS and OncoScan™) (Table 1).
Table 1.
Sample information of AYAs cancer patients
| No. AYAa | Age | Sex | Tumor type | Tissue type | Previous treatmentb | Platform | ||
|---|---|---|---|---|---|---|---|---|
| WES | WTS | OncoScan | ||||||
| #01 | 30 | M | Prostate cancer | Fresh-frozen | Docetaxel + Pd | v | v | v |
| #02 | 30 | M | Olfactory neuroblastoma | Fresh-frozen | ICE | v | v | - |
| Op | ||||||||
| PORT | ||||||||
| #04 | 33 | M | Head and neck squamous cell carcinoma | Fresh-frozen | Op | v | - | v |
| CCRT | ||||||||
| DP | ||||||||
| Cetuximab | ||||||||
| FP | ||||||||
| Op | ||||||||
| #06 | 32 | M | Urachalcarcinoma | FFPE | Op | v | - | v |
| #07 | 21 | M | Germ cell tumor | Fresh-frozen | Op | v | - | v |
| BEP | ||||||||
| IE | ||||||||
| #09 | 34 | M | Lung cancer | Pleurisy | - | - | v | - |
| #10 | 33 | M | Liposarcoma | Fresh-frozen | Op | v | v | v |
a#03: exclusion, because of no tumor sample provided; #05, 08: exclusion, because of insufficient sample to sequencing
b Pd prednisolone, ICE ifosfamide + carboplatin + etoposide, Op operation, PORT postoperative radiotherapy, CCRT concurrent chemoradiotherapy, DP docetaxel + cisplatin, FP 5-fluorouracil + cisplatin, BEP bleomycin + etoposide + cisplatin, IE ifosfamide + etoposide
We processed WES data using well-known bioinformatics tools described in Supporting Fig. 1 following previously published studies [8, 10]. From the WES data of six tumors and matched normal blood, we generated total of 95 gigabases (Gb, range 12.5–20.0/sample) and 106 Gb (range 12.7–27.0/sample) of mapped sequences, respectively. The mean target coverage was 135X (range, 101X-190X) and 150X (range, 115X-205X) for tumor samples and normal blood, respectively. The coverage of mean target bases exceeded 30X for 89.8 and 92.5 % of the tumor and normal blood samples, respectively (Additional file 2: Table S1).
Fig. 1.
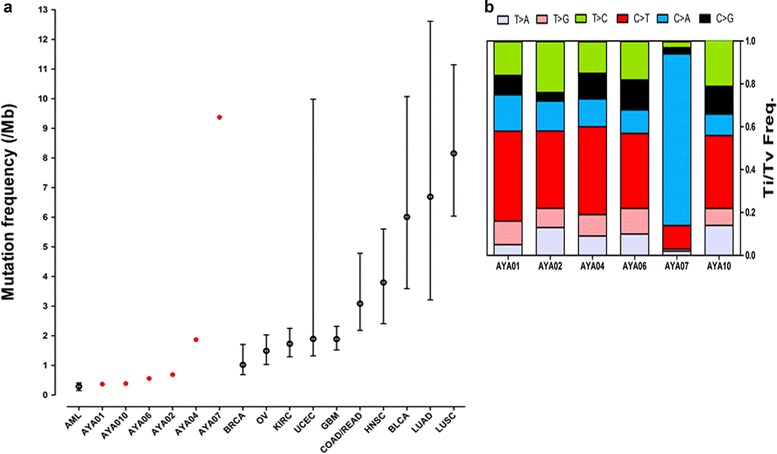
Mutation frequency and mutation spectrum of AYA cancers. a Somatic mutation frequencies of pediatric, AYA and adult cancers are shown. Mutation frequencies of AYA cancers were assessed using somatic mutations annotated as nonsynonymous SNVs, synonymous SNVs, nonsense mutations, stop-loss mutations, splicing mutations, frameshift indels, in-frame indels, and noncoding RNA. Mutation frequencies for other cancers were derived from published data from the same regions [19]. b The mutation spectrum of AYA cancers (transition and transversion frequency) was assessed using SNVs processed with MuTect in whole-exome regions
Mutation frequency and mutation spectrum
We analyzed the mutation frequency and mutation spectrum of six samples from processed WES data (Fig. 1). Except for AYA07, which contained a hypermutation, the coding regions (exon and splicing regions) of a total of 276 somatic mutations (median, 40; range, 26–133) were detected (Additional file 3: Table S2). Compared with the mutation frequency of 12 major cancer types obtained from published data, the somatic mutation frequency of most samples was low (median, 0.56/Mb; range, 0.37–1.87), except for AYA07 (Fig. 1a) [19]. This finding is consistent with data showing that somatic mutations are accumulated with age [1]. Although the mutation frequency of AYA07 was high (9.37/Mb), the overall mutation frequency of AYA cancers is low: a recent large-scale study of testicular germ cell tumors (TGCTs), the same tumor type as that of AYA07, demonstrated a low mutation frequency (mean 0.5/Mb) [25]. Additionally, three AYA cancers with higher mutation frequencies (AYA02, 04, and 07) harbored mutations in TP53 or in several DNA repair genes compared with other samples (mean, 3.98/Mb vs. 0.44/Mb, respectively), as shown in large studies [19].
Except for AYA07, the mutation spectrum of AYA cancers showed prominent C > T transitions, as has been demonstrated in many solid cancers (Fig. 1b) [26]. Interestingly, AYA07 showed a high proportion of C > A transversions (79.7 %). This result may be related to an over-representation of C > A transversions in the TCGT study [25] or multiple cycles of chemotherapy for the AYA07 patient, because increased C > A transversions were observed in all samples obtained from eight relapsed AML patients after chemotherapy [27].
Individual AYA cancer analysis: patient-specific genetic alterations
After processing the WES data, we annotated variants using ANNOVAR as described in Additional file 1: Figure S1. All annotated variants are described in Additional file 4: Table S3. We then selected driving genetic alterations using our pattern-based annotation, because it is limited to analyzing rare types of cancers using the same statistical methods to select driving genetic alterations used in large-scale studies (Additional file 3: Figure S2) [4]. By applying our approach to TCGA AML data, we could detect all candidate genes that were previously defined using statistical methods (Additional file 3: Figure S3 and Additional file 5: Table S4) [28].
Except for the hypermutations in AYA07, we focused on level-1 variants to identify driving genetic alterations of AYA cancers that are specific to each patient and may be druggable (Fig. 2 and Additional file 6: Table S5). CNVs were analyzed to identify candidate driving CNVs (OncoScan™) and chromosome-level CNVs (OncoScan™ or VarScan2) (Fig. 3 and Additional file 1: Figure S4).
Fig. 2.
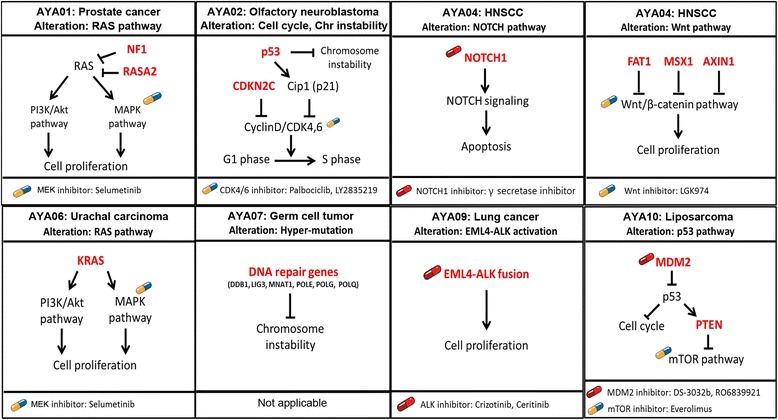
Candidate driving genetic alterations and their druggability in AYA cancers. An analysis of WES/WTS and OncoScan™ with our heuristic annotation identified level-1 candidate genetic alterations. By analyzing DAVID and DGIdb, the representative pathway of AYA cancers and druggability were also identified. The druggability is indicated by illustrations of pills; red indicates a direct inhibitor of a candidate target gene, and blue/yellow indicates an inhibitor of a pathway that includes the candidate alterations. AYA07 was excluded from the candidate gene search due to the hypermutation. All candidate genetic alterations are described in Additional file 4: Table S3
Fig. 3.
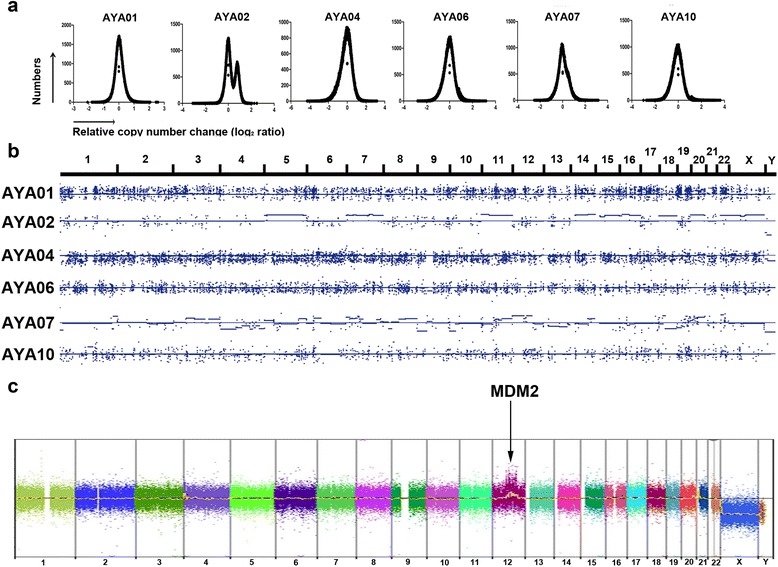
Analysis of CNVs in AYA cancers. a Distributions of relative copy number change (C) in AYA cancers, shown on a log2 scale. b Chromosome-level alterations are shown and were processed by VarScan2. Similar patterns were detected by OncoScan™ (Additional file 1: Figure S3). c OncoScan™ identified a focal amplification of MDM2 in AYA10
AYA01, prostate cancer: aberrant activation of the RAS pathway
AYA01 showed concurrent loss-of-function in genes of the RasGAP family (NF1 and RASA2). A frameshift deletion and LOH were detected in NF1, and a frameshift insertion and splicing mutation were detected in RASA2 that were validated by sequencing (Additional file 1: Figure S5). Interestingly, concurrent mutations in RasGAPs have been identified in several types of cancers according to the cBio portal (Additional file 1: Figure S6 and Additional file 7: Table S6). Furthermore, a recent study demonstrated the synergistic oncogenic effects of non-canonical Ras mutations in the context of loss-of-function in RasGAP [29]. Because RasGAPs contribute to tumorigenesis, we suggested an MEK inhibitor (as a single agent or in combination) for the treatment of AYA01 [30, 31].
AYA02, olfactory neuroblastoma: chromosome instability and loss-of-function of CDKN2C
AYA02 harbored a chromosome-level alteration with a TP53 missense mutation that contributed to chromosome instability [32]. Interestingly, AYA02 showed a double peak of a relative copy number change and arm-level alterations, which differed from other tumors (Fig. 3a and b). A loss-of-function in CDKN2C was identified with high-allelic frequency (0.667). Given the tumor suppressor function of CDKN2C in breast cancer, a loss-of-function of CDKN2C may have driven tumor formation in AYA02; therefore, we selected CDK4/6 inhibitors as a potential drug (palbociclib and LY2835219) [33].
AYA04, HNSCC: alteration of the Wnt and NOTCH pathways
AYA04 harbored TP53 mutations with alterations in FAT1, NOTCH1 and SMAD4 that have been recurrently discovered by several large-scale studies of head and neck cancer [34–36]. Specifically, AYA04 harbored concurrent mutations in Wnt pathway genes, such as FAT1, MSX1 and AXIN1, which were reported in a recent large-scale study of HNSCC with HPV (−) [34]. We suggested potential drugs (LGK974 and γ secretase inhibitor) for AYA04 based on the importance of the Wnt and NOTCH pathways.
AYA06, urachal carcinoma: alteration in noted KRAS mutation
Because AYA06 showed only one level-1 variant in KRAS (G13D) with no candidate CNVs, we selected an MEK inhibitor (selumetinib) as a potential drug. However, a missense mutation in USP6 (R133K, Lv2 OG) was detected in AYA06 and AYA04. USP6 is known to be able to initiate tumorigenesis either in cell lines or in mice via the activation of the NF-κB pathway, although the function of the R133K variant remains elusive [37].
AYA07, germ cell tumor: alteration in DNA repair genes and genome instability
AYA07 was excluded from the identification of candidate driving genetic alterations, because AYA07 showed a high mutation frequency with many CNVs may be caused by missense mutations in six DNA repair genes (DDB1, LIG3, MNAT1, POLE, POLG and POLQ) (Fig. 1 and Additional file 4: Table S3) [38]. The most frequently mutated gene, KIT, which was found in a large-scale study of TGCT, was not detected in AYA07 [25].
AYA09, lung cancer: well-known fusion, EML4-ALK
AYA09 was analyzed by WTS only because an IHC result for ALK was positive (data not shown). Because the EML4-ALK detected in our fusion processing is well known in lung cancer, crizotinib and ceritinib were recommended as potential drugs for AYA09 (Additional file 1: Figure S6 and Additional file 8: Table S7). The patient was treated with crizotinib and showed clinically significant tumor shrinkage, as expected.
AYA10, liposarcoma: amplification of MDM2 with PTEN deletion
Although level-1 SNV/indel alterations were not detected, CNVs in MDM2 and PTEN, which play a role in the p53 pathway, were identified in AYA10 (Fig. 3c). Target-specific drugs for MDM2 amplification, DS-3032b and RO6839921, plus an mTOR inhibitor, everolimus, were recommended.
Comparison of candidate driving genes from AYA cancers and cancers in other age groups
To investigate differences in the genomic profiles between AYA cancers and cancers found in other age groups, we compared candidate driving genes between AYAs and all age groups for the same cancer based on published data from analyses similar to ours (Table 2). The mutation pattern of AYA01 (prostate cancer) differed from that shown in a large-scale study analysis. Whereas AYA01 harbored alterations in the Ras pathway (NF1 and RASA2), prostate cancer from other age groups showed recurrent mutations in SPOP, TP53, and PTEN [39]. However, several commonly altered genes in HNSCC, such as TP53, FAT1, and NOTCH1, were identified in both AYA04 and in large-scale studies, whereas other mutations differed [34].
Table 2.
Comparison of candidate driving genes of same cancer type from AYAs with those from all age group
| Prostate cancer | HNSCC | |||
|---|---|---|---|---|
| AYA01 | Barbieri, et al. | AYA04 | TCGA | |
| Sequencing platform | WES (n = 1) | WES (n = 112) | WES (n = 1) | WES (n = 279) |
| Exome capture kit | Agilent SureSelect | Agilent SureSelect | Agilent SureSelect | Agilent SureSelect |
| Sequencing instrument | Illumina HiSeq | Illumina HiSeq | Illumina HiSeq | Illumina HiSeq |
| Depth (mean) | 138X | 118X | 198X | 95X |
| Age (median) | 30 | 63 | 33 | 61 |
| Bioinformatic pipeline | ||||
| Alignment | bwa (hg19) | |||
| Deduplication | Picard | |||
| Realignment | GATK | |||
| Recalibration | GATK | |||
| Variant calling | ||||
| SNVs | MuTect | |||
| Indels | Somatic Indel Detector/Indelocator | |||
| Selection of candidate SNV/Indels | Heuristic annotation | MutSig | Heuristic annotation | MutSig |
| CNVs detection | AffymetrixOncoScan | Affymetrix SNP 6.0 | AffymetrixOncoScan | Affymetrix SNP 6.0 |
| Selection of candidate CNVs | Heuristic annotation | GISTIC | Heuristic annotation | GISTIC |
| Candidate driving genes | ||||
| SNVs/Indels | NF1 | SPOP | TP53 | CDKN2A |
| RASA2 | TP53 | FAT1 | FAT1 | |
| ATAD5 | PTEN | MSX1 | TP53 | |
| FOXA1 | USP6 | CASP8 | ||
| CDKN1B | ANK2 | AJUBA | ||
| ZNF595 | CHD5 | PIK3CA | ||
| THSD7B | FOXL2 | NOTCH1 | ||
| MED12 | ITGB4 | KMT2D | ||
| NIPA2 | LECT1 | NSD1 | ||
| PIK3CA | HLA-A | |||
| C14orf9 | TGFBR2 | |||
| SCN11A | ||||
| CNVsa | NF1 | NAb | NOTCH1 | FADD |
| SUZ12 | SMAD4 | CDKN2A | ||
| SETD2 | CSMD1 | |||
| PTCH1 | SOX2 | |||
| AXIN1 | LRP1B | |||
| BAP1 | EGFR | |||
| CDH1 | FAT1 | |||
aEmphasized results from published paper (q < 0.0001)
bThere was not available emphasized result of CNVs
Discussion
Genomic profiling of AYA cancers
In this study, we described the genomic profiles of seven different rare types of AYA cancers using three different genomics platforms (WES, WTS and OncoScan™). After processing genomics data, we identified potential druggable targets for each cancer and selected existing anti-cancer drugs to treat individual patients (Fig. 2 and Additional file 6: Table S5).
We identified candidate driving genetic alterations specific to each patient using logical manual curation (pattern-based heuristic annotation) as alternative to statistical method (Additional file 9: Supplementary materials and methods and Additional file 10: Table S8). It is needed to alternative method to select candidate genes in rare type of cancers, like AYA cancers, since low number of samples is limited to the selection of candidate driving genes using statistical method as shown in large-scale studies [4].
Because the features of AYA cancers are distinct from those of other age groups, including the incidences and clinical outcomes, studying the genomic profiles of AYA cancers is important to identify the unique features of AYA cancers [5, 6]. When comparing candidate genes between AYAs and all age groups for the same cancer type, we identified different candidate genes in prostate cancer (AYA01) and a germ cell tumor (AYA07), although several common candidate genes (TP53, FAT1,and NOTCH1) were found in HNSCC (AYA04) in both AYAs and all age groups (Table 2). These results showed that AYA-specific genetic alterations may be different from those in other age groups; thus, further study is needed to define the significance of the differences in the genetic alterations between AYAs and other age groups.
Clinical implication
In this study, we analyzed individual cancer genomes and suggested potential drugs for each patient based on his or her genetic alterations. Characterizing the genomes of patients and genomics-driven knowledge enabled personalized medicine and advanced cancer genomics for clinical implications [40]. Moreover, to establish the clinical validity of genetic tests, especially for NGS data, the FDA discussed ‘post-marketing pursuit’ to define the clinical implications of variants generated from NGS, which have remained unknown [41]. Therefore, we expect many prospective genomic studies, such as our study, to link the patient to therapy as well as diagnosis, prognosis, and monitoring [42].
Conclusion
We analyzed seven different metastatic AYA cancers’ genome, and potential targets were identified. Genetic alterations in cancers from AYA and those from all age groups were varied by their cancer type.
Additional files
Figure S1. WES pipeline for our study. Figure S2. Pattern-based heuristic annotation to identify driving genetic alterations. Figure S3. Pattern-based heuristic annotation for large-scale samples. Figure S4. Chromosome-level CNVs of AYA cancers from OncoScan™ and VarScan2. Figure S5. Sequencing validation of RASA2 and NF1 in AYA01 sample. Figure S6. Concurrency of RasGAPs in large-scale studies. Figure S7. EML4-ALK validation in AYA09 cells. (DOCX 50738 kb)
Sequencing information for WES data. (PDF 95 kb)
Mutation frequency of WES data for AYA cancers. (PDF 50 kb)
Processed WES data. (PDF 635 kb)
Patient-specific genetic alterations of TCGA AML study selected by our pattern-based annotation. (PDF 156 kb)
Candidate driving genetic alterations of AYA cancers. (PDF 150 kb)
RasGAPs in large-scale studies. (PDF 234 kb)
EML4-ALK fusion in AYA09. (PDF 128 kb)
Supplementary materials and methods. (DOC 21 kb)
CVE list. (PDF 285 kb)
Acknowledgements
This study was supported by grant 03-2014-0290 from the Seoul National University Hospital Research Fund. This research was also supported by the MSIP (The Ministry of Science, ICT and Future Planning), Korea and Microsoft Research, under the ICT/SW Creative research program supervised by the NIPA (National ICT Industry Promotion Agency)“ (NIPA-2014-ITAH051014011012)”. We thank Ji-Eun Yoon and Su Jung Huh for collecting the tumor tissue samples and matched normal blood as well as for extracting genetic materials. Additionally, we thank Jiae Koh for revising the draft.
Abbreviation
- AYA
adolescent and young adult
- CNV
copy number variation
- FFPE
formalin-fixed paraffin-embedded
- GATK
the genome analysis toolkit
- HNSCC
head and neck squamous cell carcinoma
- ICGC
international cancer genome consortium
- IGV
integrative genomics viewer
- LM-PCR
ligation-mediated PCR
- NGS
next generation sequencing
- SNV
single nucleotide variant
- TCGA
the cancer genome atlas
- TGCT
testicular germ cell tumor
- WES
whole exome sequencing
- WTS
whole transcriptome sequencing
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SC, SHL, JIK designed the study. SC, SHL drafted the manuscript. SC, JL, JYS performed experiment and data analyses. JYK, SHS, BK, TMK, DWK, DSH participated in critical review of study design and data analyses, SC, JL, JYS, JYK, SHS, BK, TMK, DWK, DSH reviewed the manuscript and criticized it. All authors read and approved the final manuscript.
Contributor Information
Soojin Cha, Email: soojincha36@gmail.com.
Jeongeun Lee, Email: jeleedict@gmail.com.
Jong-Yeon Shin, Email: jongyeon.anna@gmail.com.
Ji-Yeon Kim, Email: medijiyeon@gmail.com.
Sung Hoon Sim, Email: cantusprote@gmail.com.
Bhumsuk Keam, Email: bhumsuk@snu.ac.kr.
Tae Min Kim, Email: gabriel9@snu.ac.kr.
Dong-Wan Kim, Email: kimdw@snu.ac.kr.
Dae Seog Heo, Email: heo1013@snu.ac.kr.
Se-Hoon Lee, Phone: +82-2-2072-2199, Email: sehoon.lee119@gmail.com.
Jong-Il Kim, Phone: +82-2-740-8251, Email: jongil@snu.ac.kr.
References
- 1.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458(7239):719–24. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153(1):17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505(7484):495-501. [DOI] [PMC free article] [PubMed]
- 5.Bleyer A, Barr R, Hayes-Lattin B, Thomas D, Ellis C, Anderson B, Biology, Clinical Trials Subgroups of the USNCIPRGiA, Young Adult O. The distinctive biology of cancer in adolescents and young adults. Nature reviews Cancer 2008, 8(4):288-98. [DOI] [PubMed]
- 6.Tricoli JV, Seibel NL, Blair DG, Albritton K, Hayes-Lattin B. Unique characteristics of adolescent and young adult acute lymphoblastic leukemia, breast cancer, and colon cancer. J Natl Cancer Inst. 2011;103(8):628–35. doi: 10.1093/jnci/djr094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Foster JM, Oumie A, Togneri FS, Vasques FR, Hau D, Taylor M, Tinkler-Hundal E, Southward K, Medlow P, McGreeghan-Crosby K et al. Cross-laboratory validation of the OncoScan(R) FFPE Assay, a multiplex tool for whole genome tumour profiling. BMC Med Genomics 2015, 8:5. [DOI] [PMC free article] [PubMed]
- 8.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333(6046):1157-60. [DOI] [PMC free article] [PubMed]
- 9.Van Allen EM, Wagle N, Stojanov P, Perrin DL, Cibulskis K, Marlow S, Jane-Valbuena J, Friedrich DC, Kryukov G, Carter SL et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nature medicine 2014, 20(6):682-88. [DOI] [PMC free article] [PubMed]
- 10.Ho AS, Kannan K, Roy DM, Morris LG, Ganly I, Katabi N, Ramaswami D, Walsh LA, Eng S, Huse JT et al. The mutational landscape of adenoid cystic carcinoma. Nature genetics 2013, 45(7):791-98. [DOI] [PMC free article] [PubMed]
- 11.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–760. [DOI] [PMC free article] [PubMed]
- 12.PICARD [http://broadinstitute.github.io/picard/]
- 13.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M et al: The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research 2010, 20(9):1297-303. [DOI] [PMC free article] [PubMed]
- 14.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature biotechnology 2013, 31(3):213-19. [DOI] [PMC free article] [PubMed]
- 15.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568–76. [DOI] [PMC free article] [PubMed]
- 17.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S: The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25(16):2078-79. [DOI] [PMC free article] [PubMed]
- 18.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nature biotechnology 2011, 29(1):24-6. [DOI] [PMC free article] [PubMed]
- 19.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502(7471):333-39. [DOI] [PMC free article] [PubMed]
- 20.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 21.Griffith M, Griffith OL, Coffman AC, Weible JV, McMichael JF, Spies NC, Koval J, Das I, Callaway MB, Eldred JM et al. DGIdb: mining the druggable genome. Nature methods 2013, 10(12):1209-210. [DOI] [PMC free article] [PubMed]
- 22.Ge H, Liu K, Juan T, Fang F, Newman M, Hoeck W. FusionMap: detecting fusion genes from next-generation sequencing data at base-pair resolution. Bioinformatics. 2011;27(14):1922–928. [DOI] [PubMed]
- 23.McPherson A, Hormozdiari F, Zayed A, Giuliany R, Ha G, Sun MG, Griffith M,Heravi Moussavi A, Senz J, Melnyk N et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS computational biology 2011, 7(5):e1001138. [DOI] [PMC free article] [PubMed]
- 24.Iyer MK, Chinnaiyan AM, Maher CA. ChimeraScan: a tool for identifying chimeric transcription in sequencing data. Bioinformatics. 2011;27(20):2903–904. [DOI] [PMC free article] [PubMed]
- 25.Litchfield K, Summersgill B, Yost S, Sultana R, Labreche K, Dudakia D, Renwick A, Seal S, Al-Saadi R, Broderick P et al. Whole-exome sequencing reveals the mutational spectrum of testicular germ cell tumours. Nat Commun. 2015;6:5973. [DOI] [PMC free article] [PubMed]
- 26.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–18. [DOI] [PMC free article] [PubMed]
- 27.Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506–10. [DOI] [PMC free article] [PubMed]
- 28.Cancer Genome Atlas Research N Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–74. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stites EC, Trampont PC, Haney LB, Walk SF, Ravichandran KS. Cooperation between noncanonical ras network mutations. Cell Reports. 2015 doi: 10.1016/j.celrep.2014.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McGillicuddy LT, Fromm JA, Hollstein PE, Kubek S, Beroukhim R, De Raedt T, Johnson BW, Williams SM, Nghiemphu P, Liau LM et al. Proteasomal and genetic inactivation of the NF1 tumor suppressor in gliomagenesis. Cancer Cell. 2009;16(1):44–54. [DOI] [PMC free article] [PubMed]
- 31.Min J, Zaslavsky A, Fedele G, McLaughlin SK, Reczek EE, De Raedt T, Guney I, Strochlic DE, Macconaill LE, Beroukhim R et al. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat Med. 2010;16(3):286–94. [DOI] [PMC free article] [PubMed]
- 32.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability--an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11(3):220–28. [DOI] [PubMed]
- 33.Pei XH, Bai F, Smith MD, Usary J, Fan C, Pai SY, Ho IC, Perou CM, Xiong Y. CDK inhibitor p18(INK4c) is a downstream target of GATA3 and restrains mammary luminal progenitor cell proliferation and tumorigenesis. Cancer Cell. 2009;15(5):389–401. [DOI] [PMC free article] [PubMed]
- 34.Cancer Genome Atlas N Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517(7536):576–82. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333(6046):1154–157. [DOI] [PMC free article] [PubMed]
- 36.Martin D, Abba MC, Molinolo AA, Vitale-Cross L, Wang Z, Zaida M, Delic NC, Samuels Y, Lyons JG, Gutkind JS. The head and neck cancer cell oncogenome: a platform for the development of precision molecular therapies. Oncotarget. 2014;5(19):8906–923. [DOI] [PMC free article] [PubMed]
- 37.Pringle LM, Young R, Quick L, Riquelme DN, Oliveira AM, May MJ, Chou MM. Atypical mechanism of NF-kappaB activation by TRE17/ubiquitin-specific protease 6 (USP6) oncogene and its requirement in tumorigenesis. Oncogene. 2012;31(30):3525–535. [DOI] [PMC free article] [PubMed]
- 38.Wood RD, Mitchell M, Sgouros J, Lindahl T. Human DNA repair genes. Science. 2001;291(5507):1284–289. [DOI] [PubMed]
- 39.Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44(6):685–89. [DOI] [PMC free article] [PubMed]
- 40.Garraway LA, Verweij J, Ballman KV. Precision oncology: an overview. J Clin Oncol. 2013;31(15):1803–805. [DOI] [PubMed]
- 41.Evans BJ, Burke W, Jarvik GP. The FDA and genomic tests - getting regulation right. N Engl J Med. 2015;372:2258–264. [DOI] [PMC free article] [PubMed]
- 42.Lee SH, Sim SH, Kim JY, Cha S, Song A. Application of cancer genomics to solve unmet clinical needs. Genomics Inform. 2013;11(4):174–79. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. WES pipeline for our study. Figure S2. Pattern-based heuristic annotation to identify driving genetic alterations. Figure S3. Pattern-based heuristic annotation for large-scale samples. Figure S4. Chromosome-level CNVs of AYA cancers from OncoScan™ and VarScan2. Figure S5. Sequencing validation of RASA2 and NF1 in AYA01 sample. Figure S6. Concurrency of RasGAPs in large-scale studies. Figure S7. EML4-ALK validation in AYA09 cells. (DOCX 50738 kb)
Sequencing information for WES data. (PDF 95 kb)
Mutation frequency of WES data for AYA cancers. (PDF 50 kb)
Processed WES data. (PDF 635 kb)
Patient-specific genetic alterations of TCGA AML study selected by our pattern-based annotation. (PDF 156 kb)
Candidate driving genetic alterations of AYA cancers. (PDF 150 kb)
RasGAPs in large-scale studies. (PDF 234 kb)
EML4-ALK fusion in AYA09. (PDF 128 kb)
Supplementary materials and methods. (DOC 21 kb)
CVE list. (PDF 285 kb)