

Abstract
Prescribing splicing
The unfolding of the instructions encoded in the genome is triggered by the transcription of a gene's DNA sequence into RNA, and the subsequent processing of the primary RNA transcript to generate functional messenger RNAs (mRNAs), which in turn are translated into proteins. DNA sequence variation can influence every step along this pathway, often in association with disease, but the impact of specific sequence variants is difficult to predict. On page 144 of this issue, Xiong et al. (1) provide remarkably accurate in silico predictions of the effects of sequence variants on pre-mRNA splicing, a key RNA processing step, including their impact on various human pathologies.
Splicing is the process by which intervening sequences in the primary transcript (introns) are excised and the remaining sequences (exons) concatenated together to form the mRNA sequence (see the figure). It is often invoked as the mechanism by which the human genome (and other genomes) can generate a larger number of RNAs and proteins from a limited repertoire of genes. Indeed, through the alternative combination of exons, multiple mRNA species are usually encoded in a gene's sequence. Although the global importance of alternative splicing regulation in defining cell type specificity is still unknown, many examples document the potential of this process to influence biological outcomes such as sex determination, neural differentiation, synapse formation, or programmed cell death (2). Not surprisingly, alterations in the machinery of the splicing process cause diseases such as spinal muscular atrophy, and an imbalance of alternatively spliced products can contribute to cancer progression as well as muscular and neurodegenerative pathologies (3).
Figure 1. Alternative splicing.
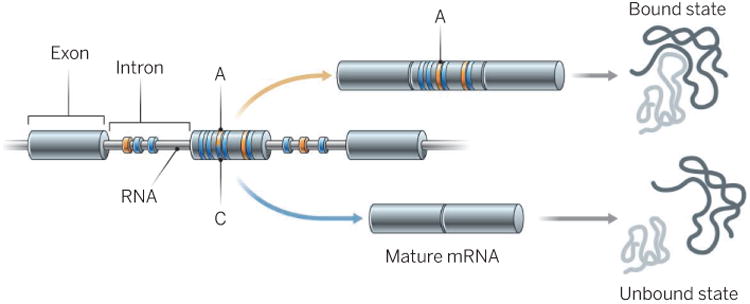
The splicing process removes introns from primary RNA and concatenates exons to generate mature mRNAs. Exons can be included or skipped, thus generating alternatively spliced products that encode different proteins (light gray). The example shows how spliced products could differ by the presence or absence of a domain that interacts with another protein (dark gray). Regulatory sequences in the exons or introns promote (orange) or prevent (blue) inclusion of the alternative exon. Nucleotide differences (e.g., A or C) can alter the function of the regulatory sequences and therefore change the balance between alternatively spliced mRNAs.
The regulation of transcription is relatively well understood, and as a consequence, mathematical models exist that can predict cell type–specific transcriptional levels of genes with great accuracy (4, 5). By contrast, the mechanisms involved in alternative splicing regulation are only partially characterized. The splice sites (relatively conserved short sequences), together with a plethora of other more transcript-specific regulatory motifs, play a role in defining intron-exon boundaries and regulating splice site selection (6). Although advances have been made in predicting differential alternative splice site choices between broad tissue types (7, 8), the algebra relating particular combinations of regulatory motifs to splice site selection in a cell type–specific manner has remained elusive.
Now, Xiong et al. have made a successful crack at this algebra. Using sophisticated machine-learning approaches, they built a mathematical model that predicts with substantial accuracy the absolute amounts of exon inclusion in different tissues. The model—a reflection of the complexity of alternative splicing regulation—depends on nearly 1400 features in the exons and neighboring introns, which often influence exon inclusion in opposite directions depending on the broader sequence context or the specific cell type in which the feature is functional. The features include splice site signals and regulatory motifs, but also exon and intron lengths, mono-, di-, and tri-nucleotide frequencies, and RNA secondary structures, among others. Xiong et al. show that the model successfully integrates this seemingly diverse set of features and predicts the individual as well as the collective effect of RNA binding proteins—the proteins that recognize the splicing regulatory motifs—in the regulation of exon inclusion.
As a demonstration of the general validity of their code, Xiong et al. show that the model can predict with substantial precision interindividual variation in exon inclusion elicited by nucleotide changes at single positions (single-nucleotide variants, SNVs) in the sequence of the splicing features. The model provides an unprecedented view of the impact of SNVs on splicing regulation by identifying more than 20,000 unique SNVs likely to affect splicing. These include synonymous changes within protein-coding sequences, generally assumed to be functionally neutral, as well as missense or nonsense changes whose effects on protein expression may be more dramatic than anticipated because of their impact on the splicing process.
This wealth of information has important medical implications. Xiong et al. 's model predicts that intronic mutations associated with disease alter splicing 9 times as often as common variants, and the authors argue that their approach is more sensitive than standard genome-wide association studies for capturing splicingrelated disease SNVs. Furthermore, they benchmark their method by correctly predicting the outcome of nearly 100 mutations in an exon of the SMN2 gene, whose level of inclusion is the target of emerging therapies for spinal muscular atrophy. In addition, their model explains disease penetrance of silent mutations in hereditary colorectal cancer, and provides interesting candidates for splicing alterations that may play a role in autism. These examples may herald a new era of personalized medicine when individual genomic sequences will be used to predict variations in splicing patterns associated with the onset or progression of pathological conditions.
As is often the case, the past is only the prologue. Although the work of Xiong et al. is indeed an important step toward understanding the complex code underlying splicing regulation, a long way remains for it to be fully deciphered. A number of features contributing to the model may reflect the effect of other steps in RNA metabolism not directly involved in splice site selection (e.g., RNA stability or translatability), thus complicating the mechanistic interpretation of the model. Also, there is emerging evidence that splicing occurs mostly cotranscriptionally (9) and that splice site selection can be influenced by the transcriptional machinery, nucleosome positioning (10, 11), epigenetic modifications (12), and possibly three-dimensional chromatin conformation. None of these features are currently captured in the model, but they could certainly be included in future developments. It is also important to remember that the truly functional unit is not the exon but the full mRNA sequence, often composed of many exons and harboring different combinations of sequence variants that affect both coding and regulatory regions. Developments in sequencing technologies and bioinformatic methods should soon make it possible to accurately quantify the cellular abundance of full mRNA species. Understanding and modeling this RNA dynamics in different cell types and along temporal processes is essential to understand how the cells work to form functional tissues and organisms—a goal toward which the study of Xiong et al. is a solid step forward.
References
- 1.Xiong HY, et al. Science. 2015;347:1254806. doi: 10.1126/science.1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nilsen TW, Graveley BR. Nature. 2010;463:457. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cooper TA, Wan L, Dreyfuss G. Cell. 2009;136:777. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karlić R, Chung HR, Lasserre J, Vlahovicek K, Vingron M. Proc Natl Acad Sci USA. 2010;107:2926. doi: 10.1073/pnas.0909344107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dong X, et al. Genome Biol. 2012;13:R53. doi: 10.1186/gb-2012-13-9-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Z, Burge CB. RNA. 2008;14:802. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barash Y, et al. Nature. 2010;465:53. doi: 10.1038/nature09000. [DOI] [PubMed] [Google Scholar]
- 8.Zhang C, et al. Science. 2010;329:439. doi: 10.1126/science.1191150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kornblihtt AR, et al. Mol Cell Biol. 2013;14:153. doi: 10.1038/nrm3525. [DOI] [PubMed] [Google Scholar]
- 10.Tilgner H, et al. Nat Struct Mol Biol. 2009;16:996. doi: 10.1038/nsmb.1658. [DOI] [PubMed] [Google Scholar]
- 11.Schwartz S, Meshorer E, Ast G. Nat Struct Mol Biol. 2009;16:990. doi: 10.1038/nsmb.1659. [DOI] [PubMed] [Google Scholar]
- 12.Luco RF, et al. Science. 2010;327:996. doi: 10.1126/science.1184208. [DOI] [PMC free article] [PubMed] [Google Scholar]