

HISTORICAL FOUNDATIONS OF MICROSCOPY
Even in medieval times it was understood that curved mirrors and hollow glass spheres filled with water had a magnifying effect. The early 17th century saw the first experiments using lenses to increase magnification. A compound telescope, with weak convex lens at one end and a concave lens as the eyepiece, was demonstrated by a Dutch spectacle maker to the court at The Hague in September 1608 (Ruestow, 1996). News quickly spread throughout Europe. Galileo made his own compound telescope in 1609, turned it on the planet Jupiter, and discovered moons. Galileo soon turned his telescope around and observed flies with it. Credit for the now standard two-convex-lens microscope goes to the son and father team of Janssen and Janssen. Naturalists Jan Swammerdam (1637–1680) and Nehemiah Grew (1641–1712), anatomist Regnier Graaf (1641–1673), and physiologist Marcello Malpighi (1628–1694) made important discoveries using magnifying lenses, especially tiny, strong single lenses (Ruestow, 1996).
Robert Hooke’s book, Micrographia, published in 1665, contains beautiful drawings based on his microscopic observations. His experimental demonstrations to the Royal Society were interrupted by the 1666 fire of London, after which he and his friend and business partner Christopher Wren had major roles in the surveying and rebuilding of the city (Jardine, 2004). Also in 1666, Sir Isaac Newton found that a prism separates white light into distinct colors, and, in another crucial, brilliant experiment, discovered that the rainbow could be recombined into white light with a second prism (Newton, 1672; 1730). In 1683, the Dutch merchant Anton van Leeuwenhoek (1632–1723), on the basis of observations performed using his own meticulously prepared lenses, published his first of many papers to the Philosophical Transactions of the Royal Society (Leewenhoek, 1683). van Leeuwenhoek’s publications describing “animalcules,” blood cells, sperm cells, and more, was the first multidecade high-throughput microscopy project. In 1773 a Danish microbiologist, Otto Muller (1730–1784), used the microscope to describe the forms and shapes of various bacteria. In 1833, Robert Brown (1773–1858) discovered the consistent presence of nuclei in plant cells. Brown also reported on the microscopic behavior of tiny, nonliving clay particles, now called Brownian motion, which Einstein discussed in one of his classic 1905 papers.
In 1856, William Perkin discovered mauve, the first useful synthetic dye. He used his first batch to stain silk. After trial and error he found he could use tannins to accomplish color-fast staining of mauve onto wool and cotton. His successful commercialization of this process revolutionized the textile industry and fashion, and rapidly led to the establishment of a modern chemical and pharmaceuticals industries (Perkin, 1906; Garfield, 2001). Mauve was a serendipitous discovery, but in 1865 F. August Kekule (1829–1896) dreamt his theory of the benzene ring, which together with an understanding of the stoichiometry of molecules and chemical reactions led to chemistry becoming a rational science.
In 1869, Friedrich Meischer isolated nucleic acids. Of the many dyes invented and used in the following decades, it may be mentioned that fluorescein was discovered and synthesized in 1871 (see Clark and Kasten, 1983, for history of staining). In 1879 to 1880, Paul Ehrlich performed a series of studies on the nature of cell and tissue staining by acidic and basic dyes and later went on to found immunology and the idea of magic bullet chemotherapies. Ernst Abbe described the mathematics of diffraction and optimal lens construction in 1876. The firm of Carl Zeiss quickly manufactured high-quality oil-immersion lenses of Abbe’s designs. In 1893, August Köhler, a German zoologist, described the principles of what we now refer to as Köhler illumination (APPENDIX 3N). This was a critical step in generating a uniform field of illumination and providing optimal image resolution. Rudolf Virchow (1821–1902) founded modern cell physiology in the midst of these twin revolutions in dye chemistry and microscope optics. Waldeyer coined the term “chromosomes” in 1888 to refer to those colored bodies he saw in dividing cells.
Köhler and Moritz von Rohr made the first ultraviolet microscope to try to take advantage of shorter wavelengths and produce better resolution. In the course of their studies they made the first fluorescence microscope. In the following decades, the firms of Zeiss and Reichert made the first fluorescence microscopes using transmitted light illumination and simple filters. Among the earliest uses of epiillumination were the observations by Singer (1932) on live tissues. While Singer preferred a carbon arc as an illumination source, (used at that time for generating therapeutic artificial sunshine) his use of epi-illumination with a heat-absorbing filter, excitation filter, mirror, and emission filter is organized along the lines of a modern fluorescence microscope. In the 1930s and 1940s, Strugger and others investigated biological fluorescence staining with acridine orange and other fluorophores. In 1934 Marrack conjugated pathogen-specific antibodies to a dye; however, the weak labeling was hard to see on the bright background of standard transmitted light microscopy. Albert Coons (1912–1978) and colleagues (see Coons, 1941, 1942, 1961) adapted Marrack’s dye-coupling idea with a fluorescent covalent labeling, introducing the concept of immunofluorescence, which enabled staining with unparalleled molecular specificity. Coons and Kaplan (1950) introduced the use of a fluorescent secondary antibody to detect the antigen-specific primary antibody. Secondary antibodies both amplified the staining intensity per antigen and made the technique generic, in that one batch-labeled secondary antibody (i.e., goat anti-rabbit) could be used to detect many different primary antibodies on different specimens (e.g., rabbit anti-pneumococcus, rabbit anti-streptococcus, etc). Immunofluorescence begat radioimmunoassays (RIA), enzyme linked immunnoassays (ELISA) for high sensitivity, high-throughput detection of almost anything in the clinical chemistry laboratory, and secondary antibody–horseradish peroxidase (Ab-HRP) amplification, peroxidase-anti-peroxidase (PAP) amplification, and alkaline phosphatase-anti-alkaline phosphatase amplification (APAAP) to amplify signals in immunofluorescence and immunohistochemistry. This culminated in a Perkin-like revolutionary discovery by Kohler and Milstein (1975) of a way to make unlimited amounts of a given antibody by fusing a B cell secreting a specific antibody with an immortal leukemia cell line. Each resulting hybridoma could then be grown as a continuous cell line producing a monospecific, monoclonal antibody (MAb). Kohler, Milstein the Medical Research Council (MRC) in the U.K. have been criticized by some (and lauded by others) for not taking out a patent on the production of monoclonal antibody–producing cell lines. Apparently the National Research Development Corporation (NRDC), the organization responsible for securing intellectual property rights for MRC inventions had not been interested (Milstein, 2000). Thirty years post-invention, monoclonal antibodies are a key component in a new wave of biologic-based chemotherapeutics. Antibodies, especially reliable, commercially available monoclonal antibodies, combined with antigen-retrieval protocols for paraffin-embedded tissue sections, revolutionized the practice of pathology in the 1990s (Shi et al., 2000).
In 1924, Feulgen and Rossenbeck reported on their use of pararosaniline, a close relative of Perkin’s mauve, and a quantitative depurination reaction to make a Schiff’s base, thereby producing a reliable way to quantify DNA in cell nuclei and bacteria. Use of this technique ultimately led to the discovery of distinct stages in the eukaryotic cell cycle and established the constancy of DNA in most mammalian cell lineages. The technique also made it possible to prove that gametogenesis involves a reduction division and stimulated many developments in quantitative microscopy in the 1930s to 1950s by Caspersson and others.
Waldeyer’s colored bodies (chromosomes) were subdivided in the 1930s by Bridges observation of banding in Drosophila chromosomes. Hsu (1952), capitalizing on a serendipitous buffer-dilution error, invented a chromosome-spreading technique based on hypotonic swelling of metaphase cells. Tjio and Levan (1956) used the hypotonic swelling/cell-dropping method to correctly enumerate the 46 human chromosomes. Lejeune et al. (1959) quickly discovered trisomy 21 in (most) Down syndrome patients. Caspersson et al. (1968) published their method of fluorescently banding chromosomes using quinacrine mustard to uniquely identify each of the 23 pairs of human chromosomes. Although most clinical chromosome banding is now done with the Perkin-era nonfluorescent dye Giemsa; fluorescent identification of chromosomes using multiple probe fluorescence hybridization combines the advances made in both cytogenetics and microscopy.
MICROSCOPY IN MODERN HUMAN GENETICS
Microscopy currently plays a crucial role in both research and diagnostic aspects of modern genetics. This typically involves the use of light microscopes for the analysis of microbiological, cytological, and pathological specimens, as well as the cytogenetic analysis of metaphase and interphase chromosomes (APPENDIX 3N). With recent advances in fluorescence technology, there has been growth, even in clinical laboratories, in the use of fluorescence microscopy. Spectral karyotyping and Multiplex-FISH instruments have made their way into many clinical laboratories, but in narrow niches. Confocal microscopy was invented by Minsky (1957, 1988) and reinvented by Egger and Petran (1967), but their achievements were not widely appreciated. The first successful confocal microscope was developed around the confocal laser scanning microscope introduced by White et al. (1987). The MRC confocal microscope’s history, in the context of cell analysis and antibody developments, has been reviewed by the inventors, Amos and White (2003). Confocal microscopy improves lateral resolution by a factor of , but, more importantly, provides optical sectioning by blocking out-of-focus light from reaching the detector (see Shotton, 1993; Diaspro, 2001). The downside is that some of the in-focus light is also blocked. Most of the fluorescence microarray readers, e.g., Affymetrix, now found in clinical core laboratories are based around confocal scanner designs. Denk et al. (1990) introduced multiphoton excitation microscopy (MPEM). MPEM is a technique with intrinsic optical-sectioning capabilities, where fluorescence only occurs in a diffraction-limited spot where a high-intensity near-infrared laser comes to a focal point. Although most MPEM microscopes are based on, and can be used as, confocal microscopes, the pinhole is typically kept open to collect more light. A few confocal microscopes are within the budget range of a well endowed clinical laboratory, but commercial multiphoton microscopes currently use expensive Ti:Sapphire pulsed lasers. The biomedical optics community is busy pushing the limits of microscopy into nanoscopy, a field reviewed by Garini (2005).
Analysis of cell types in blood and biopsy specimens, apoptosis assays in diseased tissue, cytogenetic analysis, and even surgical procedures using fluorescence have all been reported. Because this unit deals primarily with the use of microscopy and contemporary image analysis in mammalian cytogenetics, a brief explanation of some of the applications of fluorescence microscopy in this field is relevant. The ability to label nucleic acids with fluorescent molecules and detect them in situ was developed in the early 1980s (Langer-Safer et al., 1982; Manuelidis et al., 1982). Fluorescence in situ hybridization (FISH; UNIT 4.3) technology has been applied to many different areas of cytogenetic investigation (Lichter and Ward, 1990). The first use involved determination of chromosome copy number in interphase nuclei using centromere-specific fluorescent probes (Manuelidis, 1985; Cremer et al., 1986; Devilee et al., 1988). This was later extended to the identification of aberrant or marker chromosomes (Taniwaki et al., 1993; Thangavelu et al., 1994; Blennow et al., 1995) or microdeletions (Ried et al., 1990) using chromosome painting probes or single-copy probes known to map to specific chromosomes. Giemsa banding was not compatible with fluorescence hybridization techniques, thereby making it difficult to obtain simultaneous identification of the chromosomes. Methods were soon developed, however, whereby banding patterns could be obtained through the hybridization of fluorescently labeled repetitive sequences in both humans (Baldini and Ward, 1991) and mice (Boyle et al., 1990a; Arnold et al., 1992). Concurrent with these advances came the start of the Human Genome Project and the application of FISH to gene mapping (Lichter et al., 1990, 1992; Ward et al., 1991; Ried et al., 1992; Otsu et al., 1993; Trask et al., 1993). FISH mapping is a useful technique for the identification of other genes in a gene family, including functional and nonfunctional genes (Giordano et al., 1993), and for the mapping of genes across species barriers, a technique referred to as Zoo-FISH (Wienberg et al., 1990; Raudsepp et al., 1996; Fronicke and Scherthan, 1997; O’Brien et al., 1997; Chowdhary et al., 1998).
The fluorescent labeling and hybridization of entire genomes is useful for a number of different areas of investigation. Somatic cell hybrid lines (see Chapter 3) have proven very useful for isolating and mapping disease genes in both mice and humans (Harris, 1995). It is not only important to determine the genomic contribution of the species of interest in these lines during their derivation, but to continue evaluating this periodically, due to their unstable nature. This can be done by labeling the genomes of the parental species and hybridizing them to metaphases from the hybrid line to display the chromosomes contributed by each species (Durnam et al., 1985; Manuelidis, 1985; Schardin et al., 1985). The reciprocal experiment of labeling the somaticline genome and hybridizing it to metaphases from each of the parental species identifies the specific chromosome(s) segregating in the hybrid (Boyle et al., 1990b; Doucette-Stamm et al., 1991). Comparative genome hybridization (CGH) is another technique involving the fluorescent labeling of entire genomes. In this instance, the genomes are from karyotypically normal reference and mutant (i.e., tumor) populations. The genomes, labeled with different fluorophores, are pooled prior to hybridization. The hybridization ratio of the two fluorophores along the length of each chromosome is calculated to determine the gain or loss of chromosomal regions in the mutant cells (UNIT 4.6; Kallioniemi et al., 1992; Forozan et al., 1997; Ried et al., 1997). This technique is extremely useful in cases involving multiple chromosomal rearrangements for which specific bands cannot be identified by Giemsa staining, or for detecting small insertions or deletions (larger than 1 Mb).
Comparative cytogenetics is the study of changes in chromosome number and composition in different species as a function of their evolutionary divergence from one another. Chromosome painting has proven very useful in identifying homologous chromosome regions between species and has led to a better understanding of the evolutionary rearrangement of genomes (Wienberg et al., 1990). Many fluorescent dyes have now been created, each with different excitation and emission characteristics. This has allowed for the simultaneous hybridization and discernment of multiple probes on a single slide (Johnson et al., 1991; Ried et al., 1992). A natural extension of this procedure involves the labeling of different probes with various combinations of fluorophores, thereby enabling the hybridization of more probes than there are distinguishable dyes. With N fluorophores, the number of possible labeling combinations is given by 2N – 1. This combinatorial labeling of individual chromosomes using five different fluorophores is used for spectral karyotyping (SKY; UNIT 4.9; Garini et al., 1996; Liyanage et al., 1996; Schröck et al., 1996) and M-FISH (UNIT 4.9; Speicher et al., 1996; Azofeifa et al., 2000; Karhu et al., 2001; Jentsch et al., 2003) analysis of mouse and human metaphase chromosomes. 25 − 1 = 31 combinations are more than the 24 human or 21 types of mouse chromosomes. This technique has proven very useful for the identification of chromosome aberrations in human tumors and mouse models of tumorigenesis (Barlow et al., 1996; Coleman et al., 1997; Veldman et al., 1997; Ghadimi et al., 1999). SKY analysis has also been applied to evolutionary studies and will improve the analysis of genomic relationships (Schröck et al., 1996). Another approach has been to label probes not only by using combinations of dyes, but also by varying the ratios in which they are used (Nederlof et al., 1992). If K different concentrations are used for each fluorophore, and if the highest concentration used is 1 and the other concentrations are defined as (1/2)1, (1/2)2.…(1/2)K−2, and 0, the total number of possible valid combinations as described by (Garini et al., 1999) is given by:
where “valid” refers to combinations that have different fluorophore-concentration ratios. In other words, ratios of (1,1) and (0.5,0.5) for a two-dye scheme have identical concentration ratios of 1. A method that extends Nederlof’s method to enough dyes and ratios to paint each human chromosome uniquely is called combined binary ratio labeling fluorescence in situ hybridization (COBRA; Tanke et al., 1998). The method has been used for adding a unique gene (HPV) FISH probe (Szuhai et al., 2000); painting p versus q arms (Wiegant et al., 2000), and staining with total of 49 colors for all chromosome arms plus a gene FISH probe (Brink et al., 2002). Using subtelomeric COBRA-labeled BAC and PAC FISH probes, all 41 unique subtelomeres have been imaged on single human metaphase spreads (Engels et al., 2003).
Fluorescence technology is also making advances in the areas of cell biology, and more recently in studies to determine nuclear topography (Lawrence et al., 1989, 1993; Carter et al., 1993; Xing et al., 1993) and chromatin organization (Sachs et al., 1995; Yokota et al., 1995, 1997). Studies designed to analyze gene function are also incorporating advances in fluorescence microscopy, but the fluorescence in this case is not from a fluorophore conjugated to a nucleic acid, but from a green fluorescent protein (GFP) isolated from Aequorea jellyfish. By making constructs encoding the gene of interest fused to the GFP gene, researchers are able to determine the cellular sublocalization of their “glowing” gene product (Chalfie et al., 1994). GFP has also been used as a reporter gene in transgenic mice to determine the developmental stage and tissue-specific transcriptional activation of promoters (Fleischmann et al., 1998). The fusion of GFP to the CENPB gene, the product of which is known to localize to all human centromeres, has been used in conjunction with time-lapse fluorescence microscopy to follow the movement of centromeres throughout the cell cycle (Sullivan and Shelby, 1999).
Subsequent in vitro modifications of the sequence of the GFP gene protein have resulted in the development of other fluorescent proteins, including blue, cyan, and yellow, thereby enabling the simultaneous use of multiple fluorescently tagged proteins in the same living cell (reviewed by Tsien, 1998, 2005). A key discovery was made by a Russian group that cloned, from an isolate of Discosoma coral, a red fluorescent protein (DsRed; Matz et al., 1999). This was followed by characterization of the genetic diversity of the colorful fluorescent protein family (Labas et al., 2002), isolation of more useful mutants of DsRed, such as “Timer” (Terskikh et al., 2000), a monomeric version, mRFP1 (Campbell et al., 2002), and mRFP1’s fruity spectrum of mutants, from orange through far-red mPlum derivatives (Shaner et al., 2004; Wang et al., 2004).
The ability to fuse cDNAs of fluorescent proteins with researchers’ proteins of interest and/or with other fluorescent proteins has been the enabling technology for many localization and colocalization studies at the cell and organismal level. The majority of fluorescent protein FRET papers now use 37°C stable versions of cyan (Heim et al., 1994) and yellow (Ormö et al., 1996) fluorescent proteins. At the cell level, the interaction of CFP-sensor-YFP fusion with a molecule of interest, or CFP-protein1 plus YFP-protein2 interactions, can answer questions with nanometer precision. These studies take advantage of fluorescence resonance energy transfer (FRET), in which the energy released by the excitation of one fluorescent molecule in the cell can directly cause the excitation of a different fluorescent molecule if they are in close enough proximity to one another. The effective range of Förster-type energy transfer is less than 10 nm, making FRET an excellent spectroscopic ruler for molecular interactions in cells (Stryer and Haugland, 1967). For example, Miyawaki and colleagues used CFP-calmodulin-M13-YFP as a calcium sensor, called CaMeleon. In the absence of calcium, the CFP and YFP were far enough apart and/or oriented such that little FRET occurred from CFP to YFP. In the presence of calcium, the binding of four calcium ions to the calmodulin component resulted in a specific interaction between calmodulin and the M13 peptide, which resulted in CFP and YFP becoming close enough and/or orienting their dipole moments such that FRET increased (Miyawaki et al., 1997).
FRET is an excellent imaging tool when large changes occur as in the binding or unbinding of calcium and CaMeleon, or the loss of FRET on cleavage of a protease substrate in a CFP-substrate-YFP protein fusion. However, subtle features of the sensor module and/or the fluorescent protein(s) can have an enormous impact on success or failure. Making the effort to optimize the FRET system can pay off with large dynamic range sensor responses, as in the outstanding 20× dynamic range for a FRET CFP-caspase substrate-YFP sensor found by mutagenesis and flow sort screening for color derivatives of the two fluorescent proteins (Nguyen and Daugherty, 2005). This methodology has been utilized to identify in vivo molecular interactions between different proteins in transgenic organisms, such as cell-type and temporal specific expression of calcium-ion sensing CaMeleons (Hasan et al., 2004) or calpain protease activation (Stockholm et al., 2005) in mice. A comprehensive review of FRET methods and calculations has been published by Jares-Erjiman and Jovin (2003).
Fluorescent proteins have also been fused with, or otherwise made to interact with, recombinant luciferases. These recombinants recapitulate the native system of Aequorea, in which the calcium-activated blue-emitting photoprotein aequorin (a one-shot luciferase), excites noncovalently bound GFP by bioluminescence resonance energy transfer (BRET), to produce predominantly green light. Researchers have made use of BRET specifically, and luciferases in general, to monitor protein-protein interactions in homogeneous biochemical assays, live cells, and from cells inside mice (e.g., Contag et al., 1995; Xu et al., 1999; Waud et al., 2001; Ray et al., 2004; Paulmurugan and Gambhir, 2005). While luciferase- and GFP-labeled cells are not going to become general clinical tools, the ability to look at scales ranging from molecules to mice, and from nanoseconds to weeks, is called molecular imaging. For example, the ability of study transplanted stem cells and their progeny in order to differentiate in mouse models is greatly facilitated by luciferase and/or fluorescent-protein tagging. Stem cell progeny can be tracked noninvasively by bioluminescence imaging (Wang et al., 2003). At the end of the experiment, the proof of identify, and presence or lack of fusion with host cells, ideally needs to be proven by combined GFP fluorescence, cell type marker–specific fluorescence immunocytochemistry, and sex chromosome– and species-specific fluorescence in situ hybridization.
TYPES OF MICROSCOPY
One source for an excellent and detailed discussion of microscopy and live cell analysis (in addition to other topics) is Cells: A Laboratory Manual (Spector et al., 1998), or, in its condensed form, Live Cell Imaging (Goldman and Spector, 2004). In general, the ability to generate a clear image is dependent on magnification, contrast between internal and external milieu, and the ability to resolve discrete objects. There are many different microscope arrangements that can be used to enhance the contrast of specimens. The major imaging modes are epi-fluorescence, transmitted bright-field (APPENDIX 3N), phase contrast (APPENDIX 3N), polarized light, Nomarski or differential interference contrast (DIC; APPENDIX 3N), dark-field illumination, and reflected-light illumination. Most of these configurations are used in conjunction with fluorescence microscopy.
With respect to fluorescence, the older literature referred to “diascopic” fluorescence in the case of transmitted-light fluorescence, or trans-fluorescence, and “episcopic” in the case of incident-light fluorescence, or, epi-fluorescence (APPENDIX 3N), which makes use of reflected light excitation. In incident-light microscopy, the incoming light is first reflected down through the specimen and then back up through it into the objective (Fig. 4.4.1). Note that reflected light is useful for imaging thick objects that cannot be transilluminated (such as skin tumors, cells in live mice, or circulating blood cells using the handheld CytoScan from Rheologics, http://www.Rheologics.com), or on microscope slides with monochromatic polarized illumination and a crossed polarizer for imaging highly scattering silver grains or immunogold. This latter configuration has high contrast and is commercially available from microscope and filter companies as an IGS filter cube (immunogold/silver staining). Because the excitation side polarizer only reduces excitation by 50%, a custom IGS cube has been made for the authors by Chroma Technology that has UV-green excitation plus polarizer, a 10% reflection/90% transmission 45° beam splitter, and green-NIR plus cross-polarizer emission filter. This custom IGS can be used in standard IGS mode with green (546 nm) exciter, or with UV exciter for quantum dot fluorescence, or with Nomarski DIC transmitted light where the emission polarizer serves as the DIC analyzer. This has the additional advantage of automating the DIC system on the authors Leica RMRXA/RF8 (8-filter cube turret) microscope while eliminating the pixel shift of many μm introduced by the standard Leica DIC analyzer slider. The single-pass exciter filters were chosen to also enable single channel imaging with a triple DAPI/Fluorescein/Cy3 filter set in the same microscope. The custom IGS cube was inspired by the nonreflective reflector fluorescence filter cube of Sawano et al. (2002).
Figure 4.4.1.
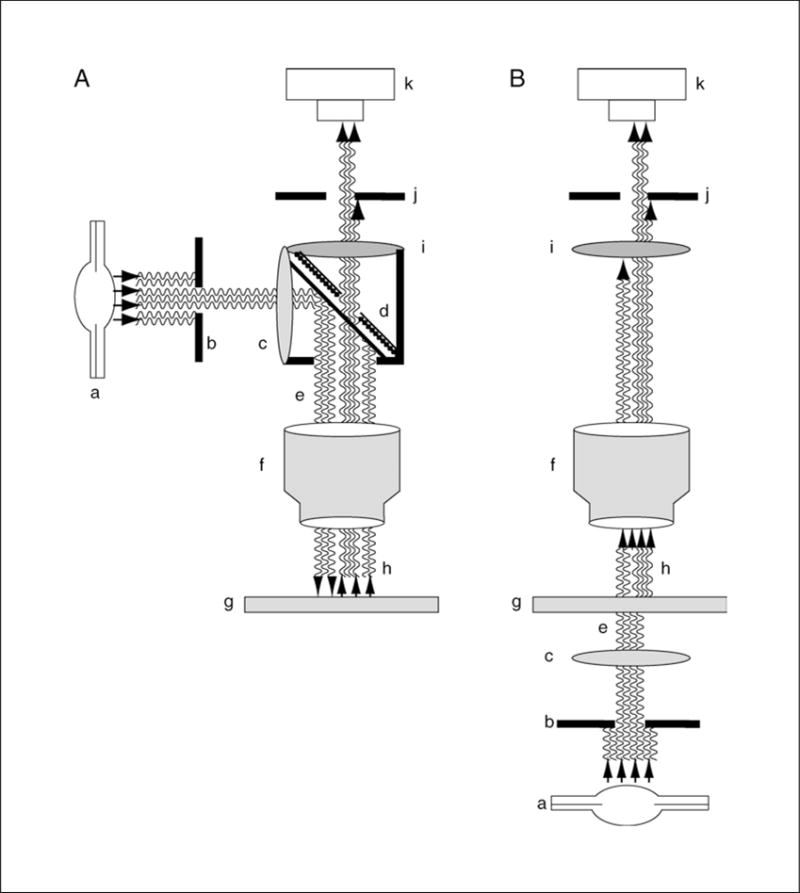
The concepts of (A) incident and (B) transmitted light as applied to fluorescence microscopy. Many of the major components are similar to those used with either type of bright-field microscopy (with the elimination of the filters which are labeled c and I here). Indicated in the diagrams are the (a) light source, (b) stage condenser, (c) excitation filter, (d) dichroic mirror, (e) selected excitation wavelength, (f) objective, (g) microscope slide with specimen, (h) reflected (in A) or transmitted (in B) illumination (short wave; blue) and emitted fluorescence (long wave; red), (i) barrier or emission filter, (j) eyepiece condenser, and (k) eyepiece or camera.
Oblique illumination, common on stereomicroscopes with a ring light or fiberoptic gooseneck, is also possible with microscope slides, using the DarkLite illuminator (Micro Video Instruments, Inc., Avon, MA, http://www.mvi-inc.com) to illuminate through the edges of the slide. The DarkLite illuminator provides barely enough light for imaging silver particles and tissue by scattered light, but is not suitable for fluorescence when used with its standard light bulb. A special type of fluorescence excitation, total internal reflection fluorescence (TIRF), uses the glass surface that the cells or molecules are on as a light guide. Essentially, the light is trapped in the high-refractive-index glass; the same principle is used in fiber optics in the communication industry. In TIRF, the light is trapped propagating parallel to the glass surface, but fluorophores within ~100 nm in the low-refractive-index mounting media (i.e., cell culture fluid) are excited by the evanescent wave in the latter medium. The advantage of TIRF is that only fluorophores very close to the glass-mounting medium interface are excited. This can include the basal plasma membrane of cells in close apposition to the interface. The evanescent wave energy falls off exponentially with distance into the mounting medium, and the depth can be controlled by adjusting the angle of light entering the glass slide. TIRF can be set up using a prism to couple the excitation light into the glass, or a high-numerical-aperture (NA) objective lens can be used, with the illumination restricted to the high-NA portion. Special TIRF lenses are commercially available from Zeiss (NA = 1.45) and Olympus (NA = 1.45 or, with special expensive coverglass and toxic immersion media, NA = 1.65). See reviews by Oheim (2001), Axelrod (2003), or Jaiswal and Simon (2003) for additional details on TIRF.
Köhler illumination (APPENDIX 3N) results in an evenly lit field of view and is used for all light microscopy. Epi-fluorescence microscopes use the objective lens for both excitation and emission. In The excitation optical path of current fluorescence microscopes are equipped with field and numerical apertures placed at appropriate conjugate optical planes for imaging. As long as they are reasonably well centered and the numerical aperture fully opened to maximize brightness, epi-Köhler illumination is assured. For a few specific applications, such as high magnification, high-numerical-aperture imaging of single metaphase spreads or interphase nuclei for FISH, the field aperture can and should be reduced to just outside the diameter of the object of interest. This will reduce glare arising from excitation of autofluorescent mounting media in the area around the object, dramatically improving contrast. Using fresh mounting medium and optimizing FISH wash steps will also reduce auto-fluorescence.
With respect to trans-Köhler illumination, there are many terms associated with microscopy that indicate the direction of the incoming light with respect to the angle at which it intercepts the sample and the side of the sample through which it first passes (Fig. 4.4.1). Bright-field microscopy (APPENDIX 3N) is the most commonly used light-microscopic technique, and is the basis for phase contrast, polarization, and Nomarski DIC contrast methods. As the name indicates, in bright-field microscopy, surrounding background is bright and the object is dark. The objects may be dark because of their scattering properties, endogenous pigments, or exogenous dyes. Light from the illumination source is transmitted along a pathway parallel to the optical axis directly through the sample into the objective. This works well with samples that scatter well, or that are naturally (i.e., chloroplasts) or artificially (i.e., Giemsa) colored. As light encounters the specimen, the intensity (or amplitude) is reduced compared to its surroundings, resulting in a darker appearance. The location of the illumination source defines the two types of bright-field microscopy. In transmitted-light bright-field microscopy, the illumination source is directly below the sample for an upright microscope. As such, the light passes through the sample only once on its way from the source to the objective. Köhler illumination is the standard configuration for transmitted illumination of nearly all biomedical microscopes. However, because the condenser can be lowered or raised and moved laterally, it is up to the user to adjust the condenser field aperture centration and focus and numerical aperture setting. Because many samples do not contain sufficient absorption properties to be discerned with normal bright-field microscopy, one can generate contrast and reveal structures with low resolution by slightly rotating the condenser turret. This technique is referred to as oblique, anaxial, or asymmetric illumination contrast (Kachar, 1985).
Both transmitted illumination and incident illumination (epi-illumination) can be used for fluorescence. Older microscopes may use transmitted fluorescence illumination (trans-fluorescence), which was pioneered a century ago by Köhler and von Rohr. Trans-fluorescence with a bright arc lamp could be hazardous to the user if full-intensity, unfiltered light was transmitted through the eyepieces. Trans-fluorescence went out of fashion in the decades after Bas Ploem introduced the Ploem-Pak filter cube (Ploem, 1967), using a wavelength-selective excitation filter, a 45° dichroic beam splitter, and emission filter. However, with modern interference filters for both excitation and emission—eliminating the need for a beam splitter or cube assembly—trans-fluorescence has uses, especially with a high-numerical aperture condenser (Tran and Chang, 2001). Trans-fluorescence may be particularly useful on microscopes that have an arc lamp with a wavelength controller, i.e., filter wheel, shutter, and the Ellis light scrambler (fiber optic or liquid light guide to homogenize illumination) for DIC or other high-resolution microscopies (Reitz and Paliaro, 1994; Inoué and Spring, 1997). With a high sensitivity CCD camera, and interference filters, even a standard tungsten-halogen lamp could serve as a light source for visible and near infra-red excitation (these lamps produce little UV, however, making them practically useless for DAPI or BFP excitation). Trans-fluorescence may be particularly useful for thick specimens, such as mouse brain slices, combined with large-Stokes-shift dyes, the idea being that the shorter-wavelength excitation light can penetrate through only part of the specimen, whereas the longer-wavelength NIR fluorescence emission can get through the specimen to the objective lens and detector. This could complement the infrared DIC-videomicroscopy method of Dodt and Zieglgansberger (1994).
Dark-field microscopy can be used to obtain resolution of objects or features that are normally below the resolution of the light microscope. This is only possible with transmitted illumination, because no direct light is allowed to enter the objective. Only incoming light diffracted, refracted, or reflected by the specimen enters the objective, resulting in bright objects on a dark background. One note of importance is that the numerical aperture of the condenser must be higher than that of the objective. This technique is incompatible with the use of phase-contrast microscopy in association with fluorescence. Dark-field microscopy also requires scrupulously clean optics and slides, because any dirt will cause light to be scattered into the objective and mar the image quality. This is because dark-field microscopy has an effectively large depth of field, and many microscope optics accumulate dust and dander if the microscope is not sealed (in order to allow heat to escape). Conversely, the large depth of field could be useful for tracking objects in relatively large volume, though most of the objects will appear out of focus (making them larger, which could in turn improve the centroid XY location precision by image measurement of the larger object).
Differential interference contrast (DIC; APPENDIX 3N; Allen et al., 1969) can be performed at high numerical apertures, gives better resolution than dark-field microscopy, and can be used in conjunction with fluorescence microscopy and live-cell imaging. DIC is referred to as an optical-sectioning technique, because for transparent objects, soon after the image leaves the focal plane it disappears. The DIC technique involves polarized light plus a pair of matched Wollaston prisms (Nomarski DIC) or similar light path splitters (Smith DIC). The incoming light is first passed through a polarizing filter that only allows waves oriented in the same direction to pass through the filter. Each plane-polarized light beam is then split into two separate beams containing perpendicularly oscillating components with a Wollaston prism (composed of two quartz prisms cemented together with their optical angles oriented at 90° with respect to each other). Thickness and refractive-index differences within the specimen generate opposing phase shifts in the two halves of the split beam. A second Wollaston prism placed after the objective recombines the halves of each split beam. Constructive or destructive interference occurs as a result of the phase shift between the two separate beams. The light then passes through another polarizing filter (analyzer) and is visualized as differences in gray-scale levels across the specimen. The bas-relief of DIC is due to differences in refractive index, and not the three-dimensional topography of the specimen. Allen et al. (1981a,b), and Inoué (1981) independently introduced the advantages of video enhanced polarization (VEC-Pol) and differential interference contrast (VEC-DIC) for imaging unstained cells. Holzwarth et al. (1997, 2000) have described polarization-modulation DIC hardware that switches the bas-relief with each image, followed by an image-difference (plus offset) mathematical operation to double the edges. As anyone who has worn polarized sunglasses knows, polarizers can reduce glare. In light microscopy, polarized light is used for imaging birefringent structures such as chromosomes and/or mitotic spindles (Inoué and Dan, 1951; see also Inoué and Spring, 1997).
Phase-contrast microscopy (APPENDIX 3N) is an alternative to DIC that converts normally invisible phase changes as light propagates into and out of interfaces of different refractive indices (e.g., culture medium versus plasma membrane, plasma membrane versus cytoplasm, cytoplasm versus organelle membrane, or organelle membrane versus organelle contents) or through thick slabs of such contents (e.g., thin versus thick cytoplasm). Phase contrast is an excellent mode for reviewing metaphase spreads on microscope slides before deciding on what areas and on which slides to carry out FISH or Giemsa staining. The invention of phase-contrast microscopy was justly honored with a Nobel Prize (Zernike, 1955). Zernike pointed out the irony that in bright-field imaging of ultrathin, transparent objects, the best focus makes the object invisible. He recognized that the light propagating through the object would delay (or advance) the wave, and worked out a way to convert phase to intensity. A phase annulus (ring) in the condenser allows only a ring of light to reach the condenser. Focusing of this ring by the condenser lens generates a hollow cone of light that is projected onto the back focal plane of the objective. Some of the light waves are retarded as they pass through the sample. This is due to absorptive differences among cellular structures and differences in refractive index or thickness. As a result, their phase is shifted relative to those waves from the original light source, which have not encountered phase-dense objects. These phase shifts are usually not sufficient, however, to generate full constructive or destructive interference visible with normal bright field microscopy. The standard DL phase ring in the back focal plane of the objective absorbs 70% to 80% of the nondiffracted rays. In the absence of an object, this only has the effect of reducing brightness slightly. The phase ring crucially shifts the phase by one quarter of the wavelength for the diffracted rays. This arrangement alters the amplitude and phase relationships of the diffracted versus nondiffracted light, thereby enhancing the contrast. Regions with a higher refractive index usually appear darker, with the standard DL (dark-light) design. Objects with too high a refractive index or thickness can result in a rather large phase shift and cause a contrast reversal (i.e., a positive phase shift of 1.5 λ would appear identical to a negative phase shift of 0.5 λ). The phase ring is made with monochromatic light of 546 nm (a mercury arc lamp line), so best results are achieved with a monochromatic green filter, though satisfactory results are obtained with a fairly broad-pass green interference filter (GIF). Either green filter will also enhance contrast of Giemsa stained chromosomes or nuclei, and can be useful for imaging hematoxylin and other absorption dyes.
Microscope manufacturers also offer limited numbers of special phase-contrast lenses, such as the DM (dark-medium) design. A key advantage for phase contrast when imaging single cells by eye is that, with the DL design, a bright phase halo surrounds the dark cell, making the invisible visible. However, the same halo results in difficulties for precise image analysis of the cells, especially in ultrathin regions such as lamellipodia or filopodia, where the contrast of edge of the cell and halo disappear due to the weakness of the phase difference. The phase ring in the objective lens absorbs some of light. This is not an issue for phase contrast bright-field illumination, where even a tungsten-halogen lamp provides plenty of light, but the ring does reduce somewhat the intensity of fluorescence.
Another technique, quantitative phase amplitude microscopy (QPm), a mathematical method using three (or more) bright-field images of different focus positions, was described by Barone-Nugent et al. (2002). QPm can be performed with either conventional bright-field microscope images or with the “free” (but not confocal optical sectioning) transmitted-light image of a confocal microscope (Cody et al., 2005), using the appropriate software. QPm can be done as off-line post processing, and produces quantitative phase maps that are independent of any absorbing features present in the specimen (provided that the entire specimen is not black). The phase maps can be used directly (refractive index measurement) or can be recombined in a user-adjustable manner with the absorbing features. The phase maps can also be digitally converted into Nomarski, DIC, or phase-contrast-like images, with or without absorbing features. The authors of this unit have not seen QPm used on metaphase chromosomes or cell nuclei, but the method may produce interesting chromosome maps or a novel form of nuclear-texture analysis. Hopefully, in the future, the refractive-index measurements can be combined with digital deconvolution algorithms to improve 3-D deconvolution fluorescence microscopy.
There are many occasions when it is useful to follow the movement of cells or their organelles as a function of time. Studies of cell division, movement of chromosomes and centrosomes, and the polymerization of mitotic tubules are a few examples. Such analysis involves the successive microscopic imaging of live cells, rather than a single image of a fixed specimen. There are two different methods for accomplishing such an analysis. The first, time-lapse microscopy, involves the acquisition of individual images at distinct time points or intervals (e.g., every 10 min over an 8-hr period). These images can then be integrated into a single composite image and displayed simultaneously for an easy comparison of changes as a function of time. Video microscopy, however, involves near-continuous imaging over a prolonged time period, as one would do with a standard video camera that has a rate of 30 frames/sec. If continuous observation is not required, a computer-controlled shutter can be added to the system and a few video images may be captured and averaged, or a single exposure with a digital CCD camera can be captured on demand. A thorough treatment of this technique, which is beyond the scope of this chapter, can be found in Spector et al. (1998), Goldman and Spector (2004), or Yuste and Konnerth (2005).
MICROSCOPE OBJECTIVES AND EYEPIECE LENSES
The compound light microscope must be equipped with the highest-quality optics (objectives and eyepieces), must be precisely aligned, and must have the proper filters installed to observe and record all relevant information from the objects under study. The steps required to align the microscope are outlined in the detailed manual available from the manufacturer of the respective microscope and in APPENDIX 3N of Current Protocols in Human Genetics. The importance of proper alignment cannot be overstated. An excellent discussion of microscopy and photography is presented in The ACT Cytogenetics Laboratory Manual (Barch et al., 1997) and in Human Cytogenetics: A Practical Approach (Rooney and Czepulkowski, 1992). Both references are invaluable resources for the cytogenetics laboratory. High-quality objective lenses are critical for obtaining maximum information in the study of biological specimens. Lenses condense light and magnify the image. The objective lens system must have high resolving power and correction for lens aberrations. The resolving power, R, of a lens is defined as the minimum distance by which two luminous points can be separated and still be discerned as distinct objects using that objective. R is described by the theory of optical diffraction as:
where λ is equal to the wavelength of the incident light and NA is the numerical aperture, a measure of the light cone entering the objective at the fixed objective distance (James and Tanke, 1991; Rawlins, 1992). The value of NA is given by:
with n equal to the refractive index of the medium between the objective and the sample, and α equal to half the vertical angle of the light cone (Fig. 4.4.2). The NA is restricted for technical reasons to a maximum of 1.35 to 1.40 for glass objectives and oil-immersion media (refractive index, 1.515) and is usually indicated on the side of the objective. High NA results in the smallest lateral resolution, smallest axial resolution, and maximum capture of light photons. This is particularly important in fluorescence microscopy, where the amount of emitted light is often very small. The brightness of the captured light is affected by many different parameters, including the concentration of the fluorophore, the transmission of light through the optics, the total magnification, and the numerical aperture of the objective (and the condenser, in the case of transmitted fluorescence). The relative image brightness in epi-fluorescence microscopy is given by the following equation:
When the so-called “object space” between the objective and cover glass contains air (as with a “dry” objective), the numerical aperture cannot exceed 0.95. However, when immersion oil with a refractive index of 1.515 is used between the two surfaces, an NA of 1.35 to 1.40 can be obtained. This is because the refractive index of the oil is identical to that of the glass slide, coverglass, and objective. This prevents the light from being refracted as it passes from the specimen through these other materials. Immersion media include various natural and synthetic oils (with varying n values), water (n = 1.333), and glycerol (n = 1.466 for 100% glycerol, 1.391 for 40% glycerol:60% water, at 23°C). Immersion objectives are usually produced for use with a specific type of immersion medium and are so labeled on the side of the objective; “dry” objectives will not function as immersion objectives. Immersion oil with low fluorescence is required for fluorescence microscopy, and the objective manufacturer can help obtain the proper type.
Figure 4.4.2.
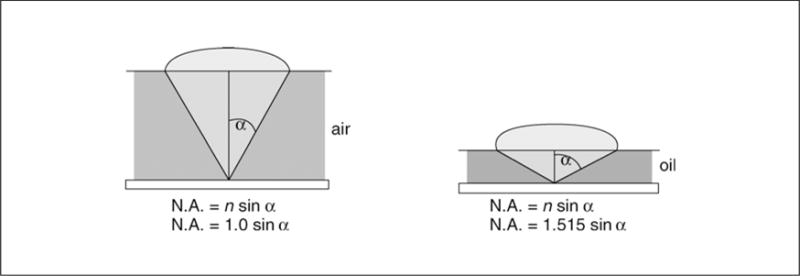
Diagrammatic explanation of numerical aperture. As α approaches 90°, sin α approaches 1.0. The refractive index of the medium between the sample and the objective is designated as n.
In addition to resolving power, which is a function of both magnification and numerical aperture, modern light microscope objectives must correct for problems of spherical and chromatic aberration. Spherical aberration is produced by failure of the curved surface(s) of a lens to direct all light rays passing through the lens to the same focal point. A cover glass of the incorrect thickness or a refractive-index mismatch (i.e., wrong immersion oil) can also cause spherical aberration and an inability to focus. Early microscopes (single or compound lenses) suffered from loss of fine detail due to a chromatic aberration, which resulted in rings of color around small objects. White light passing through the lens is broken up into its constituent colors. Different wavelengths are diffracted to different extents, and hence have different focal points. After 1820, achromatic lenses were developed, allowing great advances in biology and medicine (Kapitza, 1996; Inoué and Spring, 1997). Achromatic objectives are rather simple in that spherical aberration is corrected for the middle range of the light spectrum, thereby directing all broken-up wavelengths to the same focal point. Plan-achromatic objectives are more complex and have the advantage of less curvature-of-field aberration than ordinary achromatic objectives. Curvature of field is caused when light passing through the periphery of the objective is focused closer to the back focal plane of the lens than light passing through the center. The result is a discrepancy in focal plane between the center and periphery of the field of view. Plan-apochromatic objectives are costly, complex, flat-field objectives that offer the greatest correction for chromatic and spherical aberration. The type of correction supplied by the objective is also indicated on its side. In the past, fluorite (synthetic quartz) and plan-fluorite lenses were required for deep-UV excitation, e.g., for 340-nm excitation of Fura-2, because apochromat lenses had many UV-absorbing glass components. Modern (post-1997) plan-apochromat lenses often, but not always, transmit light down to 300 nm. If this may be important, as with Fura-2, or to truly maximize quantum dot excitation, transmission curves should be obtained from the manufacturer, or several lenses should be tested in the laboratory. As biologists expand their desired color palette, wanting to use microscopes for imaging specimens from UV (<380 nm) to near infrared (Cy5.5, Cy7, NIR quantum dots, etc) and to excite with UV (e.g., 337-nm nitrogen laser dissection) to near-infrared (two-photon and three-photon excitation, 600 to 1200 nm) lasers, it is being discovered that apochromat often means visible light only. Manufacturers are starting to supply lenses that match the present expanded palette. The addition of special accessories, such as the OptiGrid (Thales Optem, http://www.thales-optem.com) or Apotome (Zeiss), that project a grid pattern (placed at the epi-illumination field aperture) onto the specimen, quickly reveal how unchromatic most current research epi-illumination light trains are.
FLUORESCENCE MICROSCOPY
Many molecules, especially those with conjugate planar rings (e.g., benzene) absorb light. The larger the planar ring structure, the longer the wavelengths of light that can be absorbed. At room temperature, the molecules are at various vibration energies, which result in broadening of the spectra. Some molecules absorb light, boosting an outer shell electron from its ground state (S0) to the first excited singlet state (S1) and efficiently lose the acquired energy to surrounding molecules, such as water, and thus return to the ground state (S0). Other molecules absorb light energy at one wavelength and either lose all the energy to surrounding molecules or lose a part of the energy to the surroundings, but emit most of the energy as light (fluorescence) at a longer wavelength. Considering one photon and one molecule, the absorption event (now also referred to as excitation) occurs in femtoseconds (fsec; sec), the initial vibration energy loss occurs in picoseconds (psec; 10−12 sec), while fluorescence occurs in the nanosecond range (nsec; 10−9 sec). All compounds that absorb in the visible light range, such as Perkin’s mauve, are called chromophores. Those molecules that efficiently emit photons, whether in the UV (tryptophan), visible (fluorescein), or near infrared (Cy5.5), are termed fluorochromes or fluorophores. The efficiency with which molecules absorb light is characterized by the extinction coefficient, ε, whose units are M−1cm−1. The efficiency of fluorescence is the ratio of light emitted divided by light absorbed, which is termed quantum yield (QY). A convenient way to compare fluorophores is to calculate the brightness index (BI) of each, where BI = ε × QY/1000. For green-fluorescing dyes, e.g., the xanthene dye, fluorescein, ε = 90,000 M−1cm−1 and QY = 0.92 (both for pH > 8), resulting in BIfluorescein = 82.8. The cyanine dye Cy2 (Molecular Probes), has a higher extinction coefficient, ε = 150,000 M−1cm−1 and QY = 0.12 (relatively pH-independent), resulting in BICy2 = 18. A more useful cyanine dye is the orange-fluorescing Cy3 (also from Molecular Probes), with ε = 150,000 M−1cm−1 and QY = 0.15 (relatively pH-independent), resulting in BICy3 = 22.5. A newer, rigidized cyanine dye from Molecular Probes, Cy3B, has very similar extinction coefficient of ε = 120,000 M−1cm−1 but with a QY = 0.67, Cy3B’s BICy3B = 87.1. Table 4.4.1 summarizes the performance of many useful fluorophores.
Table 4.4.1.
Fluorophore Photophysics Dataa
| Fluorophore | Absorbance or emission maximum (nm) | Emission max (nm) | Extinction coefficient | Quantum yield | Brightness indexb |
|---|---|---|---|---|---|
| Acridine orange | 271 | 520 | 27,000 | 0.20 | 5.4 |
| Alexa Fluor 488 | 495 | 519 | 71,000 | 0.94 | 66.7 |
| Alexa Fluor 532 | 532 | 553 | 81,000 | 0.80 | 64.8 |
| Alexa Fluor 546 | 556 | 573 | 104,000 | 0.96 | 99.8 |
| Alexa Fluor 568 | 578 | 603 | 91,300 | 0.75 | 68.5 |
| Alexa Fluor 594 | 590 | 617 | 73,000 | 0.64 | 46.7 |
| Allophycocyanin | 650 | 660 | 700,000 | 0.68 | 476.0 |
| Atto 520 | 525 | 547 | 105,000 | 0.95 | 99.8 |
| Atto 532 | 534 | 560 | 115000 | 0.90 | 103.5 |
| Atto 565 | 566 | 590 | 120,000 | 0.97 | 116.4 |
| Atto 590 | 598 | 634 | 120,000 | 0.90 | 108.0 |
| Atto 610 | 616 | 646 | 150,000 | 0.70 | 105.0 |
| Atto 620 | 620 | 641 | 120000 | 0.50 | 60.0 |
| Atto 635 | 637 | 660 | 120000 | 0.45 | 54.0 |
| Atto 655 | 655 | 680 | 125,000 | 0.50 | 62.5 |
| Atto 680 | 675 | 699 | 125,000 | 0.40 | 50.0 |
| ATTO-Dino 1 (dsDNA) | 490 | 531 | 179,000 | 0.70 | 125.3 |
| ATTO-Dino 2 (dsDNA) | 506 | 535 | 162,000 | 0.70 | 113.4 |
| BODIPY 507/545 | 513 | 549 | 82800 | 0.73 | 60.4 |
| BODIPY FL | 504 | 510 | 70,000 | 0.90 | 63.0 |
| BODIPY TR | 588 | 616 | 68000 | 0.84 | 57.1 |
| B-phycoerythrin | 545 | 575 | 2,410,000 | 0.98 | 2,361.8 |
| Calcein | 494 | 516 | 81000 | 0.78 | 63.2 |
| Cascade Blue | 378 | 423 | 26,000 | 0.54 | 14.0 |
| Coumarin 6 | 456 | 500 | 54,000 | 0.78 | 42.1 |
| Cresyl violet perchlorate | 603 | 622 | 83,000 | 0.54 | 44.8 |
| Cy3 | 552 | 570 | 150,000 | 0.15 | 22.5 |
| Cy3B | 552 | 570 | 130,000 | 0.67 | 87.1 |
| Cy5 | 649 | 670 | 250,000 | 0.28 | 70.0 |
| Cy5.5 | 675 | 694 | 250,000 | 0.23 | 57.5 |
| Cy7 | 755 | 778 | 250,000 | 0.28 | 70.0 |
| DAPI (in DMSO) | 353 | 465 | 27,000 | 0.58 | 15.7 |
| DAPI (in H2O) | 344 | 487 | 27,000 | 0.04 | 1.2 |
| DsRed | 558 | 583 | 75,000 | 0.70 | 52.5 |
| Eosin Y | 525 | 543 | 112,000 | 0.67 | 75.0 |
| EYFP | 514 | 527 | 84,000 | 0.61 | 51.2 |
| Fluorescein | 490 | 514 | 90000 | 0.92 | 82.8 |
| FM 1-43 | 479 | 598 | 40000 | 0.30 | 12.0 |
| Hoechst 33258 (in DMF) | 354 | 486 | 46,000 | 0.35 | 16.1 |
| Hoechst 33258 (in H2O) | 345 | 507 | 46,000 | 0.03 | 1.6 |
| IRDye38 | 778 | 806 | 179,000 | 0.35 | 61.8 |
| IRDye40 | 768 | 788 | 140,000 | 0.38 | 53.2 |
| IRDye700 | 681 | 712 | 170,000 | 0.48 | 81.1 |
| IRDye78 | 768 | 796 | 220,000 | 0.31 | 68.2 |
| IRDye80 | 767 | 791 | 250,000 | 0.21 | 52.5 |
| IRDye800 | 787 | 812 | 275,000 | 0.15 | 41.3 |
| JOE | 520 | 548 | 73,000 | 0.60 | 43.8 |
| Lucifer Yellow CH | 230 | 542 | 24,200 | 0.21 | 5.1 |
| merocyanine 540 | 559 | 579 | 138,000 | 0.39 | 53.8 |
| neo-Cy5 (DMSO) | 656 | 675 | 195,000 | 0.25 | 48.8 |
| NIR1 | 761 | 796 | 268,000 | 0.23 | 61.6 |
| NIR2 | 662 | 684 | 250,000 | 0.34 | 85.0 |
| NIR3 | 750 | 777 | 275,000 | 0.28 | 77.0 |
| NIR4 | 650 | 671 | 260,000 | 0.43 | 111.8 |
| Oregon Green 488 | 496 | 516 | 76,000 | 0.90 | 68.4 |
| Oregon Green 514 | 506 | 526 | 88000 | 0.96 | 84.5 |
| Oyster – 645 (ethanol) | 651 | 669 | 250,000 | 0.40 | 100.0 |
| Oyster – 656 (ethanol) | 665 | 684 | 220,000 | 0.50 | 110.0 |
| Perylene | 253 | 435 | 38,500 | 0.94 | 36.2 |
| Phenylalanine | 222 | 279 | 195 | 0.02 | 0.0 |
| POPOP | 256 | 407 | 47,000 | 0.93 | 43.7 |
| Quinine sulfate (in 0.5M H2SO4) | 256 | 451 | 5,700 | 0.55 | 3.1 |
| Rhodamine 110 | 496 | 520 | 80000 | 0.89 | 71.2 |
| Rhodamine 6G | 530 | 552 | 116,000 | 0.95 | 110.2 |
| Rhodamine B | 543 | 565 | 106,000 | 0.70 | 74.2 |
| Rose bengal | 559 | 571 | 90,400 | 0.11 | 9.9 |
| R-Phycoerythrin | 480 | 578 | 1,960,000 | 0.68 | 1,332.8 |
| SNIR1 | 666 | 695 | 218,000 | 0.24 | 52.3 |
| SNIR3 | 667 | 697 | 245,000 | 0.24 | 58.8 |
| Sulforhodamine 101 | 576 | 591 | 139,000 | 0.90 | 125.1 |
| Texas Red | 586 | 605 | 108000 | 0.77 | 83.2 |
| Texas Red-X | 583 | 603 | 116,000 | 0.90 | 104.4 |
| TMR | 540 | 565 | 95000 | 0.68 | 64.6 |
| Trp | 287 | 348 | 6000 | 0.31 | 1.9 |
| Tyr | 275 | 303 | 1500 | 0.21 | 0.3 |
Abridged from a 4700+ entry data table Excel file posted on the Internet by the authors(http://home.earthlink.net/~fluorescentdyes/). The Web site also contains a 400+ entry fluorescent proteins data table Excel file. Spectra for many of these dyes, and commercial filters and light sources, are available through an interactive website at http://www.mcb.arizona.edu/ipc/fret/default.htm.
Brightness Index = (extinction coefficient × quantum yield)/1000.
Of the conventional fluorophores, B- and R-phycoerythrin, and allophycocyanin stand out. These molecules are very useful in flow cytometry because they exhibit bright fluorescence, excite well with the typical 488-nm laser line, and can serve as efficient FRET donors to near-infrared dyes. However, all three are based on multiple dyes in protein complexes and photobleach very rapidly in fluorescence microscopy. DAPI and Hoechst 33258 (H33342 is similar) fluoresce poorly as free dyes in aqueous media—which is a good thing for keeping the background low—and have substantially higher brightness indices in non-aqueous solvents. Both of these dyes bind well to DNA, resulting in high local concentrations, and the DNA shields these two fluorophores from water, enhancing fluorescence. As a consequence, while not having exceptional photophysics, DAPI and the Hoechst dyes are brilliant DNA counterstains. DAPI only fluoresces in AT-rich DNA, which results in the characteristic R-banding pattern.
A new class of products, fluorescent nanocrystals, or quantum dots, are highly photostable, high-absorbing molecules (typically CdSe, CdS, CdTe, and/or ZnS) with good quantum yield, encased in a biocompatible coating. These can be coupled to streptavidin and antibodies, as well as to other proteins and haptens such as biotin (Bruchez et al., 1998; Chan and Nie, 1998). Table 4.4.2 summarizes the performance of commercially available quantum dots.
Table 4.4.2.
Quantum Dots Photophysics Dataa
| Product | Emission peak | Emission full width half maximum | Extinction coefficient | Quantum yield | Brightness index |
|---|---|---|---|---|---|
| QD525a | 525 | ≤32 | 320,000 | 0.60 | 192 |
| QD565a | 565 | ≤34 | 1,100,000 | 0.40 | 440 |
| QD585a | 585 | ≤34 | 2,200,000 | 0.40 | 880 |
| QD605a | 605 | ≤27 | 2,400,000 | 0.40 | 960 |
| QD655a | 655 | ≤34 | 5,700,000 | 0.40 | 2280 |
| QD705a | 705 | wide | |||
| 567 nm – water (Larson)b | 1,100,000 | 0.58 | 638 | ||
| 581 nm – water (Larson)b | 2,200,000 | 0.68 | 1,496 | ||
| 585 nm – water (Larson)b | 2,200,000 | 0.37 | 814 | ||
| 605 nm – water (Larson)b | 2,400,000 | 0.71 | 1,704 | ||
| 609 nm – water (Larson)b | 2,400,000 | 0.66 | 1,584 | ||
| 645 nm – water (Larson)b | 5,700,000 | 0.35 | 1,995 | ||
| 646 nm – water (Larson)b | 5,700,000 | 0.37 | 2,109 | ||
| Lake Placid Bluec,d | 490±10 | <30 | 100,000 | ||
| Adirondack Greenc,d | 520±10 | <30 | 130,000 | ||
| Catskill Greenc,d | 540±10 | <30 | 160,000 | ||
| Hops Yellowc,d | 568±10 | <30 | 200,000 | ||
| Birch Yellowc,d | 580±10 | <30 | 240,000 | ||
| Fort Orangec,d | 598±10 | <30 | 300,000 | ||
| Maple Red-Orangec,d | 620±10 | <30 | 450,000 | ||
| McIntosh Redc,e | 620±10 | <40 | 200,000 | ||
| Cortland Redc,e | 640±10 | <35 | 220,000 | ||
| Rome Redc,e | 660±10 | <35 | 280,000 | ||
| Empire Redc,e | 680±10 | <35 | 330,000 |
Data from Quantum Dot Corp.
From Larson et al. (2003). Larson quantum dots are supplied by Quantum Dot Corp. The Larson et al. extinction coefficients are adapted here from the equivalent QDot product.
Evident Technologies products (Lake Placid Blue to Empire Red). Emission peaks are ±10 nm.
Lake Placid Blue through Maple Red-Orange are CdSe/ZnS (core/shell), ranging from 2.0 to 5.0 nm shell diameter;
McIntosh Red to Empire Red are CdTe/ZnS, ranging from 4.0 to 5.2 nm shell diameter. The actual diameter of capped and functionalized products may be substantially larger.
In order to grasp the principles of fluorescence, it is necessary to further understand the laws describing light. Energy behaves in accordance with Planck’s law, which states:
where E is energy, h is Planck’s constant, v is the light frequency, c is the velocity of light, and λ equals light wavelength. Thus, energy is linearly proportional to the light frequency and inversely proportional to its wavelength. The quantum of energy (E) is greater for radiations of shorter wavelengths, such as UV, than for radiations of longer wavelength, such as infrared. Wavelengths in the UV spectrum (300 to 380 nm), visible light spectrum (380 to 700 nm), and near infrared spectrum (700 nm to 1000 nm) are nowadays used in fluorescence. The spectral characteristics of individual fluorophores and fluorescent proteins are dependent upon the regions of the light spectrum where absorption (excitation) and emission of light energy occur. Stokes’ law states that the average wavelength of emitted fluorescence is longer than the average excitation wavelength for any given fluorophore. The longer wavelength of the emitted light is due to the rapid loss of some vibrational energy in the first picoseconds post-absorption (with a spectral confocal microscope and bright fluorescent objects, the anti-Stokes emission can be detected—however, this is a small fraction of the Stokes emission). Multi-photon excitation fluorescence works by having two low-energy photons, e.g., 800-nm wavelength each, reach the molecule at the same time (within femtoseconds of one another). This results in an excitation event equivalent to absorbing an ~400-nm photon and fluorescence practically identical to that of one-photon excitation. At the quantum-mechanical level, the selection rules for 1-, 2- and 3-photon excitation do differ, which leads to some dyes working poorly and others—such as quantum dots—exceptionally well, for multi-photon excitation.
DAPI (4,6-diamino-2-phenylindole) is a fluorescent dye with affinity for A-T residues in DNA. This is often used to give a chromosome banding pattern (R-banding) complementary to that obtained with Giemsa staining (UNIT 4.2). A simple digital image-processing step, “inverse contrast,” is thus able to transform a DAPI-stained metaphase spread into a Giemsa-like staining pattern. An additional sharpening step then improves the banding to near G-band quality. It is also useful as a DNA counterstain for interphase nuclear studies, where, for example, flat tissue culture cell nuclei can be identified as human or mouse on the basis of dark nucleoli versus bright centromeres, respectively (the comparison only works for human versus Mus musculus nuclei; other species, e.g., bovine and other mice, have other structures). The energy absorbed at 350 nm raises an electron to a higher excitation state. Some of the energy is then lost to vibration. When the electron falls back to its starting, or ground state, the energy is released as fluorescent light. Because the amount of energy is less than the input, the emitted light has a longer wavelength (the mode being 470 nm) and is said to be red-shifted. With fluorescence lifetime spectral imaging it is possible to demonstrate quantitatively that the molecules that emit rapidly (1 ns) tend to emit shorter wavelengths (higher energies) than those that emit slowly (5 ns). The latter are red-shifted because they have had more time to lose more vibrational energy to the surrounding molecules. Fluorophores can also give up some or all of their energy to other molecules. Many excited fluorophores can have the excited electron transition from the first excited singlet state (S1) to the slightly lower energy first triplet state (T1). This is referred to as intersystem crossing. The triplet state can be very long-lived, lasting for microseconds (10−6 sec) to milliseconds (10−3 sec), after which delayed fluorescence (if the molecule flips back to the S1 state) or phosphorescence (emission directly from the T1 to the ground state) can occur. However, when in the T1 state, the energy may be transferred to a nearby, normally triplet-state oxygen molecule (O23), which flips to the highly reactive singlet state (O21). The singlet oxygen molecule can convert to an oxygen radical, all of which are highly reactive. Since the fluorophore and the reactive oxygen molecule are in close proximity, they often react, resulting in destruction of the fluorophore. Sometimes it is possible to take advantage of this reaction, as in the reaction of superoxide with nonfluorescent hydroethidine to make a fluorescent ethidium dye (Haugland, 2004; also see http://www.probes.com/handbook/). More generally, an excited fluorophore may donate its energy to another molecule, the acceptor, which, if it is an efficient fluorophore, may then emit at its characteristic wavelength. This process, is nonradiative (the light is not emitted and then re-absorbed), and, when the transfer occurs with an efficiency that falls off as a function of distance to the sixth power, is termed Förster-type fluorescence resonance energy transfer (FRET; Förster, 1965; Jares-Erijman and Jovin, 2003).
Some fluorophores, however, may require an excitation energy that overlaps with the emission energy of another fluorophore. This fact must be considered when setting up an experiment involving the simultaneous use of more than one fluorophore. It is crucial to note that spectral properties of some fluorophores are subject to significant environmental effects and vary depending on measuring conditions—e.g., medium, pH, and substrate binding (Haugland, 1992; Mason, 1993).
Understanding the spectral properties of various fluorophores is important when choosing components of the microscope used in fluorescence microscopy. This includes the light source (Xe or Hg) as well as the filter(s). To generate and observe fluorescence requires a fluorophore bound to a molecule. The fluorophore can be bound either directly to a nucleic acid probe or indirectly through the use of haptens (such as biotin and digoxigenin) and fluorescently-conjugated moieties with which these haptens interact (avidin and anti-digoxigenin antibodies, respectively). Another means of fluorescence tagging is fusion of the green fluorescent protein (GFP) or any of its many derivatives directly to a protein of interest. A light source and optical filters are required to produce the correct wavelength(s) of light energy required for excitation of the fluorescent moiety. The light passing through or being emitted by the sample must then pass through another set of optical filters such that emitted light energy of only the desired wavelength reaches the detection system (i.e., eye or other detector; Fig. 4.4.1). Details of these requirements are elaborated in the following sections.
Light Sources
The standard light source for bright-field microscopy is a tungsten-filament bulb. The intensity of the illumination can be controlled by changing the amount of current flowing to the lamp via a rheostat dial. The light sources used in fluorescence microscopy are either mercury or xenon arc lamps. The choice of lamp is determined by the wavelengths of excitation energy needed to excite the fluorescent molecule being used as a probe. Mercury lamps have three main peaks of excitation light around 440, 550, and 580 nm, whereas xenon lamps are more uniform in their intensity across this range. An additional mercury peak around 365 nm in the UV range is important for the imaging of DAPI-stained objects (i.e., DNA). Both types of bulbs are available in a number of different wattages. Brighter lamps result in more intense fluorescence, and therefore a shorter exposure time is required. A 100-W bulb also has a longer operating life than a 50-W bulb (200 versus 100 hr). Another important parameter is the gap between the anode and cathode in the bulb itself. This is known as the arc gap. Small gaps provide a small arc that can emulate a point source for epi-Köhler illumination.
It is important to monitor the bulb use, because older bulbs result in weaker fluorescence signals. This will affect exposure settings; these are more important when using photographic film, because it is easier to retake the image with a digital-imaging device. Also, mercury bulbs should not be used >200 hr because there is a risk of explosion that can damage the microscope. Changing and aligning the bulb requires patience and skill and is often best performed by the microscope service representative.
Filters
Excitation filters (Fig. 4.4.1, component c) allow the passage of selected wavelengths of light from the illumination source that corresponds to the excitation spectrum of the fluorophore. Other wavelengths are either absorbed or reflected by this filter. A second filter, the emission or barrier filter, is necessary on the imaging side of the sample for blocking transmission of unabsorbed wavelengths of excitation light and allowing transmission of the emitted fluorescence light to the detector (Fig. 4.4.1, component i). This is important because some of the excitation light is reflected off various microscope surfaces and would be brighter than the fluorescence were it not reflected out of the emission path by the dichroic mirror and also suppressed by the barrier filter. Fluorescence microscopes with incident or epi-illumination also utilize a dichroic mirror (chromatic beam splitter) to aid in separation of fluorescence emission light from unabsorbed reflected excitation light. Dichroic mirrors have a surface coating that reflects light excitation wavelengths toward the sample and passes emission wavelengths to the detector or eyepiece (Fig. 4.4.1, component d). Choice of filters for fluorescence microscopy is determined by the fluorophore and counterstain used in sample preparation. Any list of available filters and/or filter sets would be incomplete and therefore misleading to anyone getting started with fluorescence microscopy. A list of vendors and their Web sites has therefore been included at the end of this unit (Table 4.4.3) and a continually updated version will be maintained on the Ried Lab Web site (see Table 4.4.3). A discussion with each manufacturer stating the nature of the experiment, illumination source available, and fluorophores to be used (particularly for samples labeled with multiple dyes) is highly recommended prior to the purchase of any filter or filter set. Fluorophores commonly used in fluorescence microscopy and for which filters are typically needed are DAPI, Hoechst, quinacrine, or propidium iodide (used as DNA counterstains), as well as fluorescein isothiocyanate (FITC), Texas red, rhodamine, and other fluorophores emitting in the far-red portion of the spectrum (used as probe-specific labels). Filters are usually supplied in sets, and consist of an excitation filter, a dichroic mirror, and an emission or barrier filter. These are often contained within a device known as a filter cube, which slides easily into the filter holder or turret. Excitation and emission filters may be either colored glass or the more expensive interference filters (a glass substrate carrying vacuum-deposited thin layers of metallic salt compounds). Interference filters are more efficient than their colored glass counterparts in allowing only the desired wavelength through to the specimen.
Table 4.4.3.
Useful Microscopy-Related Web Sites
Filter sets also differ in the amount of light they permit to pass. Short-pass filters allow all light shorter than a particular wavelength to pass through to the specimen, while long-pass filters only allow the passage of longer wavelengths. These types of filters are therefore not very restrictive in their transmission range. Band-pass filters are more selective in that they transmit one (or more) particular region(s) or band(s) of the light spectrum. This means that wavelengths both shorter and longer than the excitation wavelength are blocked. Narrow-band-pass filters have a much more restricted range of transmitted wavelengths compared to wide-pass (or broad-pass) filters. Broad-pass filters, because they allow more light through, result in a brighter image but include a broader spectrum of wavelengths. Band-pass filters now have high light-transmission values (>90%) and very narrow band characteristics that allow selective excitation of one or more fluorophores.
Dual- and triple-band-pass filters, which permit concurrent visualization of two or three fluorophore combinations, are available. These are useful for the simultaneous excitation and detection of multiple fluorophores hybridized to the same sample, and abrogate the need to change filters between imaging each fluorophore. The simplest application is the imaging of two gene-specific probes for mapping. To take advantage of these filter sets, one needs a means of imaging that distinguishes the different colors. This can be as simple as a camera and color film or as technologically advanced as a color CCD or video camera. An example of a triple-pass filter cube is the one used for spectral karyotyping (SKY). This contains filters that allow alternating regions of excitation and emission wavelengths. This is necessary due to the overlapping excitation and emission spectra of the five fluorophores used in the hybridization. Quadruple-pass filters do have some compromises, however, in that the brightness may be affected (P. Millman, Chroma Technology, pers. comm.). Filters and filter sets can be purchased from microscope companies or directly (and usually at a reduced cost) from filter manufacturers (e.g., Chroma Technology). Other filters are important for altering the intensity of light entering the system. Neutral-density filters decrease the overall amount of transmitted light without altering the intensity ratios of different wavelengths. This may be desired if the signal intensity is strong and the fluorophore is particularly sensitive to photo-bleaching. Heat filters can be extremely important in removing excessive heat radiating from the bulbs. KG-1, BG-38, and Hot-Mirrors (Schott Glass) are some examples of heat filters. Each has a different wavelength at which it reduces the transmitted heat. Choosing the correct heat filter depends on the wavelengths one needs for excitation of the sample. Requiring shorter excitation wavelengths allows one to choose a heat filter that prevents a larger portion of the spectrum from reaching the sample. Heat filters not only protect the specimen from damage, but are also useful for protecting sensitive elements such as polarizers and other filters.
IMAGE ACQUISITION
After identifying an object that is worthy of documenting, an image must be acquired, particular portions of the object resolved and defined, and a careful analysis performed to generate useful information. In the past, images were photographed or studied directly at the microscope. High-technology image-analysis systems are now available that allow computerized image capture, image enhancement, manipulation of captured images, mass storage and retrieval, and computer analysis. Imaging systems are now commercially available for storage and analysis of DNA gels and autoradiograms, sperm morphometry and motion analysis, and interphase and metaphase cytogenetic studies. These systems have found their places in clinical diagnostic laboratories as well as in basic research laboratories. Preparation of the mammalian karyotype has always been a time-consuming and labor-intensive process. Significant strides have been made in automating cytogenetic analysis, and several commercial imaging systems are now available that offer either semiautomatic or interactive karyotyping capabilities. These systems save time because they eliminate the darkroom work required to make photographic prints, as well as the physical cutting and pasting of chromosomes, to produce a karyotype. Each system is composed of the following basic components: a microscope with a charge-coupled device (CCD) camera, a video monitor to view the image, a computer with the appropriate image-capture and storage capabilities, and a high-resolution printer for generating a copy of the image. Because the metaphases and karyotypes are digital images, long-term storage and fading of photographs is avoided, and quick transmission of high-quality data is possible via computer networks.
Photography is sometimes useful for imaging metaphase cells, cell morphology, expression of proteins (e.g., β-galactosidase) in transfected tissue culture cells and stained histological specimens. The following section will therefore cover some of the essentials.
Film and Photography
Several types of film are suitable for black- and-white photography of metaphase chromosomes banded by a variety of methods. These include Agfapan 100, Ilford FP4, and Kodak TP 2415. Sometimes certain filters are required to increase contrast and improve object definition. Black-and-white photography of certain objects (e.g., G-banded chromosomes) is enhanced by use of a green filter in combination with panchromatic film. Suitable green filters include Wratten 58 (Eastman Kodak) or a 550-nm interference filter (Thomson and Bradbury, 1987). Kodak TP 2415 film is widely used by cytogenetics laboratories. This film has a fine grain and variable contrast influenced by the choice of photographic developer. Kodak HC110 developer allows for a wider range of contrast that is determined by the developer dilution. Kodak D19 and D76 can also be used to develop TP2415 film, but they do not provide the flexibility of HC110.
Once a film and developer are chosen, a test roll of film should be shot, varying the ASA/DIN to determine which settings provide optimum contrast. An ASA of 50 is commonly used for bright-field microscopy and 200 for fluorescence microscopy. Kodak TP-2415 is also suitable for black-and-white photography of fluorescent images to avoid expensive page charges for publication of color images. In FISH studies of interphase or metaphase cells, nucleotides modified with biotin (or digoxigenin) are incorporated into the probe. The cells are incubated with the probe, and a conjugate of avidin (or anti-digoxigenin) and a fluorophore (e.g., fluorescein, rhodamine, or Texas red) then binds to the probe, emitting fluorescence when exposed to light of appropriate wavelength and intensity. It is also possible to directly label the probe with fluorophores, thereby eliminating the need for detection with antibodies or avidin. Propidium iodide may be used as a counterstain to aid in visualization of nonfluorescent objects (i.e., chromosomes). A derivative of the fluorescent dye DAPI is used for chromosome banding to permit unequivocal chromosome identification. Dual- or triple-band-pass filters allow simultaneous visualization of different fluorophores and DAPI-stained (banded) chromosomes. If only single-band-pass filters are used, the fluorescent image is photographed, the filters are switched, and then the DAPI-banded chromosomes are photographed.
As with bright-field photomicrography, camera settings for fluorescence photomicrography will vary depending on the type of microscope, and optimal settings must be determined empirically. To reduce fading of the fluorescent signal, use the shortest exposure times that are adequate to record the image. When film is exposed for long periods of time, as is often necessary when photographing fluorescent images (e.g., mammalian chromosomes using FISH techniques), the film’s sensitivity gradually becomes lower than its labeled value, necessitating a longer exposure time than indicated. This phenomenon is known as reciprocity failure, and it varies with film type. Most camera systems have a correction setting for reciprocity failure, and this is one variable that must be assessed for optimal photography.
Because of the high cost of producing color prints, many laboratories use color slide film for photographing FISH images. There are numerous color slide films available; many investigators find that Kodak Ektachrome color slide film (HC400) works well. This high-speed film (ASA 400) results in better photographic capture of weak probe signals. Although the “fast” film produces a grainier print than does ASA 100 film, the additional graininess does not interfere with enlargements ≤8 × 10 in. It is possible to expose ASA 100 film with an ASA setting of 400 and have the development “pushed” by the color photography laboratory. Pushing increases film sensitivity, but it also results in very red chromosomes when propidium iodide is used as the counterstain, and this makes it more difficult to print accurate images.
Although film has high spatial resolution, it does suffer from low quantum efficiency. Quantum efficiency (QE) is defined as the number of photons detected divided by the number that reach the detector. All detectors have QE values that vary with wavelength, but, for historical reasons and illustrative purposes, this discussion will consider 546-nm light (the mercury arc line for which achromatic objective lenses are optically corrected). The QE of good black- and-white film is 1% (i.e., one in one hundred photons reaching the film actually contributes to the signal). The QE of color film depends on the color emulsion and the wavelength(s) of light. The QE of a typical video CCD camera (see discussion of Digital Image Acquisition) is 3%, that of an intensifier for low-light video-rate microscopy is 30%, that of a scientific-grade 12-bit digital CCD camera is 35% to 60%, and that of a very expensive “slow scan back-illuminated” 16-bit digital CCD is 80% to 90%. For comparison, the original commercial laser scanning confocal microscopes (LSCM) used a photomultiplier tube with a QE of <10%, while current confocal microscopes use detectors with QE values of 10% to 25%. In other words, digital CCD cameras detect 30 to 90 times more photons than film, and convert the intensity information of the scene into a computer-ready format for quantitation and display (Inoué and Spring, 1997).
Digital Image Acquisition
Instead of traditional photographic cameras, charge-coupled device (CCD) cameras, developed in the 1970s and 1980s, are used in digital-imaging systems. Modern high-performance CCD cameras were originally used in quantitative astronomy, where CCDs revolutionized the field. They currently find applications in security and surveillance, e.g., at military installations, airports, and banks, and are also used for radar tracking and in quality-control. CCDs are much smaller and more robust than the old tube cameras, and their household applications have become ubiquitous in the form of hand-held video camcorders and digital “photography.” One-dimensional CCD arrays are also the enabling technology for the flat-bed scanners used to digitize photographs and printed pages. CCD cameras are used increasingly in image-processing and image analysis. Several companies now market CCD cameras for diverse applications in video microscopy.
CCD cameras use various technologies for specific functions. Most CCD cameras sample an image 30 times per second, but integrating CCD cameras have the ability to delay the readout for several seconds, rather than milliseconds, thus offering good performance in low-light applications such as fluorescence microscopy. One-chip color CCD cameras are available for fluorescence microscopy. A red, green, or blue filter (striped filters) is placed in front of each pixel. These are broad-band filters and are not perfectly spectrally matched to the fluorophores. Three-chip color CCD cameras, with one color per chip, are frequently used in pathology applications. These offer a reasonably high resolution, but require ample light. They are nonintegrating and relatively insensitive.
Cooled, slow-scan CCDs seem to be the best suited for fixed-cell studies because of their high quantum yield and excellent linearity, resolution, dynamic range, and spatial fidelity. Other CCDs include intensified high-gain CCDs that can detect very dim images and video-rate CCDs with high sensitivity for use in rapid kinetic studies. CCD camera technology is progressing rapidly, and new products may offer advantages over existing cameras. For most users, a 12-bit digital CCD camera with the appropriate color filters will be the most appropriate image-capture tool. For special applications, confocal microscopy or digital deconvolution is used to obtain optical sectioning and Z-series capture, and Sagnac interferometer-based spectral imaging is used for 5-color fluorescence applications such as the 24-color spectral karyotyping (SKY; Schröck et al., 1996).
The CCD is an array of individual light sensors, each of which is a linear photometer (Aikens, 1990; Photometrics, 1995). Silicon CCDs utilize silicon crystals, whose covalent bonds are broken by incoming photons, thereby liberating electrons and generating electron hole pairs. The CCD itself contains a rectangular array (or matrix) of wells where the liberated electrons are collected and stored until their quantity (i.e., charge) is measured. The capacity of each well may vary in accordance with the manufacturer, with capacities that can range up to 1 × 106 electrons per well. Each well represents a pixel of digital information the size of which (in μm2) is related to the magnification of the objective and the size of the array. Other energy sources, such as heat, can generate charge not related to the electromagnetic energy released by the fluorophore. This “dark current” can be reduced through cooling of the silicon array.
The signal output of a CCD array is a voltage that is linearly proportional to the charge present in each pixel. But how is the charge in each well, hereafter referred to as a charge packet, measured? Think of the array as being divided into rows and columns (Fig. 4.4.3). Once the CCD is exposed to the emitted light, charge accumulates in each of the wells. The information in each well is shifted by one row, with the first row being transferred to an output node (i.e., an extra row outside the array used for the transfer of information to the signal-processing device). The packets in the node are then shifted column-by-column to the processor and counted. Once all the wells in the node have been measured, the next row is transferred and the process repeated until the entire array has been read. The matrix is then capable of being exposed again to the light. The charge packets can be transferred thousands of times without significant loss of charge. This charge transfer efficiency (CTE) measurement is an important factor in choosing a camera, especially where the charge packets are small and any loss may result in significant image degradation. Because this is a complex process, methods have been established for reducing the readout time. One such technique, called binning, combines the charge from adjacent pixels; readout time is reduced, as this effectively reduces the number of pixels that must be read. This comes at the price of reduced spatial resolution, however. One can control the amount and direction of binning. For example, 2 × 2 binning combines the energy packets of four pixels; two pixels in the horizontal direction and two pixels in the vertical direction (Fig. 4.4.3, panels D to F). CCD performance is affected by a number of factors including linearity, charge-transfer efficiency, resolution, noise, dynamic range, quantum efficiency, and signal-to-noise ratio (Aikens, 1990; Photometrics, 1995).
Figure 4.4.3.
The 4 × 4 pixel CCD array illustrated above (A) has accumulated energy packets in several of the wells. (B) The energy packets are transferred one row at a time to the serial register. (C) They are then shifted into the output node one pixel at a time and on to the processor where they are counted and recorded. The concept of 2 × 2–pixel binning is demonstrated in panels D to F. Modified with permission from Photometrics (1995).
Spurred by penetration into the industrial machine-vision market and consumer demand for all things digital, scientific digital cameras and other detectors have come a long way in the past decade. Circa 1995, most light microscopes captured images on 35-mm film or using commercial video standards. Film was processed in specialty photo stores or using an in-laboratory darkroom. Video was saved to the relatively new videocassette recorder or analog optical memory disk recorder, or digitized to a computer using an expensive frame-grabber card. Computer memory was over $50/Mb, with PCs supporting 64 Mb of memory. A 75 MHz Pentium (586 chip) was the fastest central processor unit (CPU), and hard drives as large as 100 Mb were rare. Scientific digital cameras typically used a cooled Kodak-1400 full-frame CCD sensor with 1317 × 1035 pixels, 12-bit dynamic range (4096 intensity levels), a readout rate of 500 kilopixels/sec, and a mechanical shutter. Fortunately, binning was available. The Kodak-1400 CCD had a maximum quantum efficiency of 35% for green light and over 10% from 400 to 700 nm. Specialty back-thinned CCD cameras, whose sensors, which were thinned in a costly and inefficient process, were illuminated from the non-gate side, were available in large-pixel (25 × 25 μm), 512 × 512 pixel, 16-bit dynamic range (65,536 intensity levels), 50 kilopixel/sec readout. The advantage of back-thinned cameras was that the sensors that survive the thinning manufacturing process had quantum efficiencies of 80% to 90% from 400 to 800 nm. The back-illuminated CCDs have been the mainstay of major astronomy telescopes where the camera is a fraction of the total instrument price, but were rare in light-microscopy facilities where such a camera might cost more than the rest of the instrument. An alternative approach for achieving high sensitivity was to combine an image intensifier (“night vision unit”) with a video or binned digital camera. At the time, the Gen II intensifier and video CCD was still the preferred method for physiological imaging of probes such as Fura-2, in spite of the “chicken wire” appearance of the images through the multichannel plate.
Turning to 2005, a plethora of fast camera-computer interfaces are available, such as USB2, Firewire, and Cameralink, though some specialty cameras still use proprietary digitizer cards. Standard CCD cameras have lost the mechanical shutter in favor of interline CCD readout. Computer memory is $280/Gb ($0.27/Mb), with PCs supporting 4 Gb (4096 Mb) of memory (more with Windows Server); dual 3.6-GHz Pentium 4 CPU’s are the fastest chips. Terabyte disk arrays for user desktops are available for under $2000. Gigapixel images have assembled by consumer digital photographers (http://www.tawbaware.com/maxlyons/gigapixel.htm).
In front of the CCD sensor is an array of microscopic Sony HyperHAD lenslets. HAD, a registered trademark of Sony, is the original Hole Accumulated Diode interline transfer CCD sensor. The HAD incorporated anti-blooming to prevent signal from saturated pixels from flowing into neighboring pixels. This concept is referred to as multi-phase pinning in Kodak terminology. The Hyper-HAD CCD combines an improved sensor with on-chip lens technology that focuses light onto the light-sensitive areas, not the opaque transfer registry. The most recent lenslet design, ExwaveHAD (also a registered trademark of Sony), improves upon the HyperHAD by nearly eliminating the gap between lens elements, resulting in more light being focused on the sensitive part of the CCD. In the past, the electronic gates of front-illuminated CCDs blocked some of the light, especially in the blue spectral range. Kodak invented a transparent tin oxide electronic gate that improved sensitivity in the blue range. The combination of the lenslets and the Kodak blue-enhanced gate resulted in front-illuminated cameras with quantum efficiencies of 60% at a fraction of the cost of the now somewhat faster back-thinned CCDs. The standard sensors are now 1600 × 1200 or 2048 × 2048 pixels, though the smaller pixels, often only 5 × 5 μm, still limit the useful dynamic range of most scientific cameras to 12 bits (4096 intensity levels). The sensors have increased in pixel number by 2× to 3×, but the readout speed has increased at a faster rate, to 10 or 20 Mb/sec. Binning is still available to further improve signal-to-noise ratio. The HyperHAD lenslets can also be dyed to transmit red, green, or blue, and laid out in a regular pattern on the CCD (most commonly the Bayer mask of 2 × 2 units of B, G, G, R) to produce one-chip color cameras. For high sensitivity, electron multiplication (EMCCD), electron bombardment (EBCCD), and high resolution Gen IV photon counting intensifiers are combining signal amplification, low noise, and fast readout for photon-counting imaging. These can be combined with spectral imagers and/or spinning disk confocal microscopes, OptiGrid (Thales Optem), Apotome (Carl Zeiss) or a digital micromirror device to provide fast, 3-D optical sectioning. Confocal microscopes are available from several vendors, and the patent holder for sub-picosecond pulsed multiphoton microscopes has agreed to not enforce the patent against do-it-yourself academic researchers. The pulsed lasers are still expensive, but are now have spectral tuning in software, are fiber-optically coupled to the microscope(s), and no longer require a Ph.D. physicist in attendance. Even the photomultiplier tubes that are the detectors of laser scanning confocal and multiphoton microscopes have improved quantum yield or have been packaged as spectral emission detectors (Zeiss META and Nikon C1 spectral detector).
Image Analysis
After the photons in a scene are collected on the CCD sensor, the number of electrons present at each picture element (pixel) is then quantified and read out as a signal whose intensity is proportional to the number of electrons, and hence to the number of photons. For video cameras, the data are reformatted to a broadcast standard for display on a television monitor. This video signal is then digitized to an 8-bit dynamic range (256 intensity level) image by using a video frame grabber in the computer. Some video cameras and frame grabbers can handle color video capture and transfer (“24-bit color”). For digital cameras, the camera electronics convert the number of electrons into a digital value that is sent directly to a custom board in the computer, where the value is deciphered by a custom driver. Most digital cameras read out 12-bit (4096-intensity-level) image data, because this is a good combination of cost and dynamic range. Three sequential red, green, and blue images acquired with a monochrome digital camera can be merged to give a better color image than possible with color video cameras, though, with image processing, the quality, but not sensitivity of a Bayer mask HyperHAD color CCD digital camera is often “good enough.” Often the same software can be used for acquiring video or digital images (assuming appropriate frame grabber or digital camera drivers), as well as image processing, analysis, color merging, and storage. Typical configurations include a PC running Microsoft Windows, Linux, or Mac OS X, and a graphics card that allows multiple image displays on a single monitor, networking capabilities for multiple workstations, choice of printers (including color), full-image contrasting, and, for FISH, hybridization spot enhancement and image sharpening.
Computerized imaging systems for cytogenetic applications vary in the extent of automation. Some possess slide-scanning and metaphase-finding capabilities and some identify specific chromosomes, while others require he user to point to each chromosome with a mouse or other pointing device and assign it a number. The software then places each chromosome onto a standard karyotype template. The chromosome images can be rotated, trimmed, inverted, labeled, and, in some systems, straightened. There are also variations in the degree of image enhancement or manipulation available. Software is available for the quantification of FISH signals, measurement analysis for gene mapping, and spot-counting analysis for aneuploidy detection. Color image ratio measurement is a feature of software developed for comparative genomic hybridization (UNIT 4.6). Similarly, sophisticated software is available for spectral karyotyping (SKY). This technique employs a labeling scheme (discussed earlier) in which different chromosome painting probes are labeled with various combinations of five different fluorophores. The software is able to identify each chromosome based on the different fluorescent-label-dependent patterns of the chromosomes and arrange them in a karyotype. It is also capable of quantitative analysis for hybridization intensity and simultaneous display of fluorescent, DAPI-banded, and classification chromosomes, along with an idiogram. Image annotation and zooming are features of many software programs, and case and patient databases are available for importing image files and compiling data for statistical and epidemiologic studies. Image analysis systems, both for routine karyotyping and for FISH applications, are undergoing constant improvement. Because price and features continue to change, manufacturers should be contacted directly for current information.
Spectral Imaging Systems
Spectral karyotyping (SKY) and various forms of multiplex FISH (M-FISH) have taken cytogenetics laboratories beyond monochrome, beyond RGB color, and even beyond the visible spectrum. The primary applications of SKY and M-FISH systems are for the use of five fluorescent dyes in combinations, on unique chromosome DNA, to paint each human or mouse chromosome uniquely (described in detail above) in FISH analysis. However, the question can be asked, as for confocal microscopy (Amos and White, 2003), whether spectral imaging is underutilized in cytogenetics and pathology. A few articles have been published in spectral pathology, including Ornberg et al. (1999), Tsurui et al. (2000), Rothmann et al. (2000), Macville et al. (2001), Farkas and Becker (2001), Schultz et al. (2001), Greenspan et al. (2002), and Jaganath et al. (2004). Of these, the 7-color immunofluorescence histology images of Tsurui et al. (2000), provide a way to maximize the immunological information from a pathology specimen. The work of Dickinson et al. (2001) on the Zeiss META emission spectral detector for multiphoton/confocal microscopes, shows what is possible for those who have enough money. As of 2005, spectral detectors are available from all of the major confocal microscope manufacturers. At least two more spectral imagers, the PARISS from Lightform Inc., and the Spectra-DV from Optical Insights, LLC, can be made into spectral confocal tissue section mappers by simply replacing the epi-illumination field aperture with a slit. The SKY system, and several other commercial spectral imagers (Table 4.4.3) could be made confocal by combining with an Atto Bioscience or Yokogawa spinning disk confocal unit.
Information relevant to spectral imaging in cytogenetics and pathology used with any or all of standard histology dyes, immunohistochemical dyes, and conventional fluorophores is summarized in Table 4.4.1, and corresponding data for fluorescent nanocrystal quantum dots are summarized in Table 4.4.2.
Image Measurements
The cytogenetic microscope/imaging systems described above for FISH and G-banding cytogenetics are essentially the same as those needed in a pathology laboratory to document H&E slides, immunohistochemistry, chromogenic in situ hybridization, and DNA ploidy. Most clinical DNA ploidy assays have moved to flow cytometers because of high-throughput, hands-off operation and instrument purchases leveraged through the need for CD4+ cell counting in HIV patient testing. There are still opportunities to use image cytometry in DNA ploidy, especially to exploit the feasibility of imaging nuclear texture on small numbers of cells (n < 100) with good statistics and low coefficient of variation. A good review on the image parameters that can be measured on a cytogenetics/pathology microscope imaging system can be found in Rodenacker and Bengtsson (2003). Note that circular statistics such as orientation data need to be calculated using correct statistical methods (see Batschelet, 1981). Uses in cell biology are reviewed by Price et al. (2002).
More generally, image measurements can be divided into tasks such as FISH spot counting, nuclear and/or cell area, and fluorescence intensity or dye absorption. Use of a digital camera and microscope enables image analysis and measurements to be done quickly and reproducibly. Automated image acquisition and measurements are becoming important in drug discovery and cell biology research, as discussed by Carpenter and Sabatini (2004). Tissue microarrays, invented by Kononen et al. (1998), are having an impact in pathology research. Automated metaphase finders, as adjuncts to karyotyping systems, are available from several companies (Table 4.4.3). NASA has had difficulties translating “faster, better, cheaper” into consistently excellent science; hopefully the biomedical microscopy community will do better.
Image Storage
With the increase in information per image and the decreasing cost of computer memory and storage space, image files have become larger—some on the order of 25 Mb per file. The easiest way to store files is on an external hard drive or server. These become full with time, and long-term storage becomes an issue. Popular disk storage include such media as Jazz (1000 or 2000 Mb per disk at $0.09/Mb), Zip (100 or 250 Mb at $0.10/Mb), CD ReWriteable ($0.10/Mb), or CD-Recordable ($0.003/Mb). The DVD formats (DVD+R, DVD−R, DVD+RW, DVD−RW, and high-capacity Blu-Ray) should eventually allow even higher capacity and cheaper storage. Many universities and corporations have information service departments that can provide long-term archiving of data (for a price). Make sure to save data in a broadly compatible file format so that any of the software that might be used for image acquisition, enhancement, manipulation, and presentation can read the data. The almost-universal standard is uncompressed tagged-image file format (TIFF). Image acquisition software is often specific to the microscope/camera system being used and the type of analysis to be performed (e.g., FISH, immunocytochemistry, CGH, SKY, microarray analysis, spectral imaging, or multidimensional imaging). Careful consideration is therefore recommended when purchasing software from a company other than that from which the hardware was obtained. At a minimum, be sure that the imaging software can save the images in the industry-standard TIFF format, so that one can share the data with colleagues. The JPEG and GIF file formats use data compression and are thus good formats for displaying image data on Web pages. Most of the multidimensional and spectral imaging systems save data in their own proprietary formats. Before purchasing such equipment, one should make sure that the data—preferably all the data—can be exported to a universal format, i.e., wavelength-named TIFF files, so that other software products can read the data and one can access the information even if the company’s product is discontinued or one moves to a location that does not have a software license.
CONCLUSIONS
As light microscopy moves into the 21st century, improvements in reagents, protocols, microscopes, imaging systems, and users’ knowledge, are being used in research and clinical laboratories to better understand Waldeyer’s chromosomes and their role in biology and medicine.
Acknowledgments
Frederick R. Bieber of Brigham and Women’s Hospital in Boston, Massachusetts contributed the original version of this unit, published in 1994.
Contributor Information
George McNamara, Childrens Hospital Los Angeles, Los Angeles, California; City of Hope National Medical Center, Duarte, California.
Michael J. Difilippantonio, National Cancer Institutes, Bethesda, Maryland
Thomas Ried, National Cancer Institutes, Bethesda, Maryland.
Literature Cited
- Aikens R. CCD cameras for video microscopy. In: Herman B, Jacobson K, editors. Optical Microscopy for Biology. John Wiley & Sons; New York: 1990. pp. 85–110. [Google Scholar]
- Allen RD, David GB, Nomarski G. The Zeiss-Nomarski differential interference equipment for transmitted-light microscopy. Z Wiss Mikrosk. 1969;69:193–221. [PubMed] [Google Scholar]
- Allen RD, Travis JL, Allen NS, Yilmaz H. Video-enhanced contrast polarization (AVEC-POL) microscopy: A new method applied to the detection of birefringence in the motile reticulopodial network of Allogromia laticollaris. Cell Motil. 1981a;1:275–289. doi: 10.1002/cm.970010302. [DOI] [PubMed] [Google Scholar]
- Allen RD, Allen NS, Travis JL. Video-enhanced contrast, differential interference contrast (AVEC-DIC) microscopy: A new method capable of analyzing microtubule-related motility in the reticulopodial network of Allogromia laticollaris. Cell Motil. 1981b;1:291–302. doi: 10.1002/cm.970010303. [DOI] [PubMed] [Google Scholar]
- Amos WB, White JG. How the confocal laser scanning microscope entered biological research. Biol Cell. 2003;95:335–342. doi: 10.1016/s0248-4900(03)00078-9. [DOI] [PubMed] [Google Scholar]
- Arnold N, Bhatt M, Ried T, Wienberg J, Ward DC. Fluorescence in situ hybridization on banded chromosomes. In: Kessler C, editor. Techniques and Methods in Molecular Biology: Nonradioactive Labeling and Detection of Biomolecules. Springer-Verlag; New York and Heidelberg: 1992. pp. 324–326. [Google Scholar]
- Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Methods Enzymol. 2003;361:1–33. doi: 10.1016/s0076-6879(03)61003-7. [DOI] [PubMed] [Google Scholar]
- Azofeifa J, Fauth C, Kraus J, Maierhofer C, Langer S, Bolzer A, Reichman J, Schuffenhauer S, Speicher MR. An optimized probe set for the detection of small interchromosomal aberrations by use of 24-color FISH. Am J Hum Genet. 2000;66:1684–1688. doi: 10.1086/302875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldini A, Ward DC. In situ hybridization of human chromosomes with Alu-PCR products: A simultaneous karyotype for gene mapping studies. Genomics. 1991;9:770–774. doi: 10.1016/0888-7543(91)90374-n. [DOI] [PubMed] [Google Scholar]
- Barch M, Knutsen T, Spurbeck J, editors. The AGT Cytogenetics Laboratory Manual. Raven Press; New York: 1997. [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A. Atm-deficient mice: A paradigm of ataxia telangiectasia. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Barone-Nugent ED, Barty A, Nugent KA. Quantitative phase-amplitude microscopy I: Optical microscopy. J Microsc. 2002;206:194–203. doi: 10.1046/j.1365-2818.2002.01027.x. [DOI] [PubMed] [Google Scholar]
- Batschelet E. Circular Statistics in Biology (Mathematics in Biology Series) Academic Press; New York: 1981. [Google Scholar]
- Blennow E, Nielson KB, Telenius H, Carter NP, Kristoffersson U, Holmberg E, Gillberg C, Nordenskjøld M. Fifty probands with extra structurally abnormal chromosomes characterized by fluorescence in situ hybridization. Am J Med Genet. 1995;55:85–94. doi: 10.1002/ajmg.1320550122. [DOI] [PubMed] [Google Scholar]
- Boyle AL, Ballard SG, Ward DC. Differential distribution of long and short interspersed element sequences in the mouse genome: Chromosome karyotyping by fluorescence in situ hybridization. Proc Natl Acad Sci USA. 1990a;87:7757–7761. doi: 10.1073/pnas.87.19.7757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle A, Lichter P, Ward DC. Rapid analysis of mouse-hamster hybrid cell lines by in situ hybridization. Genomics. 1990b;7:127–130. doi: 10.1016/0888-7543(90)90529-4. [DOI] [PubMed] [Google Scholar]
- Brink AA, Wiegant JC, Szuhai K, Tanke HJ, Kenter GG, Fleuren GJ, Schuuring E, Raap AK. Simultaneous mapping of human papillomavirus integration sites and molecular karyotyping in short-term cultures of cervical carcinomas by using 49-color combined binary ratio labeling fluorescence in situ hybridization. Cancer Genet Cytogenet. 2002;134:145–150. doi: 10.1016/s0165-4608(01)00620-3. [DOI] [PubMed] [Google Scholar]
- Bruchez M, Jr, Moronne M, Gin P, Weiss S, Alivisatos AP. Semiconductor nanocrystals as fluorescent biological labels. Science. 1998;281:2013–2016. doi: 10.1126/science.281.5385.2013. [DOI] [PubMed] [Google Scholar]
- Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. A monomeric red fluorescent protein. Proc Natl Acad Sci USA. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Sabatini DM. Systematic genome-wide screens of gene function. Nat Rev Genet. 2004;5:11–22. doi: 10.1038/nrg1248. [DOI] [PubMed] [Google Scholar]
- Carter KC, Bowman D, Carrington W, Fogarty K, McNeil JA, Fay FS, Lawrence JB. A three-dimensional view of precursor messenger RNA metabolism within the mammalian nucleus. Science. 1993;259:1330–1336. doi: 10.1126/science.8446902. [DOI] [PubMed] [Google Scholar]
- Caspersson T, Farber S, Foley GE, Kudynowski J, Modest EJ, Simonsson E, Wagh U, Zech L. Chemical differentiation along metaphase chromosomes. Exp Cell Res. 1968;49:219–222. doi: 10.1016/0014-4827(68)90538-7. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- Chan WCW, Nie S. Quantum dot bioconjugates for ultrasensitive nonisotopic detection. Science. 1998;281:2016–2018. doi: 10.1126/science.281.5385.2016. [DOI] [PubMed] [Google Scholar]
- Chowdhary B, Raudsepp T, Fronicke L, Scherthan H. Emerging patterns of comparative genome organization in some mammalian species as revealed by Zoo-FISH. Genome Res. 1998;8:577–589. doi: 10.1101/gr.8.6.577. [DOI] [PubMed] [Google Scholar]
- Clark G, Kasten FH. History of Staining. Williams & Wilkins; Baltimore: 1983. [Google Scholar]
- Cody SH, Xiang SD, Layton MJ, Handman E, Lam MH, Layton JE, Nice EC, Heath JK. A simple method allowing DIC imaging in conjunction with confocal microscopy. J Microsc. 2005;217:265–274. doi: 10.1111/j.1365-2818.2005.01452.x. [DOI] [PubMed] [Google Scholar]
- Coleman AE, Schröck E, Weaver Z, du Manoir S, Yang F, Ferguson-Smith MA, Ried T, Janz S. Previously hidden chromosome aberrations in T(12;15)-positive BALB/c plasmacytomas uncovered by multi-color spectral karyotyping. Cancer Res. 1997;57:4585–4592. [PubMed] [Google Scholar]
- Contag CH, Contag PR, Mullins JI, Spilman SD, Stevenson DK, Benaron DA. Photonic detection of bacterial pathogens in living hosts. Mol Microbiol. 1995;18:593–603. doi: 10.1111/j.1365-2958.1995.mmi_18040593.x. [DOI] [PubMed] [Google Scholar]
- Coons AH. The beginnings of immunofluorescence. J Immunol. 1961;87:499–503. [PubMed] [Google Scholar]
- Coons AH, Kaplan MH. Localization of antigen in tissue cells II Improvements in a method for the detection of antigen by means of a fluorescent antibody. J Exp Med. 1950;91:1–13. doi: 10.1084/jem.91.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coons AH, Creech HJ, Jones RN. Immunological properties of an antibody containing a fluorescent group. Proc Soc Biol Med. 1941;47:200. [Google Scholar]
- Coons AH, Creech HJ, Jones RN, Berliner E. The demonstration of pneumococcal antigen in tissue by the use of a fluorescent antibody. J Immunol. 1942;45:159–170. [Google Scholar]
- Cremer T, Landegent JE, Bruckner A, Scholl HP, Schardin M, Hager H-D, Devilee P, Pearson PL, van der Ploeg M. Detection of chromosome aberrations in the human interphase nucleus by visualization of specific target DNAs with radioactive and nonradioactive in situ hybridization techniques: Diagnosis of trisomy 18 with probe L1.84. Hum Genet. 1986;74:346–352. doi: 10.1007/BF00280484. [DOI] [PubMed] [Google Scholar]
- Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- Devilee P, Thierry RF, Kievits T, Kolluri R, Hopman AHN, Willard HF, Pearson PL, d Cornelisse CJ. Detection of chromosome aneuploidies in interphase nuclei from human primary breast tumors using chromosome specific repetitive DNA probes. Cancer Res. 1988;48:5825–5830. [PubMed] [Google Scholar]
- Diaspro A, editor. Confocal and Two-Photon Microscopy; Foundations, Applications and Advances. Wiley-Liss; New York: 2001. [Google Scholar]
- Dickinson ME, Bearman G, Tille S, Lansford R, Fraser SE. Multi-spectral imaging and linear unmixing add a whole new dimension to laser scanning fluorescence microscopy. Biotechniques. 2001;31:1272, 1274–6, 1278. doi: 10.2144/01316bt01. [DOI] [PubMed] [Google Scholar]
- Dodt H-U, Zieglgansberger W. Infrared videomicroscopy: A new look at neuronal structure and function. Trends Neurosci. 1994;17:453–458. doi: 10.1016/0166-2236(94)90130-9. [DOI] [PubMed] [Google Scholar]
- Doucette-Stamm LA, Riba L, Handelin B, Difilippantonio M, Ward DC, Wasmuth JJ, Gusella JF, Housman DE. Generation and characterization of irradiation hybrids of human chromosome 4. Somat Cell Mol Genet. 1991;17:471–480. doi: 10.1007/BF01233171. [DOI] [PubMed] [Google Scholar]
- Durnam DM, Gelinas RE, Myerson D. Detection of species specific chromosomes in somatic cell hybrids. Somat Cell Mol Genet. 1985;11:571–577. doi: 10.1007/BF01534722. [DOI] [PubMed] [Google Scholar]
- Egger MD, Petran M. New reflected-light microscope for viewing unstained brain and ganglion cells. Science. 1967;157:305–307. doi: 10.1126/science.157.3786.305. [DOI] [PubMed] [Google Scholar]
- Engels H, Ehrbrecht A, Zahn S, Bosse K, Vrolijk H, White S, Kalscheuer V, Hoovers JM, Schwanitz G, Propping P, Tanke HJ, Wiegant J, Raap AK. Comprehensive analysis of human subtelomeres with combined binary ratio labelling fluorescence in situ hybridisation. Eur J Hum Genet. 2003;11:643–651. doi: 10.1038/sj.ejhg.5201028. [DOI] [PubMed] [Google Scholar]
- Farkas DL, Becker D. Applications of spectral imaging: Detection and analysis of human melanoma and its precursors. Pigment Cell Res. 2001;14:2–8. doi: 10.1034/j.1600-0749.2001.140102.x. [DOI] [PubMed] [Google Scholar]
- Feulgen R, Rossenbeck H. Mikroskopisch-chemischer Nachweis einer Nucleisäure vom Typus der Thymonucleinsäure und die darauf beruhende elektive Färburg von Zellkernen in mikroskopischen Präparaten. Hoppe Seyler Z Physiol Chem. 1924;135:203–248. [Google Scholar]
- Fleischmann M, Bloch W, Kolossov E, Andressen C, Muller M, Brem G, Hescheler J, Addicks K, Fleischmann BK. Cardiac specific expression of the green fluorescent protein during early murine embryonic development. FEBS Lett. 1998;440:370–376. doi: 10.1016/s0014-5793(98)01476-8. [DOI] [PubMed] [Google Scholar]
- Forozan F, Karhu R, Kononen J, Kallioniemi A, Kallioniemi OP. Genome screening by comparative genomic hybridization. Trends Genet. 1997;13:405–409. doi: 10.1016/s0168-9525(97)01244-4. [DOI] [PubMed] [Google Scholar]
- Förster T. Delocalized excitation and excitation transfer. In: Sinanoglu O, editor. Modern Quantum Chemistry Part III: Action of light and organic Crystals. Academic Press; New York: 1965. pp. 93–137. [Google Scholar]
- Fronicke L, Scherthan H. Zoo-fluorescence in situ hybridization analysis of human and Indian muntjac karyotypes (Muntiacus muntjac vaginalis) reveals satellite DNA clusters at the margins of conserved syntenic segments. Chromosome Res. 1997;5:254–261. doi: 10.1023/B:CHRO.0000032298.22346.46. [DOI] [PubMed] [Google Scholar]
- Garfield S. Mauve: How One Man Invented a Color That Changed the World. WW Norton & Company; New York: 2001. [Google Scholar]
- Garini Y, Macville M, du Manoir S, Buckwald RA, Lavi M, Katzir N, Wine D, Bar-Am I, Schröck E, Cabib D, Ried T. Spectral karyotyping. Bioimaging. 1996;4:65–72. [Google Scholar]
- Garini Y, Gil A, Bar-Am I, Cabib D, Katzir N. Signal to noise analysis of multiple color fluorescence imaging microscopy. Cytometry. 1999;35:214–226. doi: 10.1002/(sici)1097-0320(19990301)35:3<214::aid-cyto4>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Garini Y, Vermolen BJ, Young IT. From micro to nano: Recent advances in high-resolution microscopy. Curr Opin Biotechnol. 2005;16:3–12. doi: 10.1016/j.copbio.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Ghadimi BM, Schröck E, Walker RL, Wangsa D, Jauho A, Melzer P, Ried T. Specific chromosomal aberrations and amplification of AIB1 nuclear receptor coactivator gene in pancreatic carcinomas. Am J Pathol. 1999;154:525–536. doi: 10.1016/S0002-9440(10)65298-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano SJ, Yoo M, Ward DC, Bhatt M, Overhauser J, Steggles AW. The human cytochrome b5 gene and two of its pseudogenes are located on chromosomes 18q23, 14q31–32.1 and 20p11.2, respectively. Hum Genet. 1993;92:615–618. doi: 10.1007/BF00420948. [DOI] [PubMed] [Google Scholar]
- Goldman RD, Spector DK, editors. Live Cell Imaging. Cold Spring Harbor Press; Cold Spring Harbor, NY: 2004. [Google Scholar]
- Greenspan H, Rothmann C, Cycowitz T, Nissan Y, Cohen AM, Malik Z. Classification of lymphoproliferative disorders by spectral imaging of the nucleus. Histol Histopathol. 2002;17:767–773. doi: 10.14670/HH-17.767. [DOI] [PubMed] [Google Scholar]
- Harris H. The Cells of the Body A History of Somatic Cell Genetics. Cold Spring Harbor Press; Cold Spring Harbor, NY: 1995. [Google Scholar]
- Hasan MT, Friedrich RW, Euler T, Larkum ME, Giese GG, Both M, Duebel J, Waters J, Bujard H, Griesbeck O, Tsien RY, Nagai T, Miyawaki A, Denk W. Functional fluorescent Ca(2+) indicator proteins in transgenic mice under TET control. PLoS Biol. 2004;2:E163. doi: 10.1371/journal.pbio.0020163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugland R. Molecular Probes (Invitrogen) Eugene; Oregon: 2004. The Handbook—A Guide to Fluorescent Probes and Labeling Technologies. [Google Scholar]
- Heim R, Prasher DC, Tsien RY. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc Natl Acad Sci USA. 1994;91:12501–12504. doi: 10.1073/pnas.91.26.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzwarth G, Webb SC, Kubinski DJ, Allen NS. Improving DIC microscopy with polarization modulation. J Microsc. 1997;188:249–254. [Google Scholar]
- Holzwarth G, Hill DB, McLaughlin EB. Polarization-modulated differential-interference contrast microscopy with a variable retarder. Appl Opt. 2000;39:6268–6294. doi: 10.1364/ao.39.006288. [DOI] [PubMed] [Google Scholar]
- Hooke R. Micrographia Or, Some physiological descriptions of minute bodies made by magnifying glasses, with observations and inquiries thereupon. Royal Society; London: 1665. [Google Scholar]
- Hsu TC. Mammalian chromosomes in vitro I The karyotype of man. J Hered. 1952;43:167–172. [Google Scholar]
- Inoué S. Video image processing greatly enhances contrast, quality, and speed in polarization-based microscopy. J Cell Biol. 1981;89:346–356. doi: 10.1083/jcb.89.2.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoué S, Dan K. Birefringence of the dividing cell. J Morphol. 1951;89:423. [Google Scholar]
- Inoué S, Spring KR. Video Microscopy. 2nd. Plenum; New York: 1997. [Google Scholar]
- Jaganath R, Angeletti C, Levenson R, Rimm DL. Diagnostic classification of urothelial cells in urine cytology specimens using exclusively spectral information. Cancer. 2004;102:186–191. doi: 10.1002/cncr.20302. [DOI] [PubMed] [Google Scholar]
- Jaiswal JK, Simon SM. Total internal reflection fluorescence microscopy for high-resolution imaging of cell-surface events. In: Bonifacino JS, Dasso M, Harford JB, Lippincott-Schwartz J, Yamada KM, editors. Current Protocols in Cell Biology. John Wiley & Sons; Hoboken, New Jersey: 2003. pp. 4.12.1–4.12.15. [DOI] [PubMed] [Google Scholar]
- James J, Tanke H. Biomedical Light Microscopy. Kluwer Academic Publishers; Boston: 1991. [Google Scholar]
- Jardine L. The Curious Life of Robert Hooke: The Man Who Measured London. Harper Collins; New York: 2004. [Google Scholar]
- Jares-Erijman EA, Jovin TM. FRET imaging. Nat Biotechnol. 2003;21:1387–1395. doi: 10.1038/nbt896. [DOI] [PubMed] [Google Scholar]
- Jentsch I, Geigl J, Klein CA, Speicher MR. Seven-fluorochrome mouse M-FISH for high-resolution analysis of interchromosomal rearrangements. Cytogenet Genome Res. 2003;103:84–88. doi: 10.1159/000076294. [DOI] [PubMed] [Google Scholar]
- Johnson CV, McNeil JA, Carter KC, Lawrence JB. A simple, rapid technique for precise mapping of multiple sequences in two colors using a single optical filter set. Genet Anal Tech Appl. 1991;8:24–35. doi: 10.1016/1050-3862(91)90052-s. [DOI] [PubMed] [Google Scholar]
- Kachar B. Asymmetric illumination contrast: A method of image formation for video light microscopy. Science. 1985;227:766–768. doi: 10.1126/science.3969565. [DOI] [PubMed] [Google Scholar]
- Kallioniemi A, Kallioniemi O-P, Sudar D, Rutovitz D, Gray JW, Waldman F, Pinkel D. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science. 1992;258:818–821. doi: 10.1126/science.1359641. [DOI] [PubMed] [Google Scholar]
- Kapitza H-G. Modern microscope objectives. Proc Royal Med Soc. 1996;31:24–27. [Google Scholar]
- Karhu R, Ahlstedt-Soini M, Bittner M, Meltzer P, Trent JM, Isola JJ. Chromosome arm-specific multicolor FISH. Genes Chromosomes Cancer. 2001;30:105–109. doi: 10.1002/1098-2264(2000)9999:9999<::aid-gcc1068>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Kohler G, Milstein C. Continuous culture of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- Labas YA, Gurskaya NG, Yanushevich YG, Fradkov AF, Lukyanov KA, Lukyanov SA, Matz MV. Diversity and evolution of the green fluorescent protein family. Proc Natl Acad Sci USA. 2002;99:4256–4261. doi: 10.1073/pnas.062552299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer-Safer PR, Levine M, Ward DC. Immunological method for mapping genes on Drosophila polytene chromosomes. Proc Natl Acad Sci USA. 1982;79:4381–4385. doi: 10.1073/pnas.79.14.4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson DR, Zipfel WR, Williams RM, Clark SW, Bruchez MP, Wise FW, Webb WW. Water-soluble quantum dots for multiphoton fluorescence imaging in vivo. Science. 2003;300:1434–1436. doi: 10.1126/science.1083780. [DOI] [PubMed] [Google Scholar]
- Lawrence JB, Singer RH, Marselle LM. Highly localized tracks of specific transcripts within interphase nuclei visualized by in situ hybridization. Cell. 1989;57:493–502. doi: 10.1016/0092-8674(89)90924-0. [DOI] [PubMed] [Google Scholar]
- Lawrence JB, Carter KC, Xing X. Probing functional organization within the nucleus: Is genome structure integrated with RNA metabolism? Cold Spring Harbor Symp Quant Biol. 1993;58:807–818. doi: 10.1101/sqb.1993.058.01.088. [DOI] [PubMed] [Google Scholar]
- Leewenhoek A. An Abstract of a Letter from Mr Anthony Leewenhoeck Writ to Sir C W Jan 22 1682/3 from Delft. Phil Trans. 1683;13:74–81. [Google Scholar]
- Lejeune J, Gautier M, Turpin R. Etude des chromosomes somatiques de neuf enfants mongoliens. Compt Rend. 1959;248:1721–1722. [PubMed] [Google Scholar]
- Lichter P, Ward DC. Is non-isotopic in situ hybridization finally coming of age? Nature. 1990;345:93–95. doi: 10.1038/345093a0. [DOI] [PubMed] [Google Scholar]
- Lichter P, Chang Tang C-J, Call K, Hermanson G, Evans GA, Housman D, Ward DC. High resolution mapping of human chromosome 11 by in situ hybridization with cosmid clones. Science. 1990;247:64–69. doi: 10.1126/science.2294592. [DOI] [PubMed] [Google Scholar]
- Lichter JB, Difilippantonio M, Wu J, Miller D, Ward DC, Goodfellow PJ, Kidd KK. Localization of the gene for MEN 2A. Henry Ford Hosp Med J. 1992;40:199–204. [PubMed] [Google Scholar]
- Liyanage M, Coleman C, du Manoir S, Veldman T, McCormack S, Dickson RB, Barlow C, Wynshaw-Boris A, Janz S, Wienberg J, Ferguson-Smith MA, Schröck E, Ried T. Multicolour spectral karyotyping of mouse chromosomes. Nature Genet. 1996;14:312–315. doi: 10.1038/ng1196-312. [DOI] [PubMed] [Google Scholar]
- Macville MV, Van Der Laak JA, Speel EJ, Katzir N, Garini Y, Soenksen D, McNamara G, de Wilde PC, Hanselaar AG, Hopman AH, Ried T. Spectral imaging of multi-color chromogenic dyes in pathological specimens. Anal Cell Pathol. 2001;22:133–142. doi: 10.1155/2001/740909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuelidis L. Individual interphase chromosome domains revealed by in situ hybridization. Hum Genet. 1985;71:288–293. doi: 10.1007/BF00388453. [DOI] [PubMed] [Google Scholar]
- Manuelidis L, Langer-Safer PR, Ward DC. High-resolution mapping of satellite DNA using biotin-labeled DNA probes. J Cell Biol. 1982;95:619–625. doi: 10.1083/jcb.95.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrack J. Nature of antibodies. Nature. 1934;133:292. [Google Scholar]
- Mason W. Fluorescent and Luminescent Probes for Biological Activity: A Practical Guide to Technology for Quantitative Real-Time Analysis. Academic Press; San Diego: 1993. [Google Scholar]
- Matsumoto B, editor. Cell Biological Applications of Confocal Microscopy. 2nd. Academic Press; New York: 2002. [Google Scholar]
- Matz MV, Fradkov AF, Labas YA, Savitsky AP, Zaraisky AG, Markelov ML, Lukyanov SA. Fluorescent proteins from nonbioluminescent Anthozoa species. Nat Biotechnol. 1999;17:969–973. doi: 10.1038/13657. [DOI] [PubMed] [Google Scholar]
- Milstein C. With the benefit of hindsight. Immunol Today. 2000;21:359–364. doi: 10.1016/s0167-5699(00)01660-1. [DOI] [PubMed] [Google Scholar]
- Minsky M. US patent 3013467 Microscopy apparatus. 1957
- Minsky M. Memoir on inventing the confocal scanning microscope. Scanning. 1988;10:128–138. [Google Scholar]
- Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, Ikura M, Tsien RY. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 1997;388:882–887. doi: 10.1038/42264. [DOI] [PubMed] [Google Scholar]
- Nederlof PM, van der Flier S, Vrolijk J, Tanke HJ, Raap AK. Fluorescence ratio measurements of double-labeled probes for multiple in situ hybridization by digital imaging microscopy. Cytometry. 1992;13:839–845. doi: 10.1002/cyto.990130806. [DOI] [PubMed] [Google Scholar]
- Newton I. A series of quere’s propounded by Mr Isaac Netwon, to be determin’d by experiments, positively and directly concluding his new theory of light and colours; and here recommended to the industry of the lovers of experimental philosophy, as they were generously imparted to the publisher in a letter of the said Mr Newton of July 8, 1672. Phil Trans. 1672;7:4004–5007. [Google Scholar]
- Newton I. Optiks; or, A Treatise of the Reflections, Refractions, Inflections & Colours of Light. 4th. Dover Publications; New York: 1730. (London, republished 1952). [Google Scholar]
- Nguyen AW, Daugherty PS. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat Biotechnol. 2005;23:355–360. doi: 10.1038/nbt1066. [DOI] [PubMed] [Google Scholar]
- O’Brien S, Cevario S, Martenson J, Thompson M, Nash W, Chang E, Graves J, Spencer J, Tsujimoto H, Lyons L. Comparative gene mapping in the domestic cat (Felis catus) J Hered. 1997;88:408–414. doi: 10.1093/oxfordjournals.jhered.a023127. [DOI] [PubMed] [Google Scholar]
- Oheim M. Imaging transmitter release II A practical guide to evanescent-wave imaging. Lasers Med Sci. 2001;16:159–170. doi: 10.1007/pl00011350. [DOI] [PubMed] [Google Scholar]
- Ormö M, Cubitt AB, Kallio K, Gross LA, Tsien RY, Remington SJ. Crystal structure of the Aequorea victoria green fluorescent protein. Science. 1996;273:1392–1395. doi: 10.1126/science.273.5280.1392. [DOI] [PubMed] [Google Scholar]
- Ornberg RL, Woerner BM, Edwards DA. Analysis of stained objects in histological sections by spectral imaging and differential absorption. J Histochem Cytochem. 1999;47:1307–1314. doi: 10.1177/002215549904701010. [DOI] [PubMed] [Google Scholar]
- Otsu K, Fujii J, Periasamy M, Difilippantonio M, Upender M, Ward DC, MacLennan DH. Chromosome mapping of five human cardiac and skeletal muscle sarcoplasmic reticulum protein genes. Genomics. 1993;17:507–509. doi: 10.1006/geno.1993.1357. [DOI] [PubMed] [Google Scholar]
- Paddock SW, editor. Confocal Microscopy Methods and Protocols. Human Press; Totawa, NJ: 1999. [Google Scholar]
- Paulmurugan R, Gambhir SS. Firefly luciferase enzyme fragment complementation for imaging in cells and living animals. Anal Chem. 2005;77:1295–1302. doi: 10.1021/ac0484777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawley JB, editor. Handbook of Biological Confocal Microscopy. 3rd. Plenum; New York: 2005. [Google Scholar]
- Perkin W. Address of William Henry Perkin. Science. 1906;24:488–493. doi: 10.1126/science.24.616.488. [DOI] [PubMed] [Google Scholar]
- Photometrics. SenSys User Manual for Windows. Photometrics Ltd; Tuscon, Ariz: 1995. [Google Scholar]
- Ploem JS. The use of a vertical illuminator with interchangeable dichroic mirrors for fluorescence microscopy with incidental light. Z Wiss Mikrosk. 1967;68:129–142. [PubMed] [Google Scholar]
- Price JH, Goodacre A, Hahn K, Hodgson L, Hunter EA, Krajewski S, Murphy RF, Rabinovich A, Reed JC, Heynen S. Advances in molecular labeling, high throughput imaging and machine intelligence portend powerful functional cellular biochemistry tools. J Cell Biochem. 2002;87:194–210. doi: 10.1002/jcb.10448. [DOI] [PubMed] [Google Scholar]
- Raudsepp T, Fronicke L, Scherthan H, Gustavsson I, Chowdhary B. Zoo-FISH delineates conserved chromosomal segments in horse and man. Chromosome Res. 1996;4:218–225. doi: 10.1007/BF02254963. [DOI] [PubMed] [Google Scholar]
- Rawlins D. Light Microscopy. BIOS Scientific Publishers; Oxford: 1992. [Google Scholar]
- Ray P, De A, Min JJ, Tsien RY, Gambhir SS. Imaging tri-fusion multimodality reporter gene expression in living subjects. Cancer Res. 2004;64:1323–1330. doi: 10.1158/0008-5472.can-03-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitz FB, Pagliaro L. Fibre optic scrambling in light microscopy: A computer simulation and analysis. J Microsc. 1994;176:143–151. doi: 10.1111/j.1365-2818.1994.tb03508.x. [DOI] [PubMed] [Google Scholar]
- Ried T, Mahler V, Vogt P, Blonden L, van Ommen GJB, Cremer T, Cremer M. Direct carrier detection by in situ suppression hybridization with cosmid clones for the Duchenne/Becker muscular dystrophy locus. Hum Genet. 1990;85:581–586. doi: 10.1007/BF00193578. [DOI] [PubMed] [Google Scholar]
- Ried T, Baldini A, Rand TC, Ward DC. Simultaneous visualization of seven different DNA probes using combinatorial fluorescence and digital imaging microscopy. Proc Natl Acad Sci USA. 1992;89:1388–1392. doi: 10.1073/pnas.89.4.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ried T, Liyanage M, du Manoir S, Heselmeyer K, Auer G, Macville M, Schröck E. Tumor cytogenetics revisited: Comparative genomic hybridization and spectral karyotyping. J Mol Med. 1997;75:801–814. doi: 10.1007/s001090050169. [DOI] [PubMed] [Google Scholar]
- Rodenacker K, Bengtsson E. A feature set for cytometry on digitized microscopic images. Anal Cell Pathol. 2003;25:1–36. doi: 10.1155/2003/548678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney D, Czepulkowski B. Human Cytogenetics A Practical Approach. Oxford University Press; New York: 1992. [Google Scholar]
- Rothmann C, Barshack I, Gil A, Goldberg I, Kopolovic J, Malik Z. Potential use of spectral image analysis for the quantitative evaluation of estrogen receptors in breast cancer. Histol Histopathol. 2000;15:1051–1057. doi: 10.14670/HH-15.1051. [DOI] [PubMed] [Google Scholar]
- Ruestow EG. The Microscope in the Dutch Republic. Cambridge University Press; New York: 1996. [Google Scholar]
- Sachs RK, van den Engh G, Trask B, Yokota H, Hearst JE. A random-walk/giant loop model for interphase chromosomes. Proc Natl Acad Sci USA. 1995;92:2710–2714. doi: 10.1073/pnas.92.7.2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawano A, Hama H, Saito N, Miyawaki A. Multicolor imaging of Ca2+ and protein kinase C signals using novel epifluorescence microscopy. Biophys J. 2002;82:1076–1085. doi: 10.1016/S0006-3495(02)75467-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schardin M, Cremer T, Hager HD, Lang M. Specific staining of human chromosomes in Chinese hamsters × man hybrid cell lines demonstrates interphase chromosome territories. Hum Genet. 1985;71:281–287. doi: 10.1007/BF00388452. [DOI] [PubMed] [Google Scholar]
- Schröck E, du Manoir S, Veldman T, Schoell B, Wienberg J, Ferguson-Smith MA, Ning Y, Ledbetter DH, Bar-Am I, Soenksen D, Garini Y, Ried T. Multicolor spectral karyotyping of human chromosomes. Science. 1996;273:494–497. doi: 10.1126/science.273.5274.494. [DOI] [PubMed] [Google Scholar]
- Schultz RA, Nielsen T, Zavaleta JR, Ruch R, Wyatt R, Garner HR. Hyperspectral imaging: A novel approach for microscopic analysis. Cytometry. 2001;43:239–247. doi: 10.1002/1097-0320(20010401)43:4<239::aid-cyto1056>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- Shi S-R, Gu J, Taylor CR. Antigen Retrieval Techniques: Immunohistochemistry and Molecular Morphology Biotechniques Books. Eaton Pub Co; Natick, Mass: 2000. [Google Scholar]
- Shotton D. Electronic Light Microscopy: The Principles and Practice of Video-Enhanced Contrast, Digital Intensified Fluorescence, and Confocal Scanning Light Microscopy. John Wiley & Sons; Hoboken, NJ: 1993. [Google Scholar]
- Singer E. A microscope for observation of fluorescence in living tissue. Science. 1932;75:289–291. doi: 10.1126/science.75.1941.289-a. [DOI] [PubMed] [Google Scholar]
- Spector D, Goldman R, Leinwand L, editors. Cells: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1998. Light microscopy and cell structure. [Google Scholar]
- Speicher M, Ballard S, Ward D. Karyotyping human chromosomes by combinatorial multi-fluor FISH. Nat Genet. 1996;12:1–11. doi: 10.1038/ng0496-368. [DOI] [PubMed] [Google Scholar]
- Stockholm D, Bartoli M, Sillon G, Bourg N, Davoust J, Richard I. Imaging calpain protease activity by multiphoton FRET in living mice. J Mol Biol. 2005;346:215–222. doi: 10.1016/j.jmb.2004.11.039. [DOI] [PubMed] [Google Scholar]
- Stryer L, Haugland RP. Energy transfer: A spectroscopic ruler. Proc Natl Acad Sci USA. 1967;58:719–726. doi: 10.1073/pnas.58.2.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan KF, Shelby RD. Using time-lapse confocal microscopy for analysis of centromere dynamics in human cells. Methods Cell Biol. 1999;58:183–202. [PubMed] [Google Scholar]
- Szuhai K, Bezrookove V, Wiegant J, Vrolijk J, Dirks RW, Rosenberg C, Raap AK, Tanke HJ. Simultaneous molecular karyotyping and mapping of viral DNA integration sites by 25-color COBRA-FISH. Genes Chromosomes Cancer. 2000;28:92–97. doi: 10.1002/(sici)1098-2264(200005)28:1<92::aid-gcc11>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Taniwaki M, Speicher MR, Lengauer C, Jauch A, Popp S, Cremer T. Characterization of two marker chromosomes in a patient with acute nonlymphocytic leukemia by two color fluorescence in situ hybridization. Cancer Genet Cytogenet. 1993;70:99–102. doi: 10.1016/0165-4608(93)90175-l. [DOI] [PubMed] [Google Scholar]
- Tanke HJ, Wiegant J, van Gijlswijk RP, Bezrookove V, Pattenier H, Heetebrij RJ, Talman EG, Raap AK, Vrolijk J. New strategy for multi-colour fluorescence in situ hybridisation: COBRA: COmbined Binary RAtio labelling. Eur J Hum Genet. 1999;7:2–11. doi: 10.1038/sj.ejhg.5200265. [DOI] [PubMed] [Google Scholar]
- Terskikh A, Fradkov A, Ermakova G, Zaraisky A, Tan P, Kajava AV, Zhao X, Lukyanov S, Matz M, Kim S, Weissman I, Siebert P. “Fluorescent timer”: Protein that changes color with time. Science. 2000;290:1585–1588. doi: 10.1126/science.290.5496.1585. [DOI] [PubMed] [Google Scholar]
- Thangavelu M, Pergament E, Espinosa RI, Bohlander SK. Characterization of marker chromosomes by microdissection and fluorescence in situ hybridization. Prenat Diagn. 1994;14:583–588. doi: 10.1002/pd.1970140712. [DOI] [PubMed] [Google Scholar]
- Tjio JH, Levan A. The chromosome number of man. Hereditas. 1956;42:1–6. [Google Scholar]
- Tran PT, Chang F. Transmitted light fluorescence microscopy revisited. Biol Bull. 2001;201:235–236. doi: 10.2307/1543340. [DOI] [PubMed] [Google Scholar]
- Trask B, Fertitta A, Christensen M, Youngblom J, Bergmann A, Copeland A, de Jong P, Mohrenweiser H, Olsen A, Carrano A, et al. Fluorescence in situ hybridization mapping of human chromosome 19: Cytogenetic band location of 540 cosmids and 70 genes or DNA markers. Genomics. 1993;15:133–145. doi: 10.1006/geno.1993.1021. [DOI] [PubMed] [Google Scholar]
- Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- Tsien RY. Building and breeding molecules to spy on cells and tumors. FEBS Lett. 2005;579:927–932. doi: 10.1016/j.febslet.2004.11.025. [DOI] [PubMed] [Google Scholar]
- Tsurui H, Nishimura H, Hattori S, Hirose S, Okumura K, Shirai T. Seven-color fluorescence imaging of tissue samples based on Fourier spectroscopy and singular value decomposition. J Histochem Cytochem. 2000;48:653–662. doi: 10.1177/002215540004800509. [DOI] [PubMed] [Google Scholar]
- van der Voort H, Valkenburg J, van Spronsen E, Woldringh C, Brakenhoff G. Confocal microscopy in comparison with electron and conventional light microscopy. In: Hayat M, editor. Correlative Microscopy in Biology: Instrumentation and Methods. Academic Press; San Diego: 1987. pp. 23–37. [Google Scholar]
- Veldman T, Vignon C, Schröck E, Rowley JD, Ried T. Hidden chromosomes: Abnormalities in hematological malignancies detected by multicolour spectral karyotyping. Nature Genet. 1997;15:406–410. doi: 10.1038/ng0497-406. [DOI] [PubMed] [Google Scholar]
- Wang X, Rosol M, Ge S, Peterson D, McNamara G, Pollack H, Kohn DB, Nelson MD, Crooks GM. Dynamic tracking of human hematopoietic stem cell engraftment using in vivo bioluminescence imaging. Blood. 2003;102:3478–3482. doi: 10.1182/blood-2003-05-1432. [DOI] [PubMed] [Google Scholar]
- Wang L, Jackson WC, Steinbach PA, Tsien RY. Evolution of new nonantibody proteins via iterative somatic hypermutation. Proc Natl Acad Sci USA. 2004;101:16745–16749. doi: 10.1073/pnas.0407752101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward DC, Lichter P, Boyle A, Baldini A, Menninger J, Ballard SG. Gene mapping by fluorescent in situ hybridization and digital imaging microscopy. In: Lindsten J, Petterson U, editors. Etiology of Human Diseases at the DNA Level. Raven Press; New York: 1991. [Google Scholar]
- Waud JP, Bermudez Fajardo A, Sudhaharan T, Trimby AR, Jeffery J, Jones A, Campbell AK. Measurement of proteases using chemiluminescence-resonance-energy-transfer chimaeras between green fluorescent protein and aequorin. Biochem J. 2001;357:687–697. doi: 10.1042/0264-6021:3570687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JG, Amos WB, Fordham M. An evaluation of confocal versus conventional imaging of biological structures by fluorescence light microscopy. J Cell Biol. 1987;105:41–48. doi: 10.1083/jcb.105.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegant J, Bezrookove V, Rosenberg C, Tanke HJ, Raap AK, Zhang H, Bittner M, Trent JM, Meltzer P. Differentially painting human chromosome arms with combined binary ratio-labeling fluorescence in situ hybridization. Genome Res. 2000;10:861–865. doi: 10.1101/gr.10.6.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienberg J, Jauch A, Stanyon R, Cremer T. Molecular cytotaxonomy of primates by chromosomal in situ suppression hybridization. Genomics. 1990;8:347–270. doi: 10.1016/0888-7543(90)90292-3. [DOI] [PubMed] [Google Scholar]
- Xing Y, Johnson CV, Dobner PR, Lawrence JB. Higher level organization of individual gene transcription and RNA splicing. Science. 1993;259:1326–1330. doi: 10.1126/science.8446901. [DOI] [PubMed] [Google Scholar]
- Xu Y, Piston DW, Johnson CH. A bioluminescence resonance energy transfer (BRET) system: Application to interacting circadian clock proteins. Proc Natl Acad Sci USA. 1999;96:151–156. doi: 10.1073/pnas.96.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota H, van den Engh G, Hearst JE, Sachs RK, Trask BJ. Evidence for the organization of chromatin in megabase pairsized loops arranged along a random walk path in the human G0/G1 interphase nucleus. J Cell Biol. 1995;130:1239–1249. doi: 10.1083/jcb.130.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota H, Singer MJ, van den Engh GJ, Trask BJ. Regional differences in the compaction of chromatin in human G0/G1 interphase nuclei. Chromosome Res. 1997;5:157–166. doi: 10.1023/a:1018438729203. [DOI] [PubMed] [Google Scholar]
- Yuste R, Konnerth A. Imaging in Neuroscience and Development A Laboratory Manual. Cold Spring Harbor Press; Cold Spring Harbor, NY: 2005. [Google Scholar]
- Zernike F. How I discovered phase contrast. Science. 1955;121:345–349. doi: 10.1126/science.121.3141.345. [DOI] [PubMed] [Google Scholar]