

Abstract
Classical banding methods provide basic information about the identities and structures of chromosomes on the basis of their unique banding patterns. Spectral karyotyping (SKY), and the related multiplex fluorescence in situ hybridization (M-FISH), are chromosome-specific multicolor FISH techniques that augment cytogenetic evaluations of malignant disease by providing additional information and improved characterization of aberrant chromosomes that contain DNA sequences not identifiable using conventional banding methods. SKY is based on cohybridization of combinatorially labeled chromosome-painting probes with unique fluorochrome signatures onto human or mouse metaphase chromosome preparations. Image acquisition and analysis use a specialized imaging system, combining Sagnac interferometer and CCD camera images to reconstruct spectral information at each pixel. Here we present a protocol for SKY analysis using commercially available SkyPaint probes, including procedures for metaphase chromosome preparation, slide pretreatment and probe hybridization and detection. SKY analysis requires approximately 6 d.
INTRODUCTION
Cytogenetic studies in cancer are complicated by the fact that many chromosomal aberrations are often difficult to characterize. Initially, researchers relied on classical banding (G-, Q- and C-banding) methods1–3. These methods helped distinguish individual chromosomes by visualizing physical landmarks (called bands) and provided chromosome-specific banding patterns for each species examined. Cytogenetic techniques became much more versatile through the introduction of hybridization of fluorescently labeled region-specific DNA probes. This molecular cytogenetic technique is now widely known as FISH4–6.
SKY is also based on the hybridization of fluorescently labeled DNA probes; however, the probe used is complex, generally consisting of up to 55 individually generated chromosome-specific probes. SKY has proven exceedingly valuable for the comprehensive analysis of cytogenetic abnormalities associated with malignant disease and has been applied to a large series of samples derived from hematological malignancies and solid tumors7–11. In fact, since its invention in 1996, more than 500 papers have been published that applied SKY for the analysis of various chromosomal preparations. SKY results can now be submitted to an interactive database12, which facilitates data analyses (SKY, M-FISH and CGH), storage and retrieval of recurrent chromosome breakpoints found in many cancers and constitutional disorders (http://www.ncbi.nlm.nih.gov/sky/skyweb.cgi). Alternatively, M-FISH13 can be used for characterizing karyotypes14. Although the hybridization principle is identical to SKY, M-FISH uses a set of fluorochrome-specific optic filters (five or seven different filters), rather than the single custom-designed filter used in SKY. Both SKY and M-FISH achieve the same goal—that is, identification of individual chromosomes in different colors. Both methods are more costly than conventional banding methods, as they require expensive materials (probes), equipment and software.
The systematic analysis of chromosomal abnormalities in cancer cells using SKY allows for the characterization of novel and hidden chromosomal translocations, identification of complex rearrangements15 and reconstruction of clonal evolution events during cancer progression, and it has revealed the role of unstable chromosome rearrangements, such as jumping translocations, occurring as tissue-specific genomic imbalances11. Although SKY allows for the identification of which particular piece of DNA may be contained within a chromosome aberration, the spectral image alone does not provide information as to the specific region of the chromosome localized within a rearrangement. For this, one must rely on the banding pattern of 4,6-diamidino-2-phenylindole (DAPI). Good-quality chromosome preparations are also key to the success of SKY analysis, if the chromosome preparations are of inferior quality—for example, if the slides are old or have too much cytoplasm surrounding the chromosomes—SKY analysis can be less than optimal. Thus, the strengths and limitations of SKY analysis are somewhat dependent on the skills of the user. Maximizing the amount of information gained through the use of this technique is greatly facilitated by a general knowledge and expertise in cytogenetics—that is, familiarity with the chromosomal banding patterns of the species being analyzed, as well as an understanding of the principles of how the software interprets the acquired information. For assistance in both identification of the banding patterns and determination of breakpoints of aberrant human chromosomes, researchers use the nomenclature rules defined in ref. 16. For mouse chromosomes, the nomenclature rules used can be found at http://www.informatics.jax.org/mgihome/nomen/gene.shtml.
SKY greatly assists in identifying chromosomal regions involved in homogenous staining regions and double minute chromosomes, regardless of size and numbers. However, with respect to homogenous staining regions and double minute chromosomes, again, SKY has its limitations, in that these aberrations often contain multiple genes and/or DNA regions that are tightly linked; resolving these details therefore often requires additional FISH hybridizations with either gene-loci probes or specific chromosome or chromosome-arm paints17. SKY has also proven to be very useful for the study of constitutional chromosome abnormalities arising from de novo balanced translocations (in which chromosomal regions are relocated in a reciprocal manner and no genetic material is lost) and unbalanced translocations (which are, by definition, associated with genomic imbalances) that occasionally involve chromosome exchanges between very small DNA regions, especially at the telomeres18–20. In prenatal diagnosis, the difference between the outcome of a balanced translocation and an unbalanced one may represent the difference between a normal child and one with mild-to-severe birth defects. SKY is useful in determining the origin of supernumerary marker chromosomes that are occasionally seen in prenatal specimens (appearing as unbanded small chromosome fragments using conventional banding methods). It is imperative that the chromosomal origin of these markers be determined, as genomic imbalances may result in birth defects and or mental retardation21, and some markers may have considerable prognostic significance.
SKY has also been applied for the reconstruction of chromosomal rearrangements that have occurred during the course of chromosome evolution, a field referred to as comparative cytogenetics22. By using SKY, one can determine and compare homologous regions of genomes between closely related species—for example, between human and a gibbon (Hylobates concolor)23. These analyses can be complemented by performing the reverse experiment—that is, hybridizing differentially labeled flow-sorted gibbon chromosomes onto human metaphase spreads. This is referred to as cross-species hybridization24. Recently, SKY analysis has also been used in other species, such as rat. Newly developed rat probes have been used to determine the homologous regions of rat, mouse and human chromosomes, as well as to characterize the karyotypes of rat tumors25.
The study of chromosomal aberrations in mice is extremely demanding, because the chromosomes of this species are all acro-centric (that is, the primary constriction, or centromere, is located at one end of the chromosome) and of similar size and morphology. Therefore, subtle aberrations such as insertions or translocations involving small regions are often unrecognizable. Murine models of human carcinogenesis are widely used to delineate genetic mechanisms that determine tumor initiation and progression26. SKY as a genome-scanning method for detection of chromosomal aberrations has now been applied to a wide variety of murine models of human cancer, including numerous models for hematological malignancies and solid tumors27–30. Although SKY has been applied to vertebrate organisms closely related to human or mouse, thus far commercially made SKY probes and analysis software are only available for human, mouse and rat.
Experimental design
The protocol we present uses SkyPaint probes (Applied Spectral Imaging). The SKY analysis protocol described herein consists of six parts: (i) preparation of metaphase chromosome spreads (Steps 1–20); (ii) slide pretreatment and slide and probe denaturation (Box 1 and Steps 21–26); (iii) hybridization (Steps 27–29) and (iv) detection (Steps 30–49) of the SKY probes; and (v) tips for image acquisition and (vi) image analysis (Step 50) (see Fig. 1). Preparation of metaphase chromosome spreads for SKY analysis requires 1 d. Slide pretreatment and denaturation of both the slide and the probe takes 2 h, at which point one can pause before continuing with the hybridization steps. The hybridization protocol, which includes denaturation of the SKY probe as well as that of the target metaphase chromosomes on the slide, is performed on day 1. For the actual hybridization of human or mouse SKY probes, the procedure continues for 48–72 h at 37 °C. The subsequent detection steps take approximately 5 h (see Fig. 1). Image acquisition and analysis can take place immediately after the detection steps, but can also be delayed and the slides can be stored in the dark at 4 °C. We highly recommend analyzing the slides within 1 week of completion of the SKY detection procedures.
BOX 1. SLIDE PRETREATMENT WITH PEPSIN TO REMOVE CYTOPLASM.
-
Equilibrate the slide in a coplin jar containing 2× SSC for 5 min, without shaking, at room temperature. The solution should cover all the slides.
▲ CRITICAL STEP Size of coplin jar used depends on number of slides being pretreated.
Dilute the 20 mg ml−1 RNase A stock solution (slide pretreatment solution 1) 1:200 in 2× SSC, made and kept at room temperature until use.
-
Apply 120 μl to a 24 mm × 60 mm coverglass, invert slide metaphase-spread-side down onto the liquid bearing coverglass, and reinvert slide, taking care not to scratch the slide.
▲ CRITICAL STEP The purpose of this step is to digest DNA molecules surrounding the chromosomes that would interfere with the hybridization and result in high fluorescent background. Ensure that sample is in full contact with the liquid and that air bubbles have been eliminated.
Incubate slides in a moist, lightproof, waterproof hybridization box (as described in Step 29) at 37 °C for 45 min.
Carefully remove coverglass by gently tapping the sides of slide onto a solid surface; the coverglass should slide off easily.
Rinse the slide by placing in a coplin jar containing 2× SSC for 5 min at room temperature, with shaking.
Repeat Box Step 6 two more times, with shaking (at room temperature), using fresh 2× SSC each time.
-
Add 2–30 μl pepsin stock solution (slide pretreatment solution 2) into an empty, clean 100-ml glass beaker, then add 100 ml 0.01 M HCl that has been prewarmed to 37 °C in a water bath, and adjust the pH to 2.0 using 1.0 N HCl. Mix well and pour appropriate volume into a coplin jar sufficient to cover slide.
▲ CRITICAL STEP It is very important that the pepsin be added to the clean beaker first and not directly into the HCL. If the pepsin is added to the HCL, it does not dissolve into solution.
-
Incubate slide in coplin jar for 30 s–5 min at 37 °C.
▲ CRITICAL STEP The time of pepsin treatment and amount of pepsin stock solution to be used is dependent on (i) the amount of cytoplasm surrounding the metaphase spreads, as observed with a light microscope using phase objectives before slide pretreatment, and (ii) the age of the slide—slides with excess cytoplasm, or older than 6 months, may require longer treatment with pepsin (3–5 min) and higher concentrations of pepsin (10–30 μl). After exposure to pepsin, the slide can be placed into a Petri dish containing 1× PBS and examined using an inverted microscope, to see whether longer pepsin treatment is required. If so, place the slide back into the coplin jar containing the pepsin-acid mixture.
Wash the slide in 1× PBS for 5 min at room temperature, with shaking.
Repeat Box Step 10, using fresh 1× PBS.
Wash the slide once for 5 min at room temperature in 1× PBS/MgCl2 (slide pretreatment solution 3), with shaking.
Place the slide in a coplin jar containing 1% (vol/vol) formaldehyde/1× PBS/MgCl2 (slide pretreatment solution 4) and incubate slide (not shaking) for 10 min at room temperature (when finished with procedure decant solution into an appropriate waste container).
Wash slide for 5 min in 1× PBS at room temperature, shaking.
Dehydrate slide in an alcohol series (70%, 90% and 100% ethanol), using different coplin jars, at room temperature, without shaking, for 3 min each.
Air-dry the slide until the ethanol has evaporated (1–2 min), on the workbench.
-
Check chromosomes to see whether the morphology has been preserved, and select an area for hybridization.
▲ CRITICAL STEP After slide pretreatment, the chromosomes should retain their original morphology and should not have the appearance of being overdigested by the enzyme. If they look hollow, they are not generally useful for SKY hybridization, and there is no reversal of this step. If there are enough slides available, using additional slides with different pepsin concentrations and times is highly recommended to achieve the best results. Redo the entire procedure if the pretreatment does not sufficiently remove the cytoplasm surrounding the chromosome.
? TROUBLESHOOTING
Figure 1.
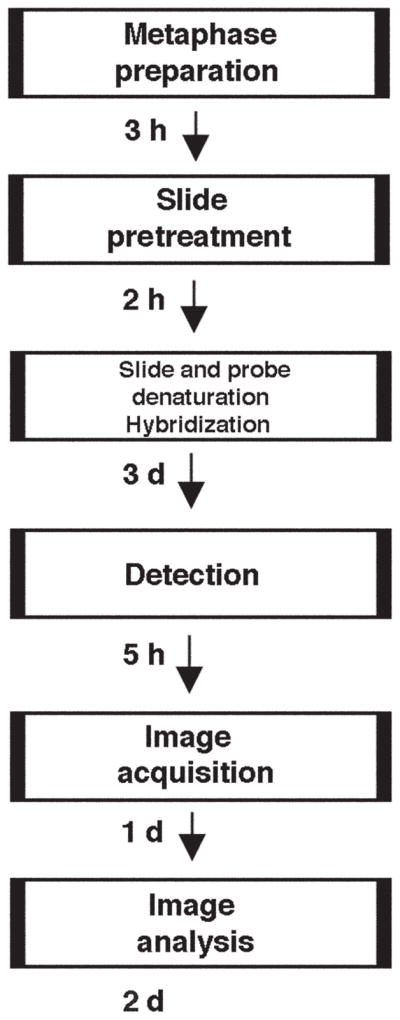
Protocol timeline flowchart. Shown are the steps in a SKY hybridization and analysis.
Preparation of metaphase spreads for SKY
This requires (i) knowledge of the ideal culture conditions of the specimen studied to obtain actively dividing cells, and (ii) recognizing the conditions necessary to yield spreads free of cytoplasm, which include (iii) the optimum humidity required for the individual chromosome preparations to ensure proper spreading of chromosomes onto glass slides and eliminate the possibility of chromosomes overlapping31. If none of these conditions are met, hybridization with SKY and or FISH probes will be suboptimal, thereby impeding an accurate analysis of the results. Excess cytoplasm surrounding the chromosomes can be reduced by pretreating the slides containing the metaphase preparations with enzymes such as trypsin or pepsin that digest the surrounding cellular proteins. This facilitates access of the DNA probe to its target sequence on the chromosome as well as reducing the amount of background signal that arises when the DNA probe nonspecifically binds the proteins (see Fig. 2). However, over-treatment of the slides can also reduce hybridization efficiency through degradation of the target DNA or excessive removal of chromosome-associated histone proteins, leading to a distortion of the chromosome morphology. Should this occur, we recommend repeating this phase of the experiment with new slides.
Figure 2.

Effects of humidity and removal of excess cytoplasm by slide pretreatment on hybridization efficiency. (a) When cells are not kept in the hypotonic solution for enough time, the chromosomes become trapped inside the cytoplasm (white arrows). Altering the humidity at which the metaphase preparation is dropped onto the microscope slide may sometimes alleviate this problem; otherwise, hybridization efficiency will be compromised. (b) When normal human metaphase spreads are pretreated with pepsin and hybridized with a whole chromosome paint probe labeled with tetramethyl rhodamine isothyocyanate (TRITC), the hybridization will be successful. (c) Here, a whole chromosome paint-labeled with TRITC was hybridized to a different normal human metaphase spread and the slide was not pretreated with pepsin. Note the weaker intensity of the hybridization signal (red color).
SKY probe generation and labeling
SKY probes consist of a cocktail of chromosome-specific painting probes of the target species. The chromosome-painting probes used for SKY analysis of human or mouse genomes are prepared by amplification of flow-sorted chromosomes using degenerate oligonucleotide–primed (DOP)-PCR32. Chromosome sorts that contain all 22 human homologs and the X and Y human chromosomes, or 19 mouse homologs and the X and Y mouse chromosomes, can be requested for a fee from the Cambridge Resource Centre for Comparative Genomics (http://www.vet.cam.ac.uk/genomics). SKY probes can either be in-house–generated probes33–35 or be purchased from Applied Spectral Imaging (ASI, http://www.spectral-imaging.com/). ASI-produced SkyPaint probes undergo stringent quality controls and their chromosome specificity is tested by hybridization of all probes individually using FISH. Probes are labeled with nucleotide analogs, either conjugated directly to fluorochromes, such as Spectrum orange, Rhodamine green or Texas Red, or indirectly via small nonfluorescent molecules—for example, biotin or digoxygenin, which are visualized using avidin- and antibody-conjugated fluorochromes. As five distinct fluorochromes are distinguishable from one another using the SKY system, each chromosome is labeled with a unique combination of these tagged nucleotides. To further enhance the chromosome specificity of the probe cocktail, the labeled chromosome-painting probes are denatured and combined with an excess of CotI DNA (which represents a fraction of the genome enriched for highly repetitive elements, such as long and short interspersed elements, Alu repeats and so forth, that are distributed throughout all the chromosomes). This step is known as preannealing and ensures that any labeled non–chromosome-specific repetitive elements in the chromosome-painting probe form double-stranded DNA with their complementary sequences found in the CotI DNA. These repetetive elements are thereby prevented from hybridizing to the metaphase chromosome spreads.
Probe hybridization and detection for SKY analysis
The principles of SKY probe hybridization and detection are very similar to those of FISH. The probe is placed in direct contact with denatured metaphase chromosomes (which are attached to glass slides), allowing for Watson-Crick base-pairing to occur between complementary sequences in the labeled probe and the metaphase chromosomes. This hybridization step is allowed to proceed for 24 h to 3 d, depending on the age and conditions of the starting material on the slide (older slides tend to require longer hybridization times). The slides are then washed to remove unhybridized probe and nonspecifically hybridized probe (that is, probe that has formed double bonds with sequences in the chromosomes that are not fully complementary). Those chromosomes labeled directly with fluorescently conjugated nucleotides do not require anything further for their visualization under the microscope. Nonfluorescensly-labeled probes, in contrast, require further detection with a molecule such as Avidin (for the detection of biotin) or an antibody, either of which has a fluorochrome attached to it. In addition to the chromosome-specific fluorescent label, we use DAPI to introduce a grayscale chromosome banding pattern that is similar to Giemsa-trypsin (G)-banding.
Image acquisition and analysis
This is accomplished by viewing the slides with an epifluorescence microscope equipped with hardware and software designed for visualizing SKY images (see Fig. 3 and ref. 36). The epifluorescence microscope should be equipped with a ×63 oil-immersion objective, a single custom-designed triple band-pass filter (SKY filter cube) that allows for the simultaneous excitation of all fluorochromes, and a DAPI filter cube. Image acquisition requires illumination from a 150-W xenon lamp. The emitted light is passed through the collection optics and then the Sagnac interferometer, where an optical path difference is created (see Fig. 3). The interferogram measures every pixel detected by the CCD camera. Using Fourier transformation, it mathematically retrieves the wavelengths of the emitted light from the combinatorially labeled chromosomes and thereby identifies a spectral signature for each pixel of the CCD camera image.
Figure 3.

SKY imaging system. (a) Diagrammatic representation of the SKY imaging system. (1) Xenon lamp source. (2) Phase filter. (3) SKY filter cube, which reflects specific wavelengths of light (blue ‘waves’). (4) ×63 objective. (5) Slide containing sample, which will fluoresce when excited (red ‘waves’). (6) BG38 filter used to reduce far-red wavelengths, which may contribute to excessive background. (7) SKY SpectraCube, which contains the Sagnac interferometer used to split the emitted light. (8) CCD camera, which captures the DAPI counter-stain as a grayscale image. (b) Photo of the SD300 SpectraCube mounted on a Leica DMXRA epifluorescence microscope. Red arrows indicate features depicted in a. The BG38 filter (6) is controlled by a lever on the right-hand side of the microscope (not visible in this image). This figure has been reproduced in part and reprinted with permission from Current Protocols in Human Genetics, Chapter 4.4 (John Wiley & Sons)36.
When beginning image acquisition, first find the area of hybridization using the SKY filter cube; next, systematically scan the slide up and down using the DAPI filter. If the fluorescence colors are bright, the acquisition time can be reduced. Conversely, if the hybridization signals are weak, the time of acquisition can be increased. When selecting metaphase spreads, do not acquire only the best-looking spreads, or the results may be biased. Metaphases with short chromosomes, with some overlaps, with debris and with large numbers of chromosomes are also informative, especially in determining whether the cells manifest chromosome instability (random gains and losses) or consist of several subclones with different karyotypes. In contrast, if the initial metaphase-spread preparations are made properly, it will not be necessary to acquire overcondensed chromosomes (too much colcemid) or incomplete metaphase spreads (too strong a hypotonic shock). Avoid imaging areas containing bright spots and nuclei, as that can also complicate the analysis. If the metaphase spread is too large to fit in the region of interest, acquire two separate images and analyze each as an individual, combining the data after analysis, but make sure to have overlapping regions so that the two images can be fit together.
Spectrum-based classification of the specific wavelengths of all chromosomes, in combination with karyotype analysis of the images, is achieved using SkyView software (ASI), following the User Manual supplied with the software. Additional information regarding the technical aspects of the ASI system can be retrieved from the ASI webpage (http://www.spectral-imaging.com/), which includes online support and tutorials. After image analysis, the chromosomes can be viewed with their specific fluorescent signature (display colors) and classification colors (pseudo-color), and their specific banding pattern (inverted DAPI) (see Figs. 4,5,6 & 7). For optimal classification, make sure there is at least one normal copy of each chromosome in the metaphase being analyzed. If there are no normal examples of a specific chromosome, attempt to locate a portion of the missing chromosome (this requires expertise in chromosome-band recognition), select the region (using the ‘lasso’ tool) containing the portion of the missing chromosome and place the piece into the correct chromosome category17. For example, if there are no normal copies of chromosome 7, look for chromosomes that have recognizable regions derived from chromosome 7 (for instance, resulting from a reciprocal translocation or unbalanced (derivative) chromosome), use the lasso tool to draw a circle around the chromosome 7 portion of the derivative chromosome and manually place that fragment into the chromosome 7 position of the karyotype (see Fig. 7). This enables the software to correctly identify the spectrum of chromosome 7, particularly as pertains to that particular hybridization. Remember to delete the chromosome 7 segment that was generated with the lasso tool after classification is complete, as the segment selected by the lasso tool is a ‘virtual’ chromosome.
Figure 4.

Normal human male (46 chromosomes, XY) metaphase spread analyzed by SKY. (a) Inverted-DAPI image of a chromosome metaphase spread. The banding pattern is similar to G-banding but accentuates the heterochromatic region of some chromosomes (for example, 1, 9, 16 and Y). (b) Same metaphase with RGB display colors. (c) Same metaphase spread with classification pseudo-colors. (d) Karyotype of the same metaphase spread. Arrows in panels a–c identify the X and Y chromosomes.
Figure 5.

Metaphase spread from a human bladder cancer cell line hybridized with a SKY probe. This aneuploid cell line, J82, contains multiple chromosomal aberrations. White arrows identify some of the chromosome translocations.
Figure 6.

Mouse spectral karyotyping. (a) SKY image of a metaphase spread from a mouse deficient for the gene mutated in the human disease ataxia telangectasia was overlayed onto the corresponding DAPI-banded image. White arrows indicate two chromosome translocations frequently observed in this mouse model system, T(12;14) and Dic(14;15). (Using mouse cytogenetic nomenclature rules, T denotes a translocation where the chromosome listed first contains the centromere; Del denotes a deletion; Dic denotes a chromosome containing two centromeres (dicentric), one from each one of the contributing chromosomes.) The arrowhead indicates the presence of a centromere at the fusion of chromosomes 14 (yellow) and 15 (blue) in the dicentric chromosome, Dic(14;15). (b) Karyotyping corresponding to the metaphase spread shown in a. Each chromosome is represented from left to right by the display color, the inverted DAPI counter-stain and the classification pseudo-color. Numbers to the right of each translocation chromosome indicate the chromosomal origin of each fragment. T(13;14) is an example of an unbalanced translocation, resulting from loss of some genomic material from chromosome 13 and the gain of a portion of chromosome 14.
Figure 7.

SKY karyotype of a metaphase spread from a pancreatoblastoma cell line. Left, inverted DAPI chromosomes; right, classification pseudo-colors. Some of the aberrations consist of multiple chromosomal segments, as indicated by numbers to right of chromosomes. Assignments of these chromosome segments were subsequently confirmed with whole chromosome–painting probes to rule out the sandwich effect. This effect is visible on the chromosome 11 aberration (asterisk). Only the chromosome 15 at the boundary of chromosome segments 11 and 4 was verified; hence, the segment identified as coming from chromosome 12 is an artifact resulting from the sandwich effect, or merging of two overlapping fluorochromes. There is no normal chromosome 7 in this karyotype. To classify any segments that involve chromosome 7, a red lasso was drawn around a chromosome 7 segment (identified by its banding pattern and spectral color) contained in the derivative chromosome 4, the chromosome 7 segment was manually placed in the chromosome 7 compartment, and finally the karyotype was reclassified.
Further intricacies of SKY analysis to consider include the following. (i) If the software cannot determine the origin of a chromosome piece, it will classify this piece as belonging to the Y chromosome. (ii) The centromeres of acrocentric chromosomes (both human and mouse) will classify as something other than the chromosomes that they reside on. (iii) The pseudo-autosomal regions of the X chromosome will show up as a translocations or insertions (assigned a different classification color). (iv) There may be other classification color(s) at the junction of the translocation breakpoint, where the fluorochromes from each contributing chromosome overlap, giving what is referred to as a ‘sandwich’ effect, which results in incorrect classification of the region (see Fig. 7). Sometimes these regions really do represent a small amount of material from a third chromosome (see Fig. 7). The definitive determination of such chromosomal aberrations requires confirmation of SKY results with chromosome-specific FISH probes17. For accurate descriptions of karyotypes and human aberrations, use the nomenclature rules of ref. 16. For nomenclature rules applying to mouse, refer to the Jackson laboratory nomenclature (http://jaxmice.jax.org/info/nomenclature_hints.html).
MATERIALS
REAGENTS
Cell-culture media
-
AlexaFluor 680 F (ab′) 2 fragment of goat anti-mouse IgG (H + L) (cat. no. A21059, Invitrogen)
▲ CRITICAL Do not repeatedly freeze and thaw antibodies used in detection protocols. Store aliquots according to manufacturer’s directions and record expiration dates. Keep antibodies out of the light when using.
Colcemid (cat. no. 295-892, Roche)
Cy5-conjugated StreptAvidin (cat. no. 016-170-084, Jackson ImmunoResearch Laboratories) ▲ CRITICAL Do not repeatedly freeze and thaw antibodies used in detection protocols Store aliquots according to manufacturers directions and record expiration dates. Keep antibodies out of the light when using.
Ethyl alcohol (ethanol), anhydrous 200 proof (cat. no. AC615090020, Fisher Scientific)
-
Hydrochloric acid (HCl), 1 N (cat. no. AC12421-0010, Fisher Scientific)
! CAUTION HCl is a corrosive acid and is irritating to respiratory system. Make all solutions in a chemical fume hood. Contact your waste management department for proper disposal protocol.
Hypotonic solution, 0.075 M KCl in H2O (cat. no. 10575-082, Invitrogen)
Monoclonal anti-digoxygenin clone DI-22 mouse ascites fluid (cat. no. D8156, Sigma-Aldrich) ▲ CRITICAL Do not repeatedly freeze and thaw antibodies used in detection protocols. Store aliquots according to manufacturer’s directions and record expiration dates.
PBS, 1×, pH 7.4, without calcium and without MgCl2 (cat. no. 10010, GIBCO, Invitrogen)
SkyPaint probe (Applied Spectral Imaging)
Trypsin (cat. no. 25200-056, Invitrogen)
EQUIPMENT
Thermotron humidity chamber (model CDS-5, Thermotron)
Clinical centrifuge (for example, Allegra 6R (equipped with the GH3.8 rotor), Beckman Coulter)
Biological safety cabinet (for example, Forma 1280 Class II, Thermo Scientific)
Reciprocal shaking water baths (2) (for example, model 15–455, Fisher Scientific)
Test tube rocker (for example, model 12-815-6, Fisher Scientific)
Slide warmer (for example, model 77, Fisher Scientific)
Thermomixers (2) (Eppendorf, model 5436, Brinkmann Instruments)
Vortex mixer (for example, Daigger vortex Genie 2, A. Daigger & Co.)
Microcentrifuge (for example, model V, VWR Scientific)
Microscope slides, glass (for example, cat no. 12-544-7, Fisher Scientific)
Microscope coverglass, 18 mm × 18 mm (for example, cat. no. 48368040, VWR Scientific)
Microscope coverglasss, 24 mm × 60 mm (for example, cat. no. 48393106, VWR Scientific)
Rubber cement (local craft or drug store)
Hybridization chamber (box), 6 in × 8 in (for example, Tupperware container, local retail)
DAPI filter cube (Chroma Technology)
SKY filter cube (Chroma Technology)
Inverted tissue-culture microscope (for example, Zeiss)
Light microscope equipped with a ×16 and a ×40 dry objective (for example, Model Axiovert, Zeiss), for observing slides during preparation of spreads.
-
Epifluorescence microscope equipped with high-end objectives (for example, Leica)
▲CRITICAL Epifluorescence microscopes from Zeiss, Nikon or Olympus would alternatively be suitable. We use a ×63 oil-immersion objective for image acquisitions.
SpectraCube (Applied Spectral Imaging)
150-W xenon lamp (for example, Opti Quip)
REAGENT SETUP
Fixative for metaphase slide preparation
Combine methanol (cat. no. 3016-02, Mallinckrodt Chemicals) and acetic acid (cat. no. V193-14, Mallinckrodt Chemicals) at a 3:1 ratio (vol/vol).
! CAUTION Methanol is a poison, and highly flammable. Use only with adequate ventilation (chemical fume hood). Wear gloves when handling this agent.
Contact your waste management department for proper disposal protocol.
▲ CRITICAL Freshly prepare before use. Can be stored before procedure (1–2 h) at 4 °C.
Slide pretreatment solution 1 (RNase A stock solution, 20 mg ml−1 % wt/vol).
Dissolve RNase A (cat. no. 109169, Roche Molecular Biochemicals) to a final concentration of 20 mg ml−1 in sterile water, boil 15 min, cool to room temperature (22 °C). Store aliquots, 100 μl each, at −20 °C.
Slide pretreatment solution 2 (pepsin stock solution, 100 mg ml−1)
Dissolve pepsin (cat. no. P-6887, Sigma-Aldrich) into sterile water, to a final concentration of 100 mg ml−1; keep on ice until aliquoted. Store aliquots, 20 μl each, at −20 °C.
Slide pretreatment solution 3 (1× PBS/MgCl2)
Add 50 ml of 1 M MgCl2 (cat. no. 351-033-060, Quality Biological) to 950 ml 1× PBS. Final volume is 1 l. Store at room temperature indefinitely.
Slide pretreatment solution 4 (1% vol/vol formaldehyde/1× PBS/MgCl2)
Add 2 ml of 37% formaldehyde (cat. no. 5016, Mallinckrodt Chemicals) to 73 ml 1× PBS/MgCl2. Make fresh for each experiment, mix well and store at room temperature until use. ! CAUTION Formaldehyde is flammable, carcinogenic and a poison. Formaldehyde should be dispensed in a chemical fume hood and only handled when wearing gloves. Contact your waste management department for proper disposal protocol.
Slide denaturation solution (70% vol/vol deionized formamide/2× SSC, or FA/SSC)
Combine 10 ml of 20× SSC (cat. no. 1666681, Roche Molecular Biochemicals), 20 ml dH2O and 70 ml deionized formamide (cat. no. 9342, Ambion). Adjust to pH 7.25 with 1.0 N HCl. Mix well; store as 1-ml aliquots at −20 °C. ! CAUTION Formamide causes eye and skin irritation and is a known mutagen. Wear gloves when handling this reagent and dispense under a chemical fume hood. Contact your waste management department for proper disposal protocol. ▲ CRITICAL Denature slides using deionized formamide, as it contains fewer impurities than conventional formamide.
Wash solution 1 (50% vol/vol formamide/2× SSC)
sCombine 100 ml formamide (cat. no. 47670, Sigma-Aldrich) with 20 ml 20× SSC and 80 ml ddH2O. Adjust pH to 7.0 using 1.0 N HCl. Mix well and use fresh; prewarm and maintain temperature in a water bath set at 45 °C until use.
Wash solution 2 (1× SSC)
Add 25 ml of 20× SSC to 475 ml dH2O. Mix well and use fresh; prewarm and maintain temperature in a water bath set at 45 °C until use.
Wash solution 3 (4× SSC/Tween 20)
Add 200 ml of 20× SSC to 799 ml of dH2O, and add 1 ml of polyoxyethylene-sorbitan monolaurate (Tween-20; cat. no. P-1379, Sigma-Aldrich). Mix well and use fresh; prewarm in a water bath at 45 °C until use.
Blocking solution (BSA)
Add 0.30 g of bovine serum albumin (BSA) Fraction V, protease-free, lyophilizate (cat. no. 03-117-332-001, Roche Molecular Biochemicals) to 10 ml of 4× SSC/Tween 20. Mix well and use fresh; prewarm and maintain temperature in a water bath set at 37 °C until use.
2×SSC. Add 10 ml of 20× SSC to 90 ml of dH2O.
Carbonate-bicarbonate buffer (pH 9.0)
Add 4 ml sodium bicarbonate, 0.5 M (pH 8.13; cat. no. S-6297, Sigma-Aldrich) to 1 ml sodium carbonate, 0.5 M (pH 11.32; cat. no. S-6139, Sigma-Aldrich), and filter-sterilize. Store, sealed at room temperature, up to 1 year.
Antifade solution
Dissolve 50 mg 1,4-phenylenediamine dihydrochloride (antifade; cat. no. P1519, Sigma-Aldrich) in 2 ml 1× PBS and adjust pH with carbonate-bicarbonate buffer to achieve pH 8.0. Add 1× PBS to 5 ml. Mix with 45 ml 86% (vol/vol) glycerol (cat. no. 11514011, Invitrogen). Leave on test tube rocker for at least 1 h. Aliquots can be stored at −20 °C. ▲ CRITICAL Add 2 ml buffer and check pH with pH paper; add dropwise 0.1 M HCl until pH approaches 8.4. As the pH approaches 8.0, check the pH with a pH meter, making sure to wash the pH meter’s probe thoroughly. If pH exceeds 8.0, the procedure must be repeated.
DAPI solution
Add 2 mg of DAPI (cat. no. 236276, Roche Molecular Biochemicals) to 10 ml dH2O for stock solution at a concentration of 0.2 mg ml−1. Store in aliquots at −20 °C. For working DAPI solution, add 40 μl of stock solution to 100 ml of 2× SSC.
! CAUTION DAPI is a known mutagen; avoid contact with eyes and skin. Protect DAPI solution from light; it can be stored at 4 °C for as long as 2 months.
EQUIPMENT SETUP
Fluorescence microscope and SKY imaging system
See Figure 3. If interested in setting up a microscope to perform SKY analysis, please directly contact Applied Spectral Imaging (http://www.spectral-imaging.com/), as they are the only suppliers of this system.
PROCEDURE
Metaphase preparation
-
1|
Grow adherent and or suspension cell cultures in their preferred media under their preferred growth conditions. In the case of adherent cell cultures, grow until they reach 60%–70% confluence (% confluence means the degree to which cells are making contact with each other). This determination is made using an inverted tissue-culture microscope.
-
2|
Add colcemid (0.1 g ml−1, 10 μl for every 1 ml medium) to cell cultures using sterile micropipettes.
-
3|
Incubate cells for 30 min–1 h.
▲ CRITICAL STEP For fast-growing cultures, reduce time of exposure to colcemid, and for slower-growing cultures, incubate for several hours with colcemid. Monitor cells throughout incubation period under the inverted tissue-culture microscope. Ideally, cultures with approximately 10–15 dividing cells per field of view (cells appear round and shiny) are ready for harvesting of metaphase chromosomes. For suspension and adherent cultures, one 75-cm2 flask should provide sufficient numbers of cells for preparation of metaphase spreads.
-
4|
Harvest either adherent cells (A) or suspension (B) cell cultures.
(A) Adherent cells
Remove medium from each flask with a sterile 10-ml pipette; save entire contents in a 50-ml conical centrifuge tube.
Add 5 ml sterile 1× PBS (pH 7.4) to each flask, rinse, and then transfer contents to 50-ml tube (the same tube as described above).
-
Add 0.5 ml sterile trypsin to each flask and incubate until cells detach. Time will vary with each cell culture, as some cells adhere to the bottom of the flask more than others, but is approximately 1–2 min. Hitting the flask may be required for firmly attached cells.
▲ CRITICAL STEP Monitor cells under inverted microscope continuously, as some cultures are sensitive to trypsin.
-
Add 5 ml of growth medium; squirt the medium with a pipette onto the surface of the flask to remove the cells from the sides of the flask or by hitting the flat part of your hand onto the bottom of the flask, and add the contents (the fresh medium, which now contains the detached cells) to the same 50-ml conical centrifuge tube used in Steps 4A(i) and 4A(ii).
▲ CRITICAL STEP The fetal bovine serum (FBS) present in the medium used to grow the cells inactivates the trypsin.
Spin cells in a clinical centrifuge at 175g (1,000 r.p.m.), for 5 min at room temperature.
Remove the supernatant carefully by vacuum aspiration, leaving 0.5 ml in the tube, and flick the tube with finger to loosen the pellet.
(B) Suspension cultures
Transfer cultures to centrifuge tubes and spin at in a clinical centrifuge at 175g (1,000 r.p.m.) for 5 min at room temperature.
Remove the supernatant carefully by vacuum aspiration, leaving 0.5 ml in the tube, and flick the tube with a finger to loosen the pellet.
-
5|
Add 2–50 ml hypotonic solution to the conical tube containing the cells in suspension and incubate at room temperature for 15–25 min.
▲ CRITICAL STEP The volume of hypotonic solution added to each tube is variable and determined specifically by the final amount of the cells in pellet. One does not need to measure the number of cells, but generally speaking, if the cell pellet is just coating the bottom of the tube, add no more than 2 ml hypotonic solution; if there appears to be 0.5–1.0 ml of cells at the bottom of the tube, add approximately 50 ml hypotonic solution. The cell-hypotonic mixture should appear slightly cloudy and not clear. Some cultures (such as lymphocytes and plasma cytoma) are more sensitive, and the time of exposure to the hypotonic solution should be approximately 15 min. Adherent cells typically require 20–25 min exposure. Chromosome metaphase preparations on the slides will have excess cytoplasm if exposure to the hypotonic time is too short (see Fig. 2).
?TROUBLESHOOTING
-
6|
Using a 1-ml pipette, add one drop of fresh methanol/acetic acid fixative (3:1 vol/vol) per ml of cell/hypotonic mixture dropwise to the tube and invert to mix (this step will stop the hypotonic process and prefix the cells).
-
7|
Spin cell suspension using a clinical centrifuge at 250g (1,200 r.p.m.) for 5 min at room temperature.
-
8|
Remove the supernatant carefully, leaving 0.5 ml liquid, and flick the tube to loosen the pellet and fully resuspend all cells.
-
9|
Add 5 ml fresh fixative down the side of the tube, very slowly, gently flicking pellet all the time.
▲ CRITICAL STEP To ensure that the final metaphase cell pellet is fully resuspended, it is crucial to add fixative very slowly down the side of the centrifuge tube; otherwise, the cell pellet will remain full of clumps, metaphase spreads will become trapped in the clumps and spreading will be compromised.
-
10|
Pellet the cells at 250g (1,200 r.p.m.) for 5 min at room temperature.
-
11|
Remove the supernatant and flick the tube to loosen the pellet and fully resuspend the cells.
-
12|
Add 5 ml fresh fixative to wash the cell preparations.
-
13|
Pellet the cells at 250g (1,200 r.p.m.) for 5 min at room temperature.
-
14|
Remove the supernatant and flick the tube to loosen the pellet and fully resuspend the cells.
-
15|
Add 1–5 ml fresh ice-cold fixative along the wall of the tube.
▲ CRITICAL STEP The final volume of fixative is determined by the desired concentration of cells on the slide. This requires a visual examination of the final solution; it should appear slightly cloudy, yet translucent, when held up to the light. If too much fixative is added, there may not be enough metaphases in the hybridization area of the slide for a thorough analysis. If the fixative volume is too small, the cell density on the slide will be high, preventing the chromosomes from spreading well and also resulting in too much cellular debris in close proximity to the metaphase spreads. This debris will nonspecifically bind the fluorescent probe, resulting in brightly fluorescent ‘spots’ that will interfere with the image acquisition (owing to pixel intensity saturation in the camera).
■ PAUSE POINT To store, tighten cap and wrap cap with laboratory film (Parafilm); store at −20 °C for short-term storage and at −80 °C for several years.
-
16|
In a humidity chamber or Thermotron set at 45% or 55% humidity at room temperature (for mouse or human cells, respectively), drop ~15 μl of the fixed cell suspension onto a clean glass microscope slide and allow the slide to fully dry by evaporation (approximately 1–2 min).
-
17|
View the dried slide with a ×16 high-dry phase objective on a light microscope to determine final cell density, extent of chromosome spreading and the presence or absence of cytoplasm.
▲ CRITICAL STEP The spreading of chromosomes and the integrity of chromosome structure are dependent on the evaporation rate of the fixative, as determined by the percent humidity, temperature and success of the hypotonic procedure. If the chromosomes are light gray in color, increase the humidity; if the chromosomes have excess cytoplasm (see Fig. 2a), appear shiny or have a bright halo around them, decrease the humidity in the chamber.
? TROUBLESHOOTING
-
18|
Dry the slides at 37 °C, or at room temperature if the humidity is low in the storage space.
▲ CRITICAL STEP Metaphase spreads on slides can be heated for 1 h at 80 °C or stored in a drying oven overnight (40 °C) before hybridization; however, slides stored for one week at 37 °C yield optimum results.
-
19|
Check the slide under a light microscope using a ×40 high-dry phase objective. If cytoplasm is present (gray particulate matter surrounding the chromosomes; see Fig. 2a), pretreat the slides with pepsin as described in Box 1. If cytoplasm is not visible (a clear light margin surrounds the metaphase chromosomes) and the chromosomes have good morphology (black in color, not phase-light or shiny), there is no need for slide pretreatment with pepsin.
? TROUBLESHOOTING
-
20|
Using a light microscope equipped with phase optics and ×10 and ×40 dry lenses, select an area for hybridization that will fit under an 18 mm × 18 mm coverglass.
▲ CRITICAL STEP Selection of the region of the slide to be hybridized requires that there be sufficient numbers of metaphase spreads to view and that the chromosome morphology has been preserved.
■ PAUSE POINT The slide(s) can be stored in a dry location until hybridization (1–3 d). Alternatively, they can be frozen at −80 °C, sealed in bags containing desiccant to eliminate moisture.
Slide and probe denaturation
-
21|
Warm SkyPaint probe in a Thermomixer set to 37 °C with shaking (1,100 r.p.m.) for 20 min, vortex and centrifuge briefly in a microcentrifuge at 80g (1,000 r.p.m.) for 1–3 s at room temperature.
-
22|
Denature the SKY probe in a Thermomixer set to 85 °C for 5 min and immediately place probe back into a different Thermomixer or water bath set to 37 °C, to allow the labeled probe DNA to preanneal with the highly repetitive genomic elements enriched in the CotI DNA fraction, which is also part of the SkyPaint probe, for approximately 1 h.
? TROUBLESHOOTING
-
23|
During the time the SKY probe is incubating, begin the slide-denaturation steps by adding 120 μl of slide-denaturation solution (formamide/2× SSC) to a 24 mm × 60 mm coverglass.
-
24|
Invert the slide onto the coverglass; reinvert the slide, metaphase-spread-side up; next, make sure it is free of air bubbles; and then place the slide onto a slide warmer set at 70 °C for mouse cells and 80 °C for human cells; incubate the slides for 30 s–1.5 min.
▲ CRITICAL STEP Denaturation of the slide is a major determinant of the success of the hybridization of SKY probes. It is important that the slide warmer is set to the right temperature: for mouse chromosomes, the slide warmer should be set to 70 °C, and for human chromosomes to 80 °C. The age of slides, amount of cytoplasm and humidity at which cells were dropped onto the slides determines the time required for denaturation. The correct timing is often determined empirically.
-
25|
Immerse the slide into ice-cold 70% (vol/vol) ethanol for 3 min (this immediately arrests the heat denaturation process, preventing the chromosomes from reannealing). Further dehydrate the slide by transferring it into 90% (vol/vol) ethanol for 3 min, and then into 100% ethanol for 3 min, and air-dry (90% and 100% ethanol are kept in separate coplin jars and held at room temperature).
▲ CRITICAL STEP Cool 70% ethanol to −20 °C by storing a tightly sealed 100-ml coplin jar inside a −20 °C freezer before use; when ready to use, keep the coplin jar inside an ice bucket.
-
26|
Examine the slide when dry under a microscope that has phase optics and determine whether the chromosome morphology is still intact. If the morphology of the chromosomes remains intact, the chromosomes will hybridize. If the chromosomes look bloated or ‘hollow’, do not hybridize this slide, as the hybridization procedure is unlikely to succeed.
Probe hybridization
-
27|
Carefully apply the entire contents of the SkyPaint probe, using a 20-μl micropipette, onto the area selected for hybridization, and gently place an 18 mm × 18 mm coverglass onto the region containing the probe.
▲ CRITICAL STEP Before adding the probe to the slide, determine the hybridization area as mentioned above, and mark it. This can be done by gently etching the underside of the glass slide with a diamond pen, marking the sides of the slide with a pencil or recording the Vernier (x and y) coordinates of the microscope stage and synchronizing these coordinates with the microscope being used for image acquisition. Keep slide away from bright lights while adding the fluorescently labeled probe.
-
28|
Remove air bubbles by gently pushing on the coverglass with a forceps, being careful not to expel large amounts of the hybridization solution. Seal the edges of the coverglass with rubber cement.
-
29|
Place the slide into a hybridization chamber that is protected from ambient light, with sufficient moisture (~1–2 ml) to prevent the rubber cement from cracking and allowing the hybridization solution to evaporate. Keep the hybridization chamber in a 37 °C incubator for 48–72 h.
▲ CRITICAL STEP The hybridization chamber must be a lightproof and a waterproof box. Keep the box moist by adding 1–2 ml water inside the box or by placing a damp paper towel inside, taking care, of course, that the slide does not come into contact with the water. Perform the hybridization procedure in an incubator. It is not necessary to have this incubator contain CO2, as is found in tissue-culture incubators. Some older samples (specimens over 1 year) may require longer incubation (4 d rather than 2 d) to achieve optimal hybridization.
Fluorescence detection
-
30|
Remove the hybridization chamber from the incubator after 48–72 h of incubation.
-
31|
Carefully remove the rubber cement from the slide, taking care not to drag the coverglass across the slide, thereby scratching the metaphase preparations.
-
32|
Place the slide into a coplin jar containing wash solution 1 (the size of the coplin jar used depends on the numbers of slide in experiment: typically, a 50-ml coplin jar is ideal for 6, and a 100-ml coplin jar can accommodate 10–12 slides). Wash slides with shaking at 45 °C for 5 min.
▲ CRITICAL STEP For all steps that indicate shaking of the slide, shake by placing the coplin jar inside a reciprocal shaking water bath set at 45 °C and 45 r.p.m. Prewarm wash solutions 1, 2 and 3 in a water bath at 45 °C.
-
33|
Repeat Step 32 two more times, with shaking, using fresh wash solution 1 each time (using the same coplin jar as in Step 32).
-
34|
After discarding wash solution 1 (using the same warm coplin jar), wash the slides with wash solution 2 a total of three times, 5 min each, with shaking.
-
35|
Discard wash solution 2 and wash once for 5 min at 45 °C with wash solution 3, with shaking.
-
36|
Dispense 120 μl of blocking solution (BSA) onto a 24 mm × 60 mm coverglass, invert the slide metaphase-spread-side down onto the liquid bearing coverglass, and immediately reinvert the slide, taking care not to scratch the slide during this process and taking care to remove air bubbles.
▲ CRITICAL STEP Prewarm the blocking solution in a water bath at 37 °C.
-
37|
Incubate slide at 37 °C for 30 min in the moist hybridization chamber (as described in Step 29).
-
38|
After incubation, remove coverglass, taking care not to scratch the slide, and place the slide in a new coplin jar with prewarmed wash solution 3. Wash by shaking for 5 min.
? TROUBLESHOOTING
-
39|
At this point, prepare the first layer of detection reagents. Use mouse anti-digoxygenin at a 1:150 dilution and StreptAvidin-Cy5 at a 1:200 dilution. Combine both reagents so that they are diluted into an appropriate volume of wash solution 3, taking into account the total number of slides that will require the detection solution.
-
40|
Place 120 μl of detection solution on a 24 mm × 60 mm coverglass for each slide. Shake off excess liquid from each slide, invert the slide metaphase-spread-side down onto the liquid bearing coverglass, and immediately reinvert the slide, taking care not to scratch the slide during this process and to gently remove air bubbles.
-
41|
Place the slide (metaphase-spread-side up) inside the moist hybridization chamber (as described in Step 29) and incubate for 1 h at 37 °C.
-
42|
Wash the slide in wash solution 3, three times, using a coplin jar, for 5 min each, with shaking.
-
43|
Prepare second layer of detection solution, which contains AlexaFluor 680 F (ab′) 2 fragment of goat anti-mouse IgG (H + L) at a 1:200 dilution in wash solution 3. The final volume (using 120 μl per slide) will be determined by the number of slides you are detecting.
-
44|
Following similar procedures as in Steps 40–42, using the second layer of detection solution.
? TROUBLESHOOTING
-
45|
Shake off excess wash solution 3 from slide. Place the slide in the DAPI solution for 5 min, with shaking at room temperature.
▲ CRITICAL STEP We do not recommend the use of other antifade products that already contain DAPI (for example, VectaShield). These products prevent photobleaching of the far-red dyes less efficiently. Keep slides out of direct light throughout DAPI staining procedure.
-
46|
Wash the slide for 5 min in a coplin jar containing 2× SSC, with shaking, at room temperature.
-
47|
Dehydrate the slide sequentially in three different coplin jars containing 70% (room temperature), 90% and 100% ethanol, respectively, without shaking, keeping the slides out of ambient light, for 3 min in each solution.
-
48|
Air-dry the slide in the dark (in a drawer, or on the workbench under a foil tent.
-
49|
Once the slides is dry, add 35 μl antifade solution to a 24 mm × 60 mm coverglass, invert the slide metaphase-spread-side down onto the liquid bearing coverglass and immediately reinvert the slide. Carefully remove any air bubbles.
■ PAUSE POINT Slides can be imaged immediately or, alternatively, can be stored at 4 °C protected from light, ideally no longer than 1 week.
Image acquisition and analysis
-
50|
The exact procedure for image capture will vary with the microscope and lamp system used. Please see the ‘Experimental design’ section of the INTRODUCTION for further information.
● TIMING
Steps 1–20 (metaphase preparation and aging of slides): 1 week
Box 1, Steps 1–17 (slide pretreatment): day 1, ~2 h
Steps 21–26 (probe and slide denaturation): day 1, ~1.5 h;
Steps 27–29 (hybridization) day 1, ~15 min;
Steps 30–49 (detection): day 4, ~4.5 h;
Step 50: image acquisition, day 5, ~10–15 min per metaphase spread (this is dictated by the acquisition time, as assesed by the ASI software); image analysis, day 6, refer to Figure 1.
? TROUBLESHOOTING
See Table 1.
Table 1.
Troubleshooting table.
| Problem | Possible reason | Solution |
|---|---|---|
| No signal | Quality of metaphases Slides are over 1 year old |
If there is a cell pellet stored, try dropping new slides with better humidity conditions and/or longer hypotonic conditions |
| Excess cytoplasm surrounding the chromosomes | Try a stronger pepsin treatment; generally, the pepsin stock solution (slide pretreatment solution 2) can be increased in 5-μl increments | |
| Probe denaturation temperature | Check the actual temperature of the equipment used to denature the probe | |
| Slide denaturation temperature | Check the actual temperature of the instrument used to heat the slides (water bath or hot plate). Always check the structural integrity of the slide after pretreatment and denaturation (before hybridization); chromosomes should not appear hollow or too shiny | |
| Detection reagents are approaching expiration | Always check the date on the detection reagents, as some expire within 3 months of purchase | |
| Detection reagents were improperly diluted | It is important to not overdilute the detection reagents; check the concentrations | |
| Antibodies were added in the wrong order | It is important to first add the mouse anti-digoxygenin and then detect that with the anti-mouse fluor | |
| The wrong filter was used during imaging | Always make sure to use the SKY filter cube for taking SKY images. | |
| Weak signal | The chromosomes were too phase-light from the start | When checking the slide, make sure to choose an area of hybridization that contains darkly stained chromosomes, not light gray ones. If no dark areas exist, try to drop new metaphases at a higher humidity |
| Forgot to use RNase and/or pepsin | These steps remove excess cytoplasm and RNA, allowing the probe to make full contact with the DNA on the slide (see Box 1) | |
| Preannealing time was too extensive | Longer preannealing times allow for unique single-copy genomic sequences to form double-stranded DNA with their complementary strands in the tube, rather than on the slide. | |
| Wash solutions were stored a long period of time | Always make up fresh solutions, especially wash 1. Make sure the pH is in the range of 7–7.4 | |
| High background/low signal specificity | Detection was not performed at the correct temperature | The stringency of the washes can be compromised by a lower temperature; therefore, check the actual temperatures of the water baths |
| Slides were not denatured long enough | Always check the appearance of chromosomes after denaturation; they should look as good as when you started the procedure. Try increasing time in 5- to 10-s increments | |
| Insufficient hybridization time | Try a 72-h incubation | |
| Excess cytoplasm surrounding the chromosomes | See Box 1 | |
| BSA was not properly washed off | Always check the BSA concentration when making wash solution 4. Also note the temperature of the wash solutions after Step 42 |
ANTICIPATED RESULTS
The results are dependent on quality of the metaphase preparations and SkyPaint probes, as described in the INTRODUCTION (also, see Fig. 2). Figures 4–6 show typical examples of SKY hybridizations of normal and abnormal human and mouse chromosome complements.
Acknowledgments
We thank E. Schröck and M. Liyange for their input into the initial development of SKY for human and mouse, respectively, T. Knutsen for editing of the original SKY protocols and B. Chen and J. Cheng for their technical assistance.
Footnotes
COMPETING INTERESTS STATEMENT: The authors declare that they have no competing financial interests.
References
- 1.Seabright M. Rapid banding technique for human chromosomes. Lancet. 1971;II:971–972. doi: 10.1016/s0140-6736(71)90287-x. [DOI] [PubMed] [Google Scholar]
- 2.Caspersson T, et al. Chemical differentiation along metaphase chromosomes. Exp Cell Res. 1968;49:219–222. doi: 10.1016/0014-4827(68)90538-7. [DOI] [PubMed] [Google Scholar]
- 3.Arrigghi FF, Hsu TC. Localization of heterochromatin in human chromosomes. Cytogenetics. 1971;10:81–86. doi: 10.1159/000130130. [DOI] [PubMed] [Google Scholar]
- 4.Landegent JE, Jansen in de Wal N, Dirks RW, Baas F, van der Ploeg M. Use of the whole cosmid cloned genomic sequences for chromosomal localization by non-radioactive in situ hybridization. Hum Genet. 1987;77:366–370. doi: 10.1007/BF00291428. [DOI] [PubMed] [Google Scholar]
- 5.Pinkel D, et al. Fluorescence in situ hybridization with human chromosome specific libraries: detection of trisomy 21 and translocation of chromosome 4. Proc Natl Acad Sci USA. 1988;85:9138–9142. doi: 10.1073/pnas.85.23.9138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cremer T, Lichter P, Borden J, Ward DC, Mannuelidis L. Detection of chromosome aberrations in metaphase and interphase tumor cells by in situ hybridization using chromosome-specific library probes. Hum Genet. 1988;80:235–246. doi: 10.1007/BF01790091. [DOI] [PubMed] [Google Scholar]
- 7.Schröck E, et al. Multicolor spectral karyotyping of human chromosomes. Science. 1996;273:494–497. doi: 10.1126/science.273.5274.494. [DOI] [PubMed] [Google Scholar]
- 8.Veldman T, Vignon C, Schrock E, Rowley JD, Ried T. Hidden chromosome abnormalities in haematological malignancies detected by multicolour spectral karyotyping. Nat Genet. 1997;15:406–410. doi: 10.1038/ng0497-406. [DOI] [PubMed] [Google Scholar]
- 9.Macville M, et al. Comprehensive and definitive molecular cytogenetic characterization of HeLa cells by spectral karyotyping. Cancer Res. 1999;59:141–150. [PubMed] [Google Scholar]
- 10.Hilgenfeld E, Padilla-Nash H, Schrock E, Ried T. Analysis of B-cell neoplasias by spectral karyotyping (SKY) Curr Top Microbiol Immunol. 1999;246:169–174. doi: 10.1007/978-3-642-60162-0_21. [DOI] [PubMed] [Google Scholar]
- 11.Padilla-Nash HM, et al. Jumping translocations are common in solid tumor cell lines and result in recurrent fusions of whole chromosome arms. Genes Chromosom Cancer. 2001;30:349–363. doi: 10.1002/gcc.1101. [DOI] [PubMed] [Google Scholar]
- 12.Knutsen T, et al. The interactive online SKY/M-FISH & CGH database and the Entrez cancer chromosomes search database: linkage of chromosomal aberrations with the genome sequence. Genes Chromosom Cancer. 2005;44:52–64. doi: 10.1002/gcc.20224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Speicher MR, Ballard SG, Ward DC. Karyotyping human chromosomes by combinatorial multi-fluor FISH. Nat Genet. 1996;12:368–375. doi: 10.1038/ng0496-368. [DOI] [PubMed] [Google Scholar]
- 14.Geigl JB, Uhrig S, Speicher MR. Multiplex-fluorescence in situ hybridization for chromosome karyotyping. Nat Prot. 2006;1:1172–1184. doi: 10.1038/nprot.2006.160. [DOI] [PubMed] [Google Scholar]
- 15.Padilla-Nash HM, et al. Molecular cytogenetic analysis of the bladder carcinoma cell line BK-10 by spectral karyotyping. Genes Chromosom Cancer. 1999;25:53–59. doi: 10.1002/(sici)1098-2264(199905)25:1<53::aid-gcc8>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 16.Shaffer LG, Tommerup N, editors. ISCN: An International System for Human Cytogenetic Nomenclature. S Karger; Basel, Switzerland: 2005. [Google Scholar]
- 17.Barenboim-Stapleton L, et al. Pediatric pancreatoblastoma: histopathologic and cytogenetic characterization of tumor and derived cell line. Cancer Genet Cytogenet. 2005;157:109–117. doi: 10.1016/j.cancergencyto.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 18.Schröck E, et al. Spectral karyotyping refines cytogenetic diagnostics of constitutional chromosomal abnormalities. Hum Genet. 1997;101:255–262. doi: 10.1007/s004390050626. [DOI] [PubMed] [Google Scholar]
- 19.Haddad BR, et al. Identification of de novo chromosomal markers and derivatives by spectral karyotyping. Hum Genet. 1998;103:619–625. doi: 10.1007/s004390050878. [DOI] [PubMed] [Google Scholar]
- 20.Ning Y, Laundon CH, Schrock E, Buchanan P, Ried T. Prenatal diagnosis of a mosaic extra structurally abnormal chromosome by spectral karyotyping. Prenat Diagn. 1999;19:480–482. [PubMed] [Google Scholar]
- 21.Cotter PD, et al. Prenatal diagnosis of minute supernumerary marker chromosomes. Gynecol Obstet Invest. 2005;60:27–38. doi: 10.1159/000083482. [DOI] [PubMed] [Google Scholar]
- 22.Schrock E, et al. Spectral karyotyping of human, mouse, rat and ape chromosomes–applications for genetic diagnostics and research. Cytogenet Genome Res. 2006;114:199–221. doi: 10.1159/000094203. [DOI] [PubMed] [Google Scholar]
- 23.Muller S, Wienberg J. “Bar-coding” primate chromosomes: molecular cytogenetic screening for the ancestral hominoid karyotype. Hum Genet. 2001;109:85–94. doi: 10.1007/s004390100535. [DOI] [PubMed] [Google Scholar]
- 24.Rens W, Fu B, O’Brien PCM, Ferguson-Smith M. Cross-species chromosome painting. Nat Prot. 2006;1:783–790. doi: 10.1038/nprot.2006.91. [DOI] [PubMed] [Google Scholar]
- 25.Buwe A, et al. Multicolor spectral karyotyping of rat chromosomes. Cytogenet Genome Res. 2003;103:163–168. doi: 10.1159/000076306. [DOI] [PubMed] [Google Scholar]
- 26.Barkan D, Montagna C, Ried T, Green JE. Mammary gland cancer. In: Holland EC, editor. Mouse Models of Human Cancer. Wiley-Liss; Hoboken, New Jersey: 2004. pp. 103–131. [Google Scholar]
- 27.Liyanage M, et al. Multicolour spectral karyotyping of mouse chromosomes. Nat Genet. 1996;14:312–315. doi: 10.1038/ng1196-312. [DOI] [PubMed] [Google Scholar]
- 28.Weaver Z, et al. Mammary tumors in mice conditionally mutant for Brca1 exhibit gross genomic instability and centrosome amplification yet display a recurring distribution of genomic imbalances that is similar to human breast cancer. Oncogene. 2002;21:5097–5107. doi: 10.1038/sj.onc.1205636. [DOI] [PubMed] [Google Scholar]
- 29.Montagna C, et al. The Septin 9 (MSF) gene is amplified and overexpressed in mouse mammary gland adenocarcinomas and human breast cancer cell lines. Cancer Res. 2003;63:2179–2187. [PubMed] [Google Scholar]
- 30.Coleman AE, et al. Previously hidden chromosome aberrations in T(12;15)-positive BALB/c plasmacytomas uncovered by multicolor spectral karyotyping. Cancer Res. 1997;57:4585–4592. [PubMed] [Google Scholar]
- 31.Barch M, Knutsen T, Spurbeck J, editors. The AGT Cytogenetics Laboratory Manual. Raven Press; New York: 1997. [Google Scholar]
- 32.Telenius H, et al. Cytogenetic analysis by chromosome painting using DOP-PCR amplified flow sorted chromosomes. Genes Chromosom Cancer. 1992;4:257–263. doi: 10.1002/gcc.2870040311. [DOI] [PubMed] [Google Scholar]
- 33.Amplification of flow sort DNA using DOP-PRC (SKY) Laboratory of Thomas Ried Protocols. 2005 http://www.riedlab.nci.nih.gov/protocols.asp.
- 34.DOP-PCR secondary (SKY) Laboratory of Thomas Ried Protocols. 2005 http://www.riedlab.nci.nih.gov/protocols.asp.
- 35.DOP-PCR labeling (SKY) Laboratory of Thomas Ried Protocols. 2005 http://www.riedlab.nci.nih.gov/protocols.asp.
- 36.McNamara G, Difilippantonio MJ, Ried T. Microscopy and image analysis. In: Miranker L, editor. Current Protocols in Human Genetics. John Wiley & Sons; New York: 2005. pp. 4.4.1–4.4.34. [DOI] [PMC free article] [PubMed] [Google Scholar]