

Abstract
Background
Despite advances in targeted therapy for lung cancer, survival for patients remains poor and lung cancer remains the leading cause of cancer-related deaths worldwide. The type I insulin-like growth factor receptor (IGF-IR) has emerged as a potential target for lung cancer treatment, however, clinical trials to date have provided disappointing results. Further research is needed to identify if certain patients would benefit from IGF-IR targeted therapies and the ideal approach to incorporate IGF-IR targeted agents with current therapies.
Methods
The dual IGF-IR/insulin receptor inhibitor, BMS-754807, was evaluated alone and in combination with platinum-based chemotherapeutics in two human non-small cell lung cancer (NSCLC) cell lines. Cell survival was determined using WST-1 assays and drug interaction was evaluated using Calcusyn software. Proliferation and apoptosis were determined using immunofluorescence for phospho-histone H3 and cleaved caspase 3, respectively.
Results
Treatment with BMS-754807 alone reduced cell survival and wound closure while enhancing apoptosis in both human lung cancer cell lines. These effects appear to be mediated through IGF-IR/IR signaling and, at least in part, through the PI3K/AKT pathway as administration of BMS-754807 to A549 or NCI-H358 cells significantly suppressed IGF-IR/IR and AKT phosphorylation. In addition of BMS-754807 enhanced the cytotoxic effects of carboplatin or cisplatin in a synergistic manner when given simultaneously to A549 cells.
Conclusions
BMS-754807 may be an effective therapeutic agent for the treatment of NSCLC, particularly in lung cancer cells expressing high levels of IGF-IR.
Keywords: BMS-754807, IGF-IR, Chemotherapy, Lung cancer, Proliferation, Apoptosis, Migration
Background
Lung cancer remains the leading cause of cancer-related deaths worldwide [1], and non-small cell lung cancer (NSCLC) represents ~80 % of the cases of lung cancer [2]. Lung cancer is often not diagnosed until later stages [3, 4] when it is no longer responsive to the standard treatment which is based on platinum chemotherapeutics and radiation [5–7]. Despite the development of a number of new targeted therapies against epidermal growth factor receptor (EGFR), vascular endothelial growth factor (VEGF), and anaplastic lymphoma kinase (ALK), overall survival rates remain poor [7–10]. There is a need to develop new treatment strategies including new drugs and understand the most effective way to incorporate them into current treatments. Several type I insulin-like growth factor receptor (IGF-IR) targeting agents have been in clinical development, including monoclonal antibodies and tyrosine kinase inhibitors, which are reviewed by Scagliotti and Novello [11] and Chen and Sharon [12]. Figitumumab, a monoclonal antibody against IGF-IR, reached a phase III clinical trial in non-adenocarcinoma NSCLC that was ended due to poor outcomes [13]. This disappointing outcome leaves researchers asking if a better understanding of the role of IGF-IR in lung cancer would identify certain biomarkers, histological subtypes, and drug combinations or sequences that could improve the effectiveness of targeting the IGF-IR as part of lung cancer treatment.
There is strong evidence implicating the IGF-IR in lung cancer. Molecular studies using lung cancer cell lines have shown that disruption of IGF-IR signaling will decrease proliferation, cell survival and migration in several lung cancer cell lines [14–16]. IGF-IR is also involved in tumor initiation since overexpression of the IGF-IR in type II alveolar cells or Clara cells was sufficient to induce spontaneous tumor formation in mice [17]. Several studies have found the IGF-IR to be frequently over-expressed in NSCLC, ranging from 30 to 84 % of patients [18–29]. While many of these studies did not find high IGF-IR expression to be associated with overall survival, it has been associated with progressive disease [18, 29–31], larger tumor size [23, 26], recurrence [24], and brain metastasis [32]. Additionally, high IGF-1 and low IGFBP-3 have been associated with poor outcome [33–39]. This suggests that increased activation of the IGF axis contributes to lung cancer progression; therefore, targeting the receptor would be an effective therapeutic strategy. Furthermore, activation of the IGF-axis via the IGF-IR has been identified as a mechanism of resistance to EGFR targeted therapies [40–45] and to cisplatin [46–48].
The IGF-IR is a tyrosine kinase receptor which exists as a dimer (α2β2): two monomers consisting of an alpha subunit and a beta subunit that are joined by disulfide bonds [49]. The IGF-IR signals through two main pathways, the PI3K/AKT pathway and the MAPK/ERK pathway [50, 51]. The IGF-IR will form heterodimers with the closely related insulin receptor (IR), which shares 60 % structural similarity [52–54]. The IGF-IR responds to ligand stimulation by IGF-I, IGF-II, and insulin. While the IR is primarily activated by insulin, it will also bind to IGF-II and can bind both IGF-I and IGF-II when part of a heterodimer with IGF-IR [55–60]. Since both IGF-I and IGF-II are reported to be important activators of IGF-IR signaling in lung cancer [61–64], there is a good rationale for targeting the IR as well as the IGF-IR in order to achieve complete inhibition of the IGF axis. Additionally, in breast cancer cells and Ewing sarcoma the IR was identified as a mechanism of resistance to IGF-IR specific monoclonal antibodies [65, 66]. There may be a therapeutic benefit to dual inhibition of both receptors over IGF-IR specific monoclonal antibodies. BMS-754807 is an ATP competitive inhibitor that targets both the IGF-IR and the IR [67, 68].
This study evaluated the efficacy of BMS-754807 alone, and in combination with cisplatin or carboplatin, on two NSCLC cell lines in vitro. Our findings showed that BMS-754807 significantly inhibited IGF-IR/IR, AKT and ERK1/2 phosphorylation and reduced the survival of both NSCLC cell lines evaluated. BMS-754807 significantly increase apoptosis in both A549 and NCI-H358 cells and significantly reduced proliferation in A549 cells. In A549 but not NCI-H358 cells, BMS-754807 induced synergistic cytotoxicity when combined with cisplatin or carboplatin. These findings demonstrate the benefit of inhibiting IGF-IR/IR signaling in a pre-clinical setting and suggest inhibiting IGF-IR/IR signaling should be further evaluated as a potential therapeutic strategy for the treatment of NSCLC.
Methods
Cell lines and reagents
Human lung cancer cell lines, A549 and NCI-H358, were purchased from American Type Culture Collection (ATCC, Manassas, VA). A549 and NCI-H358 cells were cultured in RPMI 1640 medium supplemented with 10 % FBS and 1 % antibiotic/antimycotic (v/v) (Life Technologies, Burlington, ON). Cell were maintained in incubators at 37 °C and 5 % carbon dioxide. Cell identity was confirmed through STR profiling (The Centre for Applied Genomics at The Hospital for Sick Children, Toronto, ON).
BMS-754807 was purchased from Sellek Chemicals (Houston, TX). Cisplatin and carboplatin were both purchased from Sigma-Aldrich (Oakville, ON). BMS-754807 was dissolved in DMSO (Sigma-Aldrich, Oakville, ON) and stored at −20 °C while cisplatin was dissolved in saline and carboplatin was dissolved in water. Cisplatin and carboplatin solutions were made fresh for each experiment, and stored at 4 °C for the duration of the experiment.
Western blotting
Following treatment with BMS-745807, adherent cells were lysed, protein was isolated and western blots performed as previously described [69]. Primary antibodies were used against IGF-IRβ (1:1000; cat# 3027), phospho-IGF-IR (Tyr1135/1136)/IR (Tyr1150/1151) (1:500; cat# 3025), phospho-Akt(Ser473) (1:1000; cat# 4060), phospho-ERK1/2 (Thr202/Tyr204) (1:500; cat# 4377), and β-actin (1:5000; cat# 4970) (Cell Signaling Technology, Danvers, MA) overnight at 4 °C. An anti-rabbit secondary antibody (1:2000; cat# 7074) (Cell Signaling Technology, Danvers, MA) was used for 1 h at room temperature. Membranes were visualized using chemiluminescence substrate (PerkinElmer, Inc., Waltham, MA) and a FluorChem 8800 gel documentation system (Alpha Innotech—ProteinSimple, Toronto, ON) or a ChemiDoc XRS + imaging system (Bio-Rad Laboratories, Mississauga, ON). For western blots captured using the FluorChem 8800 gel documentation, densitometry of the bands was determined using AlpahEase FC software (Alpha Innotech—ProteinSimple, Toronto, ON) while western blots captured using the ChemiDoc XRS + gel documentation, densitometry of the bands was determined using ImageLab 5.2.1 software (Bio-Rad Laboratories, Mississauga, ON).
WST-1 Assay
Cells were plated in a 96 well plate at a density of 2500 cells/well, and allowed to adhere for 4 h. BMS-754807, cisplatin, or carboplatin were added either alone or in combination or a solvent only control was added (0.1 % of total volume). Drugs and media were refreshed every 24 h. At 72 h, cell viability reagent WST-1 (Roche, Mississauga, ON) was added and absorbance was read using Universal Microplate Reader EL800 (Bio-Tek Insturments, Inc., Winooski, VT) after a 3 h incubation.
Analysis of single and combination drug response
Cell viability determined with WST-1 (Roche, Mississauga, ON) assay after single agent and combination treatments with BMS-754807, cisplatin, and carboplatin were used to determine the IC50 and Combination Indices (CI) as calculated using Calcusyn software (Biosoft, Cambridge, UK). For combination effects: a synergistic effect is considered for a CI < 1; an additive effect is considered for a CI = 1; and an inhibitory effect is considered for a CI > 1.
Immunofluorescence
Cells were plated on coverslips in 6-well dishes at a density of 0.5–1 × 105 cells/well, allowed to adhere overnight, then treated with 0.5 μM of BMS-754807), or DMSO only (to 0.1 % v/v of total volume). Cells were fixed at 24 h post-treatment in 10 % buffered formalin (Fisher Scientific, Ottawa, ON) for 1 h. Cells were washed in PBS, permeabilized in 0.5 % Triton-X in PBS, and blocked in 5 % BSA in PBS. Then the cells were incuabated in primary antibodies against the proliferation marker Ki67 (1:400; cat# Ab15580) (Abcam, Toronto, ON) or apoptosis marker cleaved caspase 3 (1:200; cat# Ab3623) (Millipore, Etobicoke, ON) overnight at 4 °C. Cells were incubated with a fluorescent anti-rabbit secondary antibody (1:200; cat# A-21207) (Life Technologies, Burlington, ON) for 1 h at room temperature. Dapi nuclear stain (Life Technologies, Burlington, ON) was used as a counterstain, and coverslips were mounted using Prolong-gold (Life Technologies, Burlington, ON). Images were captured using Olympus BX61 microscope and Metamorph software (Molecular Devices, Sunnyvale, CA). Proliferation and apoptosis rates within cell populations were quantified by manual counts of the number of cells staining positive for Ki67 or cleaved caspase 3, respectively, and expressed as percentages relative to the total number of cells present as revealed by nuclear DAPI staining.
Wound healing assay
Cells were plated on 12 well plates at a density of 2.0 × 105 cells/well and cells typically reached 90–95 % confluency the following day. A wound was made through the centre of the well and cells were treated with 0.5 μM of BMS-754807, or solvent only (to 0.1 % v/v of total volume). Drugs and media were refreshed and wound closure was measured every 24 h. Images were captured using Olympus IX71 microscope and Q-Capture software (Q-Imaging, Surrey, BC).
Statistics
All experiments were repeated in triplicate or quadruplicate. Statistical analysis was performed using Graphpad Prism 5 (Graphpad Software, Inc., La Jolla, CA). Means were compared using a one-way ANOVA and post hoc Dunnett’s test when multiple groups were compared back to a control value or a paired Student’s T test when two means were compared. Error is represented by Standard Error of Measurement (SEM). Statistical significance is noted as p < 0.05.
Results
IGF-IR Activity is Inhibited by BMS-754807
A549 and NCI-H358 cells were treated with the dual IGF-IR/IR inhibitor BMS-754807 under regular growth conditions. When BMS-754807 was used at concentrations of 0.25 and 0.5 μM, the levels of activated IGF-IR/IR (pIGF-IR/IR) were reduced within 4 h, and a significant reduction in pIGF-IR/IR was observed at 24 h in both cell lines suggesting BMS-754807 inhibited IGF-IR/IR activation (Fig. 1a–h). Furthermore, treatment with BMS-754807 caused a concurrent decrease in phosphorylated AKT in both lung cancer cell lines (Fig. 1a–h). ERK phosphorylation was significantly reduced following administration of 0.5 μM but not 0.25 μM BMS-754807 (Fig. 1a–h).
Fig. 1.
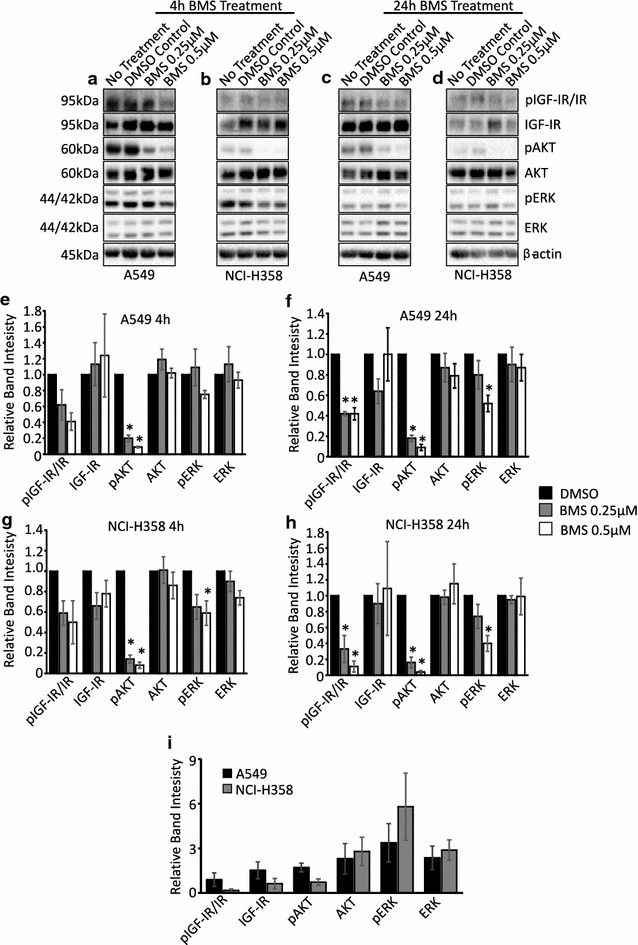
IGF-IR is expressed in lung cancer cells and its activity is inhibited by treatment with BMS-754807. Western blots of pIGF-IR/IR, IGF-IR, pAKT, AKT, pERK and ERK in A549 (a, c) and NCI-H358 (b, d) cells 4 (a, b) or 24 (c, d) hours after administration of 0.25 or 0.5 μM of BMS-754807. The bar graphs (e–h) represent the quantification of three independent western blots with the bars representing the means and the error bars representing SEM. The protein levels were normalized to the DMSO control group for each protein; the no treatment group was not quantified. β-actin was used as a loading control in the western blots and *p < 0.05 as determined by a Dunnett’s test. The bands from the 4 h No Treatment groups were quantified and are presented as the mean ± SEM for each protein relative to β-actin to illustrate the differences in protein levels in A549 and NCI-H358 cells (i)
To compare protein levels in A549 and NCI-H358 cells protein bands from the 4 h No Treatment groups were quantified. The IGF-IR protein was detected in both cell lines with A549 cells expressing more IGF-IR protein than NCI-H358 cells (Fig. 1a, b, i). A549 cells also possessed higher levels of phosphorylated IGF-IR/IR (pIGF-IR/IR) and phosphorylated AKT (pAKT) than NCI-H358 cells (Fig. 1a, b, i).
Treatment with BMS-754807 inhibits proliferation and induces apoptosis
Concentration–response curves were generated to determine the sensitivity of each lung cancer cell line to BMS-754807. BMS-754807 administration was sufficient to reduce cell viability in both A549 and NCI-H358 cells (Fig. 2a). The IC50 values were determined using Calcusyn software (Table 1) to be 0.76 μM for NCI-H358 cells and 1.08 μM for A549 cells. In order to determine the specific cause of this reduction of cell viability, proliferation and apoptosis rates were determined in A549 and NCI-H358 cells following treatment with 0.5 μM BMS-754807 by immunoflourescence for Ki67 (Fig. 2b) or cleaved caspase 3 (Fig. 2c), respectively. BMS-754807 significantly inhibitied proliferation in A549 cells (Fig. 2d) but not NCI-H358 cells (Fig. 2e). Treatment with 0.5 μM BMS-754807 significantly increased apoptosis in both A549 (Fig. 2f) and NCI-H358 cells (Fig. 2g).
Fig. 2.
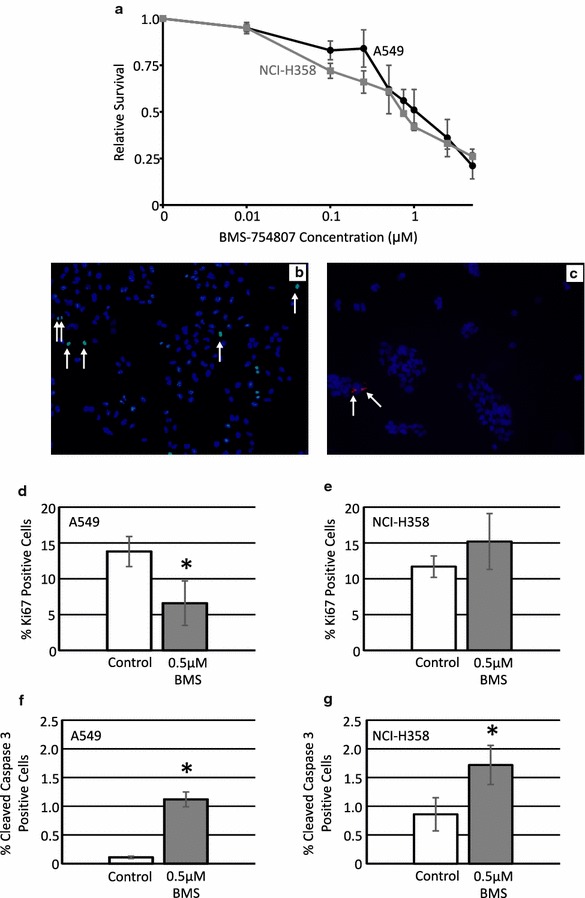
Treatment with BMS-754807 reduces cell viability in a concentration dependent manner. A549 (black line) and NCI-H358 (grey line) cells were treated with various concentrations of BMS-754807 every 24 h for 72 h. Cell viability was then measured with a WST-1 assay (a; n = 3). Immunofluorescence for Ki67 and cleaved caspase 3 were used to evaluate the impact of BMS-754807 on these cellular properties. A representative image of Ki67 (b; green color) and cleaved caspase 3 (c; red color) are presented as overlay images of cell nuclei stained with 4′,6-diamidino-2-phenylindole (DAPI, blue color). Arrows highlight some of the positive cells in each image. The number of Ki67 positive cells (d, e) and cleaved caspase 3 positive cells (f, g) along with the total number of cells were counted 24 h after treatment with 0.5 μM BMS-754807 and are presented as relative proliferation (d, e) or relative apoptosis (f, g) in A549 (d, f) and NCI-H358 (e, g) cells. The data is presented as mean ± SEM (n = 4) and the percentage of positive cells have been normalized to the DMSO control. *p < 0.05 as determined by a paired Student’s T-test
Table 1.
IC50 concentrations for BMS-754805, cisplatin, and carboplatin
| Cell line | Drug (µM) | ||
|---|---|---|---|
| BMS-754808 | Cisplatin | Carboplatin | |
| A549 | 1.08 | 3.33 | 65.29 |
| NCI-H358 | 0.76 | 14.89 | 202.72 |
BMS-754807 impairs wound closure
To determine whether inhibition of IGF-IR/IR signaling influences wound closure, wound assays were performed following BMS-754807 treatment. Representative images of A549 cells shown at time 0 h (Fig. 3a, c) and 48 h (Fig. 3b, d) after treatment with the DMSO control (Fig. 3a, b) or 0.5 μM BMS-754807 (Fig. 3c, d). There was a minor, but statistically significant, impairment of wound closure in NCI-H358 and A549 cells treated with 0.5 μM BMS-754807 (Fig. 3e, f).
Fig. 3.
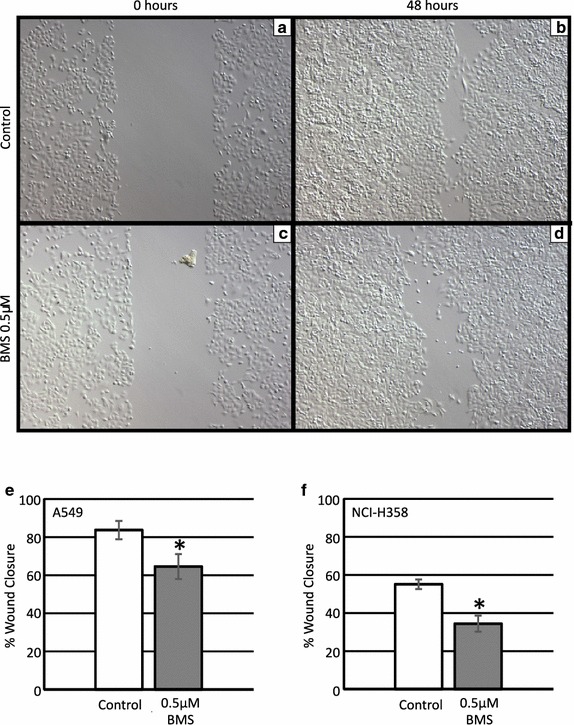
BMS-754807 reduces wound closure of lung cancer cells. Scratch wounds were made on near confluent plates of lung cancer cells. Cells were treated with BMS-754807 every 24 h, and wound closure was measured over 48–72 h. Representative images from A549 cells at 0 h (a, c) or 48 h (b, d) after treatment with either an appropriate concentration of DMSO or 0.5 μM of BMS-754807. Quantification of the relative scratch wound closure at 48 h in A549 (e) or at 72 h in NCI-H358 (f) cells was determined by measuring the remaining area not filled with cells and presenting this relative to the area of the scratch at time 0 h. Data represents mean ± SEM (n = 4), *p < 0.05 as determined by a paired Student’s T-test
Treatment with BMS-754807 or platinum chemotherapeutics reduces cell survival
Concentration–response curves were generated to determine the sensitivity of each lung cancer cell line to cisplatin and carboplatin when they were used as single agents. Both cell lines were sensitve to growth inhibition by cisplatin (Fig. 4a) and carboplatin (Fig. 4b) in a concentration dependant manner. The IC50 values were determined using Calcusyn software (Table 1). A549 cells were more sensitive to platinum chemotherapeutics than NCI-H358 cells, with IC50 values of 3.3 and 14.9 μM for cisplatin and 65.3 and 202.7 μM for carboplatin, respectively. Both cell lines were more sensitive to cisplatin than to carboplatin as reflected by the lower IC50 values for cisplatin compared to carboplatin.
Fig. 4.
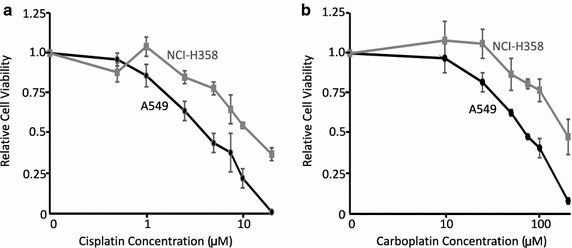
Treatment with platinum chemotherapeutics reduced cell viability in a concentration dependent manner. A549 (black line) and NCI-H358 (grey line) cells were treated with various concentrations of cisplatin (a), or carboplatin (b). Media and drugs were refreshed every 24 h for 72 h, then cell viability was measured with a WST-1 assay. All values are relative to the DMSO control and are presented as mean ± SEM (n = 3)
BMS-754807 synergizes with platinum chemotherapeutics to reduce A549 Cell viability
To determine if the addition of BMS-754807 could enhance the anticancer effects of platinum based chemotherapeutics at low concentrations, BMS-754807 was combined simultaneously with either cisplatin or carboplatin and the effects on cell viability were compared to those of each single agent. Three concentrations of BMS-754807 (0.01, 0.1, 0.25 μM in A549 cells and 2.5, 5.0, 10 µM in NCI H358 cells) were combined with three concentrations of cisplatin (0.5, 1, 2.5 μM) or carboplatin (10, 25, 50 μM in A549 cells and 50, 75 and 100 µM in NCI H358 cells), which based on the single agent concentrations were responsible for no more than approximately 25–30 % decrease in cell viability. At all concentrations of cisplatin and carboplatin used, the addition of BMS-754807 enhanced the effects of cisplatin and carboplatin (Fig. 5a, b). Combination indices were calculated using Calcusyn software to determine the nature of the interaction between cisplatin and BMS-754807 or carboplatin and BMS-754807. Representative plots of these interactions are presented in Fig. 5c, d. Combination indices <1 suggest synergistic interaction between the two drugs while combination indices of approximately 1 suggest additive drug interactions and combination indices >1 suggest antagonistic drug interactions. Based on the data presented in Fig. 5c, d, the combination of 0.25 μM BMS-754807 with cisplatin or carboplatin resulted in a synergistic reduction of cell survival in A549 cells (black symbols) but only additive or antagonistic interactions in NCI-H358 cells (white symbols). A complete list of the drug interactions for all the drug combinations tested are presented in Tables 2 and 3.
Fig. 5.
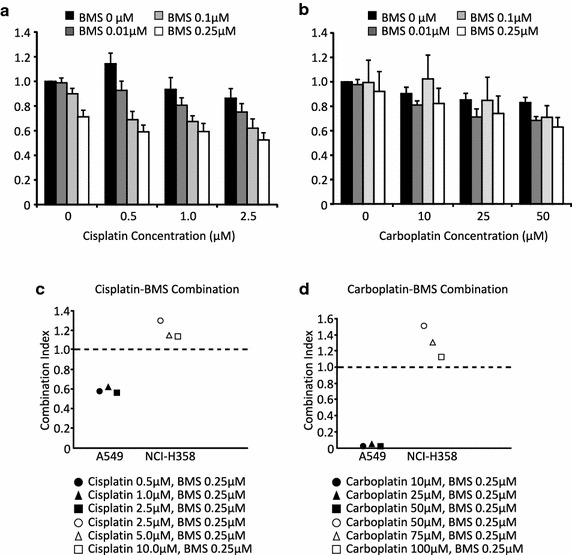
BMS-754807 synergizes with platinum based chemotherapeutics to enhance killing of A549 cells. Lung cancer cells were treated with a combination of BMS-754807 and either cisplatin (a, c) or carboplatin (b, d). Media and drugs were refreshed every 24 h for 72 h, then cell viability was measured using a WST-1 assay. Data illustrated in (a, b) are representative graphs of A549 cells treated BMS-754807 in combination with cisplatin (a) or carboplatin (b). This data is presented as mean ± SEM (n = 4). Combination indices were calculated using Calcusyn software and the data for cisplatin in combination with 0.25 μM of BMS-754807 is presented in (e) while the data for carboplatin in combination with 0.25 μM of BMS-754807 is presented in (f). A549 cells are plotted as black symbols while NCI-H358 cells are plotted as white symbols. The complete list of the interactions of all BMS-754807 concentrations with either cisplatin or carboplatin are presented in Tables 2 and 3
Table 2.
Drug interaction between cisplatin and BMS-754807
| Cell line | Cisplatin (μM) | BMS-754807 (µM) | Interactiona |
|---|---|---|---|
| A549 | 0.5 | 0.01 | Synergistic |
| A549 | 1.0 | 0.01 | Synergistic |
| A549 | 2.5 | 0.01 | Synergistic |
| A549 | 0.5 | 0.1 | Synergistic |
| A549 | 1.0 | 0.1 | Synergistic |
| A549 | 2.5 | 0.1 | Synergistic |
| A549 | 0.5 | 0.25 | Synergistic |
| A549 | 1.0 | 0.25 | Synergistic |
| A549 | 2.5 | 0.25 | Synergistic |
| NCI-H358 | 2.5 | 0.001 | Antagonistic |
| NCI-H358 | 5.0 | 0.001 | Antagonistic |
| NCI-H358 | 7.5 | 0.001 | Antagonistic |
| NCI-H358 | 2.5 | 0.1 | Additive |
| NCI-H358 | 5.0 | 0.1 | Additive |
| NCI-H358 | 7.5 | 0.1 | Additive |
| NCI-H358 | 2.5 | 0.25 | Antagonistic |
| NCI-H358 | 5.0 | 0.25 | Additive |
| NCI-H358 | 7.5 | 0.25 | Additive |
aCombination index <1 = synergistic interaction, combinations index ~1
= additive interaction, combination index >1 = antagonistic interaction
Table 3.
Drug interaction between carboplatin and BMS-754807
| Cell line | Carboplatin (µM) | BMS-754807 (µM) | Interactiona |
|---|---|---|---|
| A549 | 10 | 0.01 | Synergistic |
| A549 | 25 | 0.01 | Antagonistic |
| A549 | 50 | 0.01 | Synergistic |
| A549 | 10 | 0.1 | Synergistic |
| A549 | 25 | 0.1 | Additive |
| A549 | 50 | 0.1 | Synergistic |
| A549 | 10 | 0.25 | Synergistic |
| A549 | 25 | 0.25 | Synergistic |
| A549 | 50 | 0.25 | Synergistic |
| NCI-H358 | 50 | 0.001 | Antagonistic |
| NCI-H358 | 75 | 0.001 | Antagonistic |
| NCI-H358 | 100 | 0.001 | Additive |
| NCI-H358 | 50 | 0.1 | Antagonistic |
| NCI-H358 | 75 | 0.1 | Antagonistic |
| NCI-H358 | 100 | 0.1 | Additive |
| NCI-H358 | 50 | 0.25 | Antagonistic |
| NCI-H358 | 75 | 0.25 | Antagonistic |
| NCI-H358 | 100 | 0.25 | Additive |
acombination index <1 = synergistic interaction, combinations index ~1
= additive interaction, combination index >1 = antagonistic interaction
Discussion
The IGF-IR is frequently expressed at high levels in NSCLC and thus represents a potential therapeutic target. One compound that targets IGF-IR (as well as IR) is BMS-754807 and this drug was evaluated in two human non-small cell lung cancer cell lines, A549 and NCI-H358. Both A549 and NCI-H358 cells express IGF-IR protein and the IGF-IR is phosphorylated in both cell lines under normal growth conditions with A549 cells containing higher levels of IGF-IR and phosphorylated IGF-IR than the NCI-H358 cells. BMS-754807 signficantly reduced, IGF-IR/IR, AKT and ERK phosphorylation in both cell lines with the most dramatic effect being the reduction in AKT phosphorylation. When Wittman et al. [67] first reported the development of BMS-754807 they reported it to be more effective at ERK inhibition than AKT with IC50 of 13 and 22 nM, respectively. However, subsequent reports in cell lines suggest the BMS-754807 is a more potent inhibitor of AKT than ERK phosporylation [68] which is consistent with our results. Furthermore, BMS-754807 was also reported to preferentialy inhibit the AKT pathway in breast cancer [70] and prostate cancer cells [71].
As a single agent, BMS-754807 effectively suppressed the survival of both A549 and NCI-H358 cells. This decrease in survival likely results from a significant increase in apoptosis induced by BMS-754807 in both A549 and NCI-H358 cells. BMS-754807 also significantly reduced proliferation in A549 but not NCI-H358 cells. This is the first report of the effects of BMS-754807 on survival, proliferation and apoptosis in A549 and NCI-H358 cells. BMS-754807 action has been evaluated in several other human NSCLC lines with similar IC50 values (0.5–5 µM range) however proliferation and apoptosis were not specifically assessed [68]. Another novel finding of this study was that BMS-754807, as a single agent, could suppress wound closure. Since several studies have demonstrated an important role for IGF-IR in the motility and invasion of lung cancer cells [72–76] it is possible that BMS-874807 can negatively impact lung cancer cell migration and potentially metastais. However, this finding of reduced wound closure requires confirmation as alterations in proliferation and apoptosis can influence the rate of wound closure independent of migration.
Platinum-based chemotherapeutic agents are commonly employed in the treatment of unresectable NSCLC [77]. Therefore, we evaluated the efficacy of cisplatin and carboplatin in A549 and NCI-H358 cells and found that both cell lines were more sensitive to cisplatin than carboplatin. This is consistent with the study by Mahalingham et al. [8] that reported cisplatin was more effective than carboplatin in the treatment of NSCLC. To evaluate whether BMS-754807 could enhance the effects of platinum chemotherpeutics, A549 and NCI-H358 cell survival was assessed following simultaneous administration of either BMS-754807 and cisplatin or BMS-754807 and carboplatin. In almost all the combinations tested, administration of BMS-754807 with cisplatin or carboplatin resulted in a syngersitic reduction in cell survival in A549 cells. In NCI-H358 cells, combinations of BMS-754807 with cisplatin or carboplatin resulted in additive or antagonistic drug interactions with respect to cell survival. It remains unclear why the combinations of BMS-754807 with the platinum agents was more effective in the A549 cells. A549 cells express higher levels of IGF-IR than NCI-H358 cells and thus it is possible that A549 cells are more reliant on IGF-IR signaling for proliferation and survival. It is also possible that the intrinsic insensitivity of NCI-H358 cells to platinum chemotherapeutics (IC50 for both cisplatin and carboplatin were considerably higher for NCI-H358 cells compared to A549 cells) impacted the interactions at the concentrations evaluated. A final potential explanation is that the different mutational profile of the two cells could influence drug interaction. While both cell lines harbour a mutation in K-ras, A549 cells have a mutation in Cdkn2a but contain wild type p53 while NCI-H358 express mutant p53 but wild type Cdkn2a (atcc.org). The only other study evaluating BMS-754807 in combination with chemotherapy in NSCLC found that BMS-754807 in combination with gefitinib resulted in synergistic reduction in cell survival in the human NSCLC cell line, NCI-H292 [78]. In small cell lung cancer (SCLC) targeting the IGF-IR using the monoclonal antibody NVP-ADW742 sensitizes SCLC cell lines to the effects of etoposide and carboplatin [79].
Conclusions
In summary, this research demonstrates for the first time, the efficacy of BMS-754807 as a single agent in A549 and NCI-H358 cells and in combination with platinum-based chemotherapeutic agents in A549 cells. Therefore, BMS-754807 may be an effective therapeutic agent for the treatment of lung cancer, particularly in patients with lung tumors expressing high levels of IGF-IR.
Authors’ contributions
SEF performed the majority of the experiments and wrote the manuscript. RJ assisted with the drug combination assays while RB performed the wound closure assays on NCI-H358 cells. PM assisted with the immunofluorescence and RAM ran the project and edited the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This work was funded by a Canadian Cancer Society (grant #20105) awarded to RAM. The Canadian Cancer Society had no role in the study design, data collection, data analysis, data interpretation, the writing of the manuscript or the decision to submit this article for publication.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
S. Elizabeth Franks, Email: sfranks@uoguelph.ca.
Robert A. Jones, Email: rjones12@uoguelph.ca
Ritesh Briah, Email: rbriah@uoguelph.ca.
Payton Murray, Email: paytonmu@uoguelph.ca.
Roger A. Moorehead, Phone: 519-824-4120 x54950, Email: rmoorehe@uoguelph.ca
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Devesa SS, Bray F, Vizcaino AP, Parkin DM. International lung cancer trends by histologic type: male:female differences diminishing and adenocarcinoma rates rising. Int J Cancer. 2005;117:294–299. doi: 10.1002/ijc.21183. [DOI] [PubMed] [Google Scholar]
- 3.Hansen HH. Treatment of advanced non-small cell lung cancer. BMJ. 2002;325:452–453. doi: 10.1136/bmj.325.7362.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morgesztern D, Ng SH, Gao F, Govindan R. Trends in stage distribution for patients with non-small cell lung cancer: a national cancer database survey. J Thorac Oncol. 2010;5:29–33. doi: 10.1097/JTO.0b013e3181c5920c. [DOI] [PubMed] [Google Scholar]
- 5.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 6.Johnson ML, Patel JD. Chemotherapy and targeted therapeutics as maintenance of response in advanced non-small cell lung cancer. Semin Oncol. 2014;41:93–100. doi: 10.1053/j.seminoncol.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 7.Carnio S, Novello S, Mele T, Levra MG, Scagliotti GV. Extending survival of stage IV non-small cell lung cancer. Semin Oncol. 2014;41:69–92. doi: 10.1053/j.seminoncol.2013.12.013. [DOI] [PubMed] [Google Scholar]
- 8.Mahalingam D, Mita A, Mita MM, Nawrocki ST, Giles FJ. Targeted therapy for advanced non-small cell lung cancers: historical perspective, current practices, and future development. Curr Probl Cancer. 2009;33:73–111. doi: 10.1016/j.currproblcancer.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 9.Horn L, Carbone DP. Reducing lung cancer mortality: 2014 update. Semin Oncol. 2014;41:3–4. doi: 10.1053/j.seminoncol.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 10.Forde PM, Ettinger DS. Targeted therapy for non-small-cell lung cancer: past, present and future. Expert Rev Anticancer Ther. 2013;13:745–758. doi: 10.1586/era.13.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scagliotti GV, Novello S. The role of the insulin-like growth factor signaling pathway in non-small cell lung cancer and other solid tumors. Cancer Treat Rev. 2012;38:292–302. doi: 10.1016/j.ctrv.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 12.Chen HX, Sharon E. IGF-1R as an anti-cancer target–trials and tribulations. Chin J Cancer. 2013;32:242–252. doi: 10.5732/cjc.012.10263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Langer CJ, Novello S, Park K, Krzakowski M, Karp DD, Mok T, Benner RJ, Scranton JR, Olszanski AJ, Jassem J. Randomized, phase III trial of first-line figitumumab in combination with paclitaxel and carboplatin versus paclitaxel and carboplatin alone in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2014;32:2059–2066. doi: 10.1200/JCO.2013.54.4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cosaceanu D, Carapancea M, Alexandru O, Budiu R, Martinsson HS, Starborg M, Vrabete M, Kanter L, Lewensohn R, Dricu A. Comparison of three approaches for inhibiting insulin-like growth factor I receptor and their effects on NSCLC cell lines in vitro. Growth Factors. 2007;25:1–8. doi: 10.1080/08977190600702865. [DOI] [PubMed] [Google Scholar]
- 15.Dong AQ, Kong MJ, Ma ZY, Qian JF, Xu XH. Down-regulation of IGF-IR using small, interfering, hairpin RNA (siRNA) inhibits growth of human lung cancer cell line A549 in vitro and in nude mice. Cell Biol Int. 2007;31:500–507. doi: 10.1016/j.cellbi.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 16.Samani AA, Chevet E, Fallavollita L, Galipeau J, Brodt P. Loss of tumorigenicity and metastatic potential in carcinoma cells expressing the extracellular domain of the type 1 insulin-like growth factor receptor. Cancer Res. 2004;64:3380–3385. doi: 10.1158/0008-5472.CAN-03-3780. [DOI] [PubMed] [Google Scholar]
- 17.Linnerth NM, Siwicky MD, Campbell CI, Watson KLM, Petrik JJ, Whitsett JA, Moorehead RA. Type I insulin-like growth factor receptor induces pulmonary tumorigenesis. Neoplasia. 2009;11:672–682. doi: 10.1593/neo.09310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cappuzzo F, Tallini G, Finocchiaro G, Wilson RS, Ligorio C, Giordano L, Toschi L, Incarbone M, Cavina R, Terracciano L, Roncalli M, Alloisio M, Varella-Garcia M, Franklin WA, Santoro A. Insulin-like growth factor receptor 1 (IGF1R) expression and survival in surgically resected non-small-cell lung cancer (NSCLC) patients. Ann Oncol. 2010;21:562–567. doi: 10.1093/annonc/mdp357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fidler MJ, Basu S, Buckingham L, Walters K, McCormack S, Batus M, Coon JI, Bonomi P, Bukingham L. Utility of insulin-like growth factor receptor-1 expression in gefitinib-treated patients with non-small cell lung cancer. Anticancer Res. 2012;32:1705–1710. [PubMed] [Google Scholar]
- 20.Gately K, Forde L, Cuffe S, Cummins R, Kay EW, Feuerhake F, O’Byrne KJ. High coexpression of both EGFR and IGF1R correlates with poor patient prognosis in resected non-small-cell lung cancer. Clin Lung Cancer. 2014;15:58–66. doi: 10.1016/j.cllc.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 21.Gong Y, Yao E, Shen R, Goel A, Arcila M, Teruya-Feldstein J, Zakowski MF, Frankel S, Peifer M, Thomas RK, Ladanyi M, Pao W. High expression levels of total IGF-1R and sensitivity of NSCLC cells in vitro to an anti-IGF-1R antibody (R1507) PLoS ONE. 2009;4:e7273. doi: 10.1371/journal.pone.0007273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kikuchi R, Sonobe M, Kobayashi M, Ishikawa M, Kitamura J, Nakayama E, Menju T, Miyahara R, Huang C-L, Date H. Expression of IGF1R is associated with tumor differentiation and survival in patients with lung adenocarcinoma. Ann Surg Oncol. 2012;19(Suppl 3):S412–S420. doi: 10.1245/s10434-011-1878-x. [DOI] [PubMed] [Google Scholar]
- 23.Ludovini V, Bellezza G, Pistola L, Bianconi F, Di Carlo L, Sidoni A, Semeraro A, Del Sordo R, Tofanetti FR, Mameli MG, Daddi G, Cavaliere A, Tonato M, Crinò L. High coexpression of both insulin-like growth factor receptor-1 (IGFR-1) and epidermal growth factor receptor (EGFR) is associated with shorter disease-free survival in resected non-small-cell lung cancer patients. Ann Oncol. 2009;20:842–849. doi: 10.1093/annonc/mdn727. [DOI] [PubMed] [Google Scholar]
- 24.Nakagawa M, Uramoto H, Oka S, Chikaishi Y, Iwanami T, Shimokawa H, So T, Hanagiri T, Tanaka F. Clinical significance of IGF1R expression in non-small-cell lung cancer. Clin Lung Cancer. 2012;13:136–142. doi: 10.1016/j.cllc.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 25.Reinmuth N, Kloos S, Warth A, Risch A, Muley T, Hoffmann H, Thomas M, Meister M. Insulin-like growth factor 1 pathway mutations and protein expression in resected non-small cell lung cancer. Hum Pathol. 2014;45:1162–1168. doi: 10.1016/j.humpath.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 26.Tsuta K, Mimae T, Nitta H, Yoshida A, Maeshima AM, Asamura H, Grogan TM, Furuta K, Tsuda H. Insulin-like growth factor-1 receptor protein expression and gene copy number alterations in non-small cell lung carcinomas. Hum Pathol. 2013;44:975–982. doi: 10.1016/j.humpath.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 27.Zhang X, Sun J, Wang H, Lou Y, Zhang Y, Sha H, Feng J, Han B. IGF-1R and Bmi-1 expressions in lung adenocarcinoma and their clinicopathologic and prognostic significance. Tumour Biol. 2014;35:739–745. doi: 10.1007/s13277-013-1100-9. [DOI] [PubMed] [Google Scholar]
- 28.Vilmar A, Santoni-Rugiu E, Cillas JG-F, Huarriz M, Sørensen JB. Insulin-like growth factor receptor 1 mrna expression as a prognostic marker in advanced non-small cell lung cancer. Anticancer Res. 2014;34:2991–2996. [PubMed] [Google Scholar]
- 29.Dziadziuszko R, Merrick DT, Witta SE, Mendoza AD, Szostakiewicz B, Szymanowska A, Rzyman W, Dziadziuszko K, Jassem J, Bunn PA, Jr, Varella-Garcia M, Hirsch FR, Bunn PA. Insulin-like growth factor receptor 1 (IGF1R) gene copy number is associated with survival in operable non-small-cell lung cancer: a comparison between IGF1R fluorescent in situ hybridization, protein expression, and mRNA expression. J Clin Oncol. 2010;28:2174–2180. doi: 10.1200/JCO.2009.24.6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hurbin A, Wislez M, Busser B, Antoine M, Tenaud C, Rabbe N, Dufort S, de Fraipont F, Moro-Sibilot D, Cadranel J, Coll J-L, Brambilla E. Insulin-like growth factor-1 receptor inhibition overcomes gefitinib resistance in mucinous lung adenocarcinoma. J Pathol. 2011;225:83–95. doi: 10.1002/path.2897. [DOI] [PubMed] [Google Scholar]
- 31.Peled N, Wynes MW, Ikeda N, Ohira T, Yoshida K, Qian J, Ilouze M, Brenner R, Kato Y, Mascaux C, Hirsch FR. Insulin-like growth factor-1 receptor (IGF-1R) as a biomarker for resistance to the tyrosine kinase inhibitor gefitinib in non-small cell lung cancer. Cell Oncol (Dordr) 2013;36:277–288. doi: 10.1007/s13402-013-0133-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu P-F, Huang W-C, Yang JC-H, Lu Y-S, Shih J-Y, Wu S-G, Lin C-H, Cheng A-L. Phosphorylated insulin-like growth factor-1 receptor (pIGF1R) is a poor prognostic factor in brain metastases from lung adenocarcinomas. J Neurooncol. 2013;115:61–70. doi: 10.1007/s11060-013-1194-3. [DOI] [PubMed] [Google Scholar]
- 33.Chang YS, Gong K, Sun S, Liu D, El-Naggar AK, Khuri FR, Hong WK, Lee H-Y, Kong G. Clinical significance of insulin-like growth factor-binding protein-3 expression in stage I non-small cell lung cancer. Clin Cancer Res. 2002;8:3796–3802. [PubMed] [Google Scholar]
- 34.Chang YS, Wang L, Liu D, Mao L, Hong WK, Khuri FR, Lee H-Y. Correlation between insulin-like growth factor-binding protein-3 promoter methylation and prognosis of patients with stage I non-small cell lung cancer. Clin Cancer Res. 2002;8:3669–3675. [PubMed] [Google Scholar]
- 35.Kim J-S, Kim ES, Liu D, Lee JJ, Solis L, Behrens C, Lippman SM, Hong WK, Wistuba II, Lee H-Y. Prognostic implications of tumoral expression of insulin like growth factors 1 and 2 in patients with non-small-cell lung cancer. Clin Lung Cancer. 2014;15:213–221. doi: 10.1016/j.cllc.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vollebergh M a, Kappers I, Klomp HM, Buning-Kager JC, Korse CM, Hauptmann M, de Visser KE, van den Heuvel MM, Linn SC. Ligands of epidermal growth factor receptor and the insulin-like growth factor family as serum biomarkers for response to epidermal growth factor receptor inhibitors in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2010, 5:1939–48. [DOI] [PubMed]
- 37.Masago K, Fujita S, Togashi Y, Kim YH, Hatachi Y, Fukuhara A, Nagai H, Irisa K, Sakamori Y, Mio T, Mishima M. Clinical significance of epidermal growth factor receptor mutations and insulin-like growth factor 1 and its binding protein 3 in advanced non-squamous non-small cell lung cancer. Oncol Rep. 2011;26:795–803. doi: 10.3892/or.2011.1354. [DOI] [PubMed] [Google Scholar]
- 38.Unsal E, Köksal D, Yurdakul AS, Atikcan S, Cinaz P. Analysis of insulin like growth factor 1 and insulin like growth factor binding protein 3 levels in bronchoalveolar lavage fluid and serum of patients with lung cancer. Respir Med. 2005;99:559–565. doi: 10.1016/j.rmed.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 39.Fu S, Tang H, Liao Y, Jiang W, Xu Q, Deng Y, Fu X. Association of preoperative serum IGF- I concentration with clinicopathological parameters in patients with non-small cell lung cancer. J Huazhong Univ Sci Technolog Med Sci. 2013;33:224–227. doi: 10.1007/s11596-013-1101-3. [DOI] [PubMed] [Google Scholar]
- 40.Chen B, Xiao F, Li B, Xie B, Zhou J, Zheng J, Zhang W. The role of epithelial-mesenchymal transition and IGF-1R expression in prediction of gefitinib activity as the second-line treatment for advanced nonsmall-cell lung cancer. Cancer Invest. 2013;31:454–460. doi: 10.3109/07357907.2013.820315. [DOI] [PubMed] [Google Scholar]
- 41.Cortot AB, Repellin CE, Shimamura T, Capelletti M, Zejnullahu K, Ercan D, Christensen JG, Wong K-K, Gray NS, Jänne PA. Resistance to irreversible EGF receptor tyrosine kinase inhibitors through a multistep mechanism involving the IGF1R pathway. Cancer Res. 2013;73:834–843. doi: 10.1158/0008-5472.CAN-12-2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guix M, Faber AC, Wang SE, Olivares MG, Song Y, Qu S, Rinehart C, Seidel B, Yee D, Arteaga CL, Engelman JA. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Clin Invest. 2008;118:2609–2619. doi: 10.1172/JCI34588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jameson MJ, Beckler AD, Taniguchi LE, Allak A, Vanwagner LB, Lee NG, Thomsen WC, Hubbard MA, Thomas CY. Activation of the insulin-like growth factor-1 receptor induces resistance to epidermal growth factor receptor antagonism in head and neck squamous carcinoma cells. Mol Cancer Ther. 2011;10:2124–2134. doi: 10.1158/1535-7163.MCT-11-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morgillo F, Kim WY, Kim ES, Ciardiello F, Hong WK, Lee HY. Implication of the insulin-like growth factor-IR pathway in the resistance of non-small cell lung cancer cells to treatment with gefitinib. Clin Cancer Res. 2007;13:2795–2803. doi: 10.1158/1078-0432.CCR-06-2077. [DOI] [PubMed] [Google Scholar]
- 45.Suda K, Mizuuchi H, Sato K, Takemoto T, Iwasaki T, Mitsudomi T. The insulin-like growth factor 1 receptor causes acquired resistance to erlotinib in lung cancer cells with the wild-type epidermal growth factor receptor. Int J Cancer. 2014;135:1002–1006. doi: 10.1002/ijc.28737. [DOI] [PubMed] [Google Scholar]
- 46.Cortés-Sempere M, de Miguel MP, Pernía O, Rodriguez C, de Castro Carpeño J, Nistal M, Conde E, López-Ríos F, Belda-Iniesta C, Perona R, de Caceres II. IGFBP-3 methylation-derived deficiency mediates the resistance to cisplatin through the activation of the IGFIR/Akt pathway in non-small cell lung cancer. Oncogene. 2013;32:1274–1283. doi: 10.1038/onc.2012.146. [DOI] [PubMed] [Google Scholar]
- 47.Ferté C, Loriot Y, Clémenson C, Commo F, Gombos A, Bibault J-E, Fumagalli I, Hamama S, Auger N, Lahon B, Chargari C, Calderaro J, Soria J-C, Deutsch E. IGF-1R targeting increases the antitumor effects of DNA-damaging agents in SCLC model: an opportunity to increase the efficacy of standard therapy. Mol Cancer Ther. 2013;12:1213–1222. doi: 10.1158/1535-7163.MCT-12-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun Y, Zheng S, Torossian A, Speirs CK, Schleicher S, Giacalone NJ, Carbone DP, Zhao Z, Lu B. Role of insulin-like growth factor-1 signaling pathway in cisplatin-resistant lung cancer cells. Int J Radiat Oncol Biol Phys. 2012;82:e563–e572. doi: 10.1016/j.ijrobp.2011.06.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ward CW, Garrett TP, McKern NM, Lou M, Cosgrove LJ, Sparrow LG, Frenkel MJ, Hoyne PA, Elleman TC, Adams TE, Lovrecz GO, Lawrence LJ, Tulloch PA. The three dimensional structure of the type I insulin-like growth factor receptor. Mol Pathol. 2001;54:125–132. doi: 10.1136/mp.54.3.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Siddle K. Signalling by insulin and IGF receptors: supporting acts and new players. J Mol Endocrinol. 2011;47:R1–R10. doi: 10.1530/JME-11-0022. [DOI] [PubMed] [Google Scholar]
- 51.Frasca F, Pandini G, Sciacca L, Pezzino V, Squatrito S, Belfiore A, Vigneri R. The role of insulin receptors and IGF-I receptors in cancer and other diseases. Arch Physiol Biochem. 2008;114:23–37. doi: 10.1080/13813450801969715. [DOI] [PubMed] [Google Scholar]
- 52.Benyoucef S, Surinya KH, Hadaschik D, Siddle K. Characterization of insulin/IGF hybrid receptors: contributions of the insulin receptor L2 and Fn1 domains and the alternatively spliced exon 11 sequence to ligand binding and receptor activation. Biochem J. 2007;403:603–613. doi: 10.1042/BJ20061709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pandini G, Vigneri R, Costantino A, Frasca F, Ippolito A, Fujita-Yamaguchi Y, Siddle K, Goldfine ID, Belfiore A. Insulin and insulin-like growth factor-I (IGF-I) receptor overexpression in breast cancers leads to insulin/IGF-I hybrid receptor overexpression: evidence for a second mechanism of IGF-I signaling. Clin Cancer Res. 1999;5:1935–1944. [PubMed] [Google Scholar]
- 54.Pandini G, Frasca F, Mineo R, Sciacca L, Vigneri R, Belfiore A. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J Biol Chem. 2002;277:39684–39695. doi: 10.1074/jbc.M202766200. [DOI] [PubMed] [Google Scholar]
- 55.Boucher J, Tseng YH, Kahn CR. Insulin and insulin-like growth factor-1 receptors act as ligand-specific amplitude modulators of a common pathway regulating gene transcription. J Biol Chem. 2010;285:17235–17245. doi: 10.1074/jbc.M110.118620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Denley A, Bonython ER, Booker GW, Cosgrove LJ, Forbes BE, Ward CW, Wallace JC. Structural determinants for high-affinity binding of insulin-like growth factor II to insulin receptor (IR)-A, the exon 11 minus isoform of the IR. Mol Endocrinol. 2004;18:2502–2512. doi: 10.1210/me.2004-0183. [DOI] [PubMed] [Google Scholar]
- 57.Kanou T, Oneyama C, Kawahara K, Okimura A, Ohta M, Ikeda N, Shintani Y, Okumura M, Okada M. The transmembrane adaptor Cbp/PAG1 controls the malignant potential of human non-small cell lung cancers that have c-src upregulation. Mol Cancer Res. 2011;9:103–114. doi: 10.1158/1541-7786.MCR-10-0340. [DOI] [PubMed] [Google Scholar]
- 58.Morcavallo A, Gaspari M, Pandini G, Palummo A, Cuda G, Larsen MR, Vigneri R, Belfiore A. Research resource: new and diverse substrates for the insulin receptor isoform a revealed by quantitative proteomics after stimulation with IGF-II or insulin. Mol Endocrinol. 2011;25:1456–1468. doi: 10.1210/me.2010-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pandini G, Conte E, Medico E, Sciacca L, Vigneri R, Belfiore A. IGF-II binding to insulin receptor isoform A induces a partially different gene expression profile from insulin binding. Ann N Y Acad Sci. 2004;1028:450–456. doi: 10.1196/annals.1322.053. [DOI] [PubMed] [Google Scholar]
- 60.Denley A, Brierley GV, Carroll JM, Lindenberg A, Booker GW, Cosgrove LJ, Wallace JC, Forbes BE, Roberts CT., Jr Differential activation of insulin receptor isoforms by insulin-like growth factors is determined by the C domain. Endocrinology. 2006;147:1029–1036. doi: 10.1210/en.2005-0736. [DOI] [PubMed] [Google Scholar]
- 61.Kim W-Y, Kim M-J, Moon H, Yuan P, Kim J-S, Woo J-K, Zhang G, Suh Y-A, Feng L, Behrens C, Van Pelt CS, Kang H, Lee JJ, Hong W-K, Wistuba II, Lee H-Y. Differential impacts of insulin-like growth factor-binding protein-3 (IGFBP-3) in epithelial IGF-induced lung cancer development. Endocrinology. 2011;152:2164–2173. doi: 10.1210/en.2010-0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Izycki T, Chyczewska E, Naumnik W, Talalaj J, Panek B, Ossolinska M. Serum levels of IGF-I and IGF-II in patients with lung cancer during chemotherapy. Exp Oncol. 2004;26:316–319. [PubMed] [Google Scholar]
- 63.Moorehead RA, Sanchez OH, Baldwin RM, Khokha R. Transgenic overexpression of IGF-II induces spontaneous lung tumors: a model for human lung adenocarcinoma. Oncogene. 2003;22:853–857. doi: 10.1038/sj.onc.1206188. [DOI] [PubMed] [Google Scholar]
- 64.Pavelić J, Pavelić L, Karadza J, Krizanac S, Unesić J, Spaventi S, Pavelić K. Insulin-like growth factor family and combined antisense approach in therapy of lung carcinoma. Mol Med. 2002;8:149–157. [PMC free article] [PubMed] [Google Scholar]
- 65.Ulanet DB, Ludwig DL, Kahn CR, Hanahan D. Insulin receptor functionally enhances multistage tumor progression and conveys intrinsic resistance to IGF-1R targeted therapy. Proc Natl Acad Sci U S A. 2010;107:10791–10798. doi: 10.1073/pnas.0914076107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Garofalo C, Manara MC, Nicoletti G, Marino MT, Lollini PL, Astolfi A, Pandini G, Lopez-Guerrero JA, Schaefer KL, Belfiore A, Picci P, Scotlandi K. Efficacy of and resistance to anti-IGF-1R therapies in Ewing’s sarcoma is dependent on insulin receptor signaling. Oncogene. 2011;30:2730–2740. doi: 10.1038/onc.2010.640. [DOI] [PubMed] [Google Scholar]
- 67.Wittman MD, Carboni JM, Yang Z, Lee FY, Antman M, Attar R, Balimane P, Chang C, Chen C, Discenza L, Frennesson D, Gottardis MM, Greer A, Hurlburt W, Johnson W, Langley DR, Li A, Li J, Liu P, Mastalerz H, Mathur A, Menard K, Patel K, Sack J, Sang X, Saulnier M, Smith D, Stefanski K, Trainor G, Velaparthi U, et al. Discovery of a 2,4-disubstituted pyrrolo[1,2-f][1,2,4]triazine inhibitor (BMS-754807) of insulin-like growth factor receptor (IGF-1R) kinase in clinical development. J Med Chem. 2009;52:7360–7363. doi: 10.1021/jm900786r. [DOI] [PubMed] [Google Scholar]
- 68.Carboni JM, Wittman M, Yang Z, Lee F, Greer A, Hurlburt W, Hillerman S, Cao C, Cantor GH, Dell-John J, Chen C, Discenza L, Menard K, Li A, Trainor G, Vyas D, Kramer R, Attar RM, Gottardis MM. BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol Cancer Ther. 2009;8:3341–3349. doi: 10.1158/1535-7163.MCT-09-0499. [DOI] [PubMed] [Google Scholar]
- 69.Jones RA, Campbell CI, Gunther EJ, Chodosh LA, Petrik JJ, Khokha R, Moorehead RA. Transgenic overexpression of IGF-IR disrupts mammary ductal morphogenesis and induces tumor formation. Oncogene. 2007;26:1636–1644. doi: 10.1038/sj.onc.1209955. [DOI] [PubMed] [Google Scholar]
- 70.Hou X, Huang F, Macedo LF, Harrington SC, Reeves KA, Greer A, Finckenstein FG, Brodie A, Gottardis MM, Carboni JM, Haluska P. Dual IGF-1R/InsR inhibitor BMS-754807 synergizes with hormonal agents in treatment of estrogen-dependent breast cancer. Cancer Res. 2011;71:7597–7607. doi: 10.1158/0008-5472.CAN-11-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dayyani F, Parikh NU, Varkaris AS, Song JH, Moorthy S, Chatterji T, Maity SN, Wolfe AR, Carboni JM, Gottardis MM, Logothetis CJ, Gallick GE. Combined Inhibition of IGF-1R/IR and Src family kinases enhances antitumor effects in prostate cancer by decreasing activated survival pathways. PLoS ONE. 2012;7:e51189. doi: 10.1371/journal.pone.0051189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Oh SH, Jin Q, Kim ES, Khuri FR, Lee H-YY. Insulin-like growth factor-I receptor signaling pathway induces resistance to the apoptotic activities of SCH66336 (lonafarnib) through Akt/mammalian target of rapamycin-mediated increases in survivin expression. Clin Cancer Res. 2008;14:1581–1589. doi: 10.1158/1078-0432.CCR-07-0952. [DOI] [PubMed] [Google Scholar]
- 73.Ma Z, Dong A, Kong M, Qian J. Silencing of the type 1 insulin-like growth factor receptor increases the sensitivity to apoptosis and inhibits invasion in human lung adenocarcinoma A549 cells. Cell Mol Biol Lett. 2007;12:556–572. doi: 10.2478/s11658-007-0022-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Qian J, Dong A, Kong M, MA Z, Fan J, Jiang G. Suppression of type 1 Insulin-like growth factor receptor expression by small interfering RNA inhibits A549 human lung cancer cell invasion in vitro and metastasis in xenograft nude mice. Acta Biochim Biophys Sin (Shanghai) 2007;39:137–147. doi: 10.1111/j.1745-7270.2007.00257.x. [DOI] [PubMed] [Google Scholar]
- 75.Liu C-H, Bao H-G, Ge Y-L, Wang S-K, Shen Y, Xu L. Celecoxib inhibits insulin-like growth factor 1 induced growth and invasion in non-small cell lung cancer. Oncol Lett. 2013;5:1943–1947. doi: 10.3892/ol.2013.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Long L, Rubin R, Brodt P. Enhanced invasion and liver colonization by lung carcinoma cells overexpressing the type 1 insulin-like growth factor receptor. Exp Cell Res. 1998;238:116–121. doi: 10.1006/excr.1997.3814. [DOI] [PubMed] [Google Scholar]
- 77.Pfister DG, Johnson DH, Azzoli CG, Sause W, Smith TJ, Baker S, Jr, Olak J, Stover D, Strawn JR, Turrisi AT, Somerfield MR. American Society of Clinical Oncology treatment of unresectable non-small cell lung cancer guideline: upate 2003. J Clin Oncol. 2004;22:330–353. doi: 10.1200/JCO.2004.09.053. [DOI] [PubMed] [Google Scholar]
- 78.Lee SJ, Kim EJ, Lee HJ, Kim SY, Oh SJ, Ryu JS, Moon DH, Ahn J-H, Kim S-W. A pilot study for the early assessment of the effects of BMS-754807 plus gefitinib in a H292 tumor model by [18F]fluorothymidine-positron emission tomography. Invest New Drug. 31:506–15. [DOI] [PubMed]
- 79.Warshamana-Greene GS, Litz J, Buchdunger E, Garcia-Echeverria C, Hofmann F, Krystal GW, García-Echeverría C. The insulin-like growth factor-I receptor kinase inhibitor, NVP-ADW742, sensitizes small cell lung cancer cell lines to the effects of chemotherapy. Clin Cancer Res. 2005;11:1563–1571. doi: 10.1158/1078-0432.CCR-04-1544. [DOI] [PubMed] [Google Scholar]