

ABSTRACT
AMP-activated protein kinase (AMPK) is a serine/threonine kinase that is well conserved during evolution. AMPK activation inhibits production of reactive oxygen species (ROS) in cells via suppression of NADPH oxidase. However, the role of AMPK during the process of Brucella infection remains unknown. Our data demonstrate that B. abortus infection induces AMPK activation in HeLa cells in a time-dependent manner. The known AMPK kinases LKB1, CAMKKβ, and TAK1 are not required for the activation of AMPK by B. abortus infection. Instead, this activation is dependent on the RNase activity of inositol-requiring enzyme 1 (IRE1). Moreover, we also found that B. abortus infection-induced IRE1-dependent activation of AMPK promotes B. abortus intracellular growth with peritoneal macrophages via suppression of NADPH-derived ROS production.
IMPORTANCE Previous studies showed that B. abortus infection does not promote any oxidative burst regulated by NADPH oxidase. However, the underlying mechanism remains elusive. We report for the first time that AMPK activation caused by B. abortus infection plays important role in NADPH oxidase-derived ROS production.
INTRODUCTION
Brucella is an intracellular bacterial pathogen that causes abortion and infertility in animals and leads to recurrent fever and debilitating musculoskeletal, cardiac, and neurological complications at the chronic stage of the infection in humans. Its virulence depends on properties for survival and replication in host cells (1).
A prominent characteristic of Brucella is its ability to maintain its intracellular growth via using a stealthy strategy to avoid activation of host cells to produce cellular bactericidal substances (2). Reactive oxygen species (ROS), which are part of a number of bactericidal substances, are chemically reactive molecules containing oxygen. In the mammalian host, ROS are induced as an antimicrobial defense. Treatment of murine macrophages with methylene blue, an electron carrier, and ROS inhibitors increased and decreased the number of intracellular B. abortus organisms, respectively (3). This result is compatible with a previous report that production of superoxide by activated macrophages with gamma interferon (IFN-γ) limits intracellular growth of B. abortus (4). In addition, B. abortus barely activates human polymorphonuclear neutrophils (PMNs) and resists the killing mechanisms of these phagocytes via prematurely killing PMNs due to low ROS formation (5). Therefore, ROS production plays an important role in mediating B. abortus intracellular survival.
NADPH oxidases are primary sources of ROS and can be induced or activated by stresses, hormones, and cytokines (6). Elimination of NADPH oxidase in macrophages in vitro increased the intracellular survival of wild-type B. abortus (4). In addition, an ultrastructural study examining the localization of NADPH oxidase in B. abortus-infected macrophages found that Brucella-containing vacuoles (BCVs) tended not to be associated with NADPH oxidase (7), which facilitates bacterial replication in BCVs. In the present study, we sought to determine whether the B. abortus infection-induced proteins also contribute to evasion of NADPH oxidase-mediated control of bacterial growth during B. abortus infection of macrophages.
AMP-activated protein kinase (AMPK) is a serine/threonine kinase that has been highly conserved during evolution and is a critical regulator of energy metabolic homeostasis at the cellular and whole-organism levels (8). AMPK is composed of α, β, and γ subunits, each of which has at least two isoforms. The α subunit contains the catalytic activity, while the β and γ regulatory subunits maintain the stability of the heterotrimer complex. The phosphorylation of Thr172 in the activation loop of the α subunit of AMPK is required for its activation. Three major AMPK kinases that phosphorylate the α subunit of AMPK have been identified to date, i.e., the tumor suppressor gene product LKB1, the transforming growth factor β-activated kinase (TAK1), and the Ca2/calmodulin-dependent protein kinase kinase β (CaMKKβ) (9). Interestingly, AMPK activation is able to suppress NADPH oxidase-derived ROS production in cells (10, 11). Considering that production of ROS is suppressed in B. abortus-infected host cells (12), we hypothesized that B. abortus infection might elicit AMPK activation, which contributes to the maintenance of B. abortus intracellular survival via suppression of ROS.
Brucella infection can cause endoplasmic reticulum (ER) stress and activate the unfolded protein response (UPR) (13–15). The UPR includes three trans-ER membrane proteins: activating transcription factor 6 (ATF6), eukaryotic translation initiation factor 2-alpha kinase 3 (PERK), and inositol-requiring enzyme 1 (IRE1) (16). However, Brucella infection of macrophages or HeLa cells induces activation of only IRE1 in the case of B. abortus (14, 15). IRE1, which has been identified to be a necessary protein for B. abortus replication (17), is both a kinase and an endoribonuclease. ER stress is able to induce IRE1-dependent AMPK activation in response to nitric oxide (18). However, the role of IRE1 RNase in mediating AMPK activation during B. abortus infection remains unclear.
This study provides evidence that B. abortus infection-induced activity of IRE1 leads to AMPK activation in host cells and shows that AMPK activation promotes bacterial growth within macrophages via suppressing NADPH oxidase-derived ROS.
MATERIALS AND METHODS
Materials.
Anti-pAMPK (Thr172), AMPK, phosphatidylinositol 3-kinase (PI3K), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), IRE1, TAK1, LKB1, and all secondary antibodies were purchased from Cell Signaling Technology (Danvers, MA). Anti-NADPH oxidase subunits (p47phox and p67phox), UCP2, and CAMKKβ were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-pIRE1 antibody was from Abcam. Fluorescein isothiocyanate (FITC)-dextran was from Sigma-Aldrich (Shanghai, China). Dihydroethidium (DHE) was purchased from Molecular Probes. Mitochondrial complex II was from Invitrogen (Shanghai, China). All PCR primers were purchased from Invitrogen. Other chemicals, if not otherwise indicated, were from Sigma-Aldrich (Shanghai, China). All drug concentrations are expressed as the final molar concentration in the working buffer.
Mice.
Eight-week-old female C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME). The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by Jilin University.
Peritoneal macrophage preparation.
The resident peritoneal macrophages from C57BL/6 mice were extracted by washing the cavity with 10 ml of cold phosphate-buffered saline (PBS). Cell suspensions were washed twice with cold complete tissue culture medium (alpha minimal essential medium [αMEM] with 10% fetal calf serum and 50 μg/ml gentamicin). Isolated peritoneal cells were counted with 0.4% trypan blue and plated in complete tissue culture medium at a concentration of 2.0 × 105 cells per well in a 24-well plate. After incubation for 60 min at 37°C in a 5% CO2 atmosphere, the nonadherent cells were removed by washing them five times in 1 ml PBS hold at 37°C. The peritoneal macrophages were incubated in fresh αMEM supplemented with 50 μg/ml of gentamicin (19).
Cell infection and survival assay.
HeLa cells or mouse peritoneal macrophages were plated in 24-well plates in complete tissue culture medium without antibiotics at a concentration of 2.0 × 105 cells per well and incubated overnight at 37°C with 5% CO2. The cells were infected with B. abortus strain 2308 in triplicate wells of a 24-well plate at a multiplicity of infection (MOI) of 100:1 by centrifuging bacteria onto cells at 400 × g for 10 min at 4°C. Following 15 min of incubation at 37°C in an atmosphere containing 5% CO2, the cells were washed three times with αMEM to remove extracellular bacteria and incubated for an additional 60 min in medium supplemented with 50 μg/ml gentamicin to kill extracellular bacteria. To monitor B. abortus intracellular survival, infected cells were lysed with 0.1% Triton X-100 in phosphate-buffered saline (PBS) at certain time points, and serial dilutions of lysates were rapidly plated onto tryptic soy agar plates to enumerate CFU (20, 21).
RNA interference.
HeLa cells were transfected with small interfering RNA (siRNA) and Lipofectamine 2000 in 6-well plates according to the manufacturer's protocol. All transfections contained 100 pmol siRNA (Invitrogen, Shanghai, China). The siRNA nucleotide sequences (LKB1, CCACCAAUGGCACACUCAA; TAK1, GCAACCCAAAGCGCUAAUU; CAMKKβ, CCGACAUAGCUGAGGACUU; and IRE1, CCACACAACAUCCUCAUAU) corresponding to the coding regions of human LKB1, TAK1, CAMKKβ, or IRE1 were selected. The negative control was set up using the 21-nucleotide RNA oligonucleotide that corresponded to the coding sequence of luciferase. Suppression of expression of endogenous LKB1, TAK1, CAMKKβ, or IRE1 by the siRNAs was determined by immunoblotting.
Measurement of ROS.
Intracellular ROS were measured using the dihydroethidium fluorescence/high-pressure liquid chromatography (HPLC) assay with minor modifications (22). Briefly, HeLa cells were incubated with 0.5 μM dihydroethidium for 30 min. After incubation, the cells were harvested and extracted with methanol. Oxyethidium (a product of dihydroethidium and superoxide anions) and ethidium (a product of dihydroethidium autoxidation) were separated and quantified by HPLC on a C18 column (mobile phase, gradient of acetonitrile and 0.1% trifluoroacetic acid) coupled with a fluorescence detector. Superoxide anion production was determined by the conversion of dihydroethidium into oxyethidium.
Measurement of NADPH oxidase activity.
NADPH oxidase activity was measured as described previously (23). Briefly, 20 mg protein was incubated with dihydroethidium (10 μM) and DNA (1.25 μg/ml) in PBS with the addition of NADPH (50 μM) in a final volume of 120 μl. Incubations were performed for 30 min at 37°C in the dark. Fluorescence intensity was recorded in a microplate reader (excitation, 490 nm; emission, 590 nm).
Western blotting.
Macrophages were lysed in ice-cold radioimmunoprecipitation assay (RIPA) buffer. The protein content was assayed with the bicinchoninic acid (BCA) protein assay reagent (Pierce, USA). Twenty micrograms of protein was loaded for SDS-PAGE and then transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was incubated with a 1:1,000 dilution of primary antibody, followed by a 1:2,000 dilution of horseradish peroxidase-conjugated secondary antibody. Protein bands were visualized by ECL (GE Healthcare). The band densities were measured by densitometry (model GS-700 imaging densitometer; Bio-Rad). The background was subtracted from the calculated area (24).
Analysis of mitochondrial UCP2.
Cells cultured in six-well plates were infected with B. abortus strain 2308 as described in “Cell infection and survival assay” above. Cell lysis of the cytosolic fraction was done with a Mitochondria/Cytosol Fractionation kit (Qiagen) according to the manufacturer's instructions. Uncoupling protein 2 (UCP2) expression in mitochondria was examined by Western blotting.
Construction of cDNA plasmids and mutagenesis.
The plasmid pcDNA3-Myc-human IRE1 was constructed by subcloning human IRE1 cDNA (accession no. NM_001433). IRE1 (K559A) and IRE1 (K907A) were generated by site-directed mutagenesis using a mutagenesis kit manufactured by Stratagene.
Cell culture and transfection.
Cell culture and transfection were performed as previously described (25). Briefly, HeLa cells were maintained in Dulbecco modified Eagle medium (DMEM) plus 10% fetal bovine serum (FBS) at 37°C and 5% CO2. The cells were cultured to 90% confluence before transfection. For transfection, the plasmid DNA (1 to 2 μg/60-mm dish) was premixed with Lipofectamine 2000 (Invitrogen) and incubated at room temperature for 15 min. The cells were incubated in the plasmid DNA-Lipofectamine 2000 mixture for 5 h. After 48 h of transfection, the cells were used for the experiment.
Statistical analysis.
Statistical analysis was performed using SPSS10.0 software. Data are expressed as means ± standard errors of the means (SEM). The statistical significance of differences was evaluated by using one-way analysis of variance (ANOVA) followed by the Student t test. A P value of <0.05 was considered statistically significant (25).
RESULTS
B. abortus infection induces AMPK activation in HeLa cells.
To examine whether B. abortus infection induced AMPK activation in HeLa cells, the level of AMPK with phosphorylation at threonine 172 was determined by immunoblotting with anti-pAMPK antibody. The results showed that B. abortus infection enhanced the phosphorylation of AMPK in HeLa cells in a time-dependent manner, with significant increases sustained from 8 to 36 h (Fig. 1A).
FIG 1.

B. abortus infection induces AMPK activation. (A) HeLa cells were infected with B. abortus for the indicated times and then lysed for immunoblot analysis of AMPK and pAMPK (T172). (B) HeLa cells were mock transfected or transfected with siRNA for LKB1, TAK1, or CAMKKβ for 48 h, followed by infection with B. abortus for 8 h. Cell lysates were prepared and analyzed by immunoblotting with anti-AMPK and -pAMPK (T172) antibody. The efficacies of silencing of LKB1, TAK1, and CAMKKβ were quantified using Bio-Rad densitometry software. GAPDH was used for normalization. Results are representative of three independent experiments. Con, control.
LKB1, TAK1, and CAMKKβ have been shown to function as activators of AMPK (26–28). To determine the mechanism by which B. abortus infection stimulates AMPK activation, siRNAs were used to knock down endogenous LKB1, TAK1, or CAMKKβ in HeLa cells. Considering that AMPK activation was increased significantly at 8 h postinfection (p.i.) (Fig. 1A), activation of AMPK at this time point was measured to show the regulatory role of B. abortus infection in cells. As shown in Fig. 1B, these siRNAs substantially reduced steady-state levels of LKB1 TAK1, or CAMKKβ protein. However, LKB1, TAK1, or CAMKKβ knockdown did not influence B. abortus infection-induced AMPK phosphorylation at 8 h postinfection. These data suggested that B. abortus infection-induced AMPK activation could be independent of LKB1, TAK1, or CAMKKβ.
The RNase activity of IRE1 is required for B. abortus infection induced-AMPK activation.
IRE1 has been reported to play an important role in mediating AMPK activation in response to ER stress (18). In addition, Brucella intracellular survival can trigger ER stress in host cells (13–15). Therefore, we hypothesized that B. abortus-stimulated AMPK activation might require IRE1 activation. To test this hypothesis, we used siRNA to knock down IRE1 expression prior to B. abortus infection. IRE1 expression was successfully suppressed compared to that in control siRNA-treated cells (Fig. 2A). Compared to control siRNA-treated cells, which showed with robust phosphorylation of AMPK, IRE1 knockdown prevented B. abortus-stimulated AMPK phosphorylation at 8 h (Fig. 2A).
FIG 2.

AMPK activation caused by B. abortus infection depends on the activity of IRE1 RNase. (A) HeLa cells were mock transfected or transfected with siRNA for IRE1 for 48 h, followed by infection with B. abortus for 8 h. Cell lysates were prepared and analyzed by immunoblotting with anti-AMPK and -pAMPK (T172) antibody. The efficacy of silencing of IRE1 was quantified using Bio-Rad densitometry software. (B) HeLa cells were infected with B. abortus for the indicated times and then lysed for immunoblot analysis of pIRE1. (C) HeLa cells transfected with empty vector or with IRE1(K599A) (kinase-deficient) or IRE1(K907A) (RNase-deficient) mutants were infected with B. abortus for 8 h, and AMPK phosphorylation was examined by Western blotting. (D) Cultured HeLa cells were pretreated with 4μ8C (30 nM) for 30 min and then infected with B. abortus for 8 h, and AMPK phosphorylation was examined by Western blotting. DMSO, dimethyl sulfoxide. GAPDH was used for normalization. Results are representative of three independent experiments.
Considering the potential role(s) of the RNase/kinase IRE1 (18), we determined whether enzyme activity of IRE1 is required for B. abortus-stimulated AMPK phosphorylation. First, HeLa cells were infected with B. abortus, and the phosphorylation state of IRE1 was analyzed by Western blotting. B. abortus infection triggered the phosphorylation of IRE1 (pIRE1) as early as 4 h after infection (Fig. 2B). These data suggest that B. abortus infection leads to activation of IRE1.
To determine the mechanisms by which activation of IRE1 leads to AMPK phosphorylation, we performed site-directed mutagenesis of IRE1 to generate kinase or RNase domain mutants. HeLa cells expressing IRE1(K599A) (kinase deficient) or IRE1(K907A) (RNase deficient) were infected with B. abortus for 8 h, and AMPK activation was examined. As shown in Fig. 2C, mutation of the RNase activity of IRE1 (K907A) markedly attenuated AMPK phosphorylation. In contrast, mutation of the kinase activity of IRE1 (K599A) did not prevent B. abortus-induced AMPK phosphorylation. To further confirm that IRE1 RNase activates AMPK, HeLa cells were pretreated with 4μ8C (an inhibitor of IRE1 RNase) before B. abortus infection, and AMPK phosphorylation was determined at 8 h p.i. Consistent with our previous result, inhibition of the RNase activity of IRE1 significantly abolished B. abortus-induced AMPK activation (Fig. 2D). Taken together, these data demonstrate that B. abortus infection stimulates AMPK activation through IRE1 RNase activity.
B. abortus infection-induced AMPK activation downregulates NADPH oxidase activity.
AMPK is a physiological suppressor of NADPH oxidase in multiple cell systems (6). Therefore, we investigated whether B. abortus infection-induced AMPK activation suppresses NADPH oxidase activity. To study the role of AMPK activation in NADPH oxidase activity, we measured NADPH oxidase activity in HeLa cells after infection with B. abortus for 2, 4, and 8 h. As shown in Fig. 3A, the activity of NADPH oxidase was markedly decreased by 4 to 8 h after infection. Compound C, a potent pharmacologic inhibitor of AMPK, significantly increased the B. abortus infection-induced NADPH oxidase activity (Fig. 3B). Consistent with the results for the compound C-treated group, B. abortus infection also enhanced NADPH oxidase activity in HeLa cells transfected with IRE1(K907A) (Fig. 3C). Taken together, these data confirm that B. abortus infection induces activation of IRE1, which in turn activates AMPK that suppresses NADPH oxidase.
FIG 3.

B. abortus infection-induced AMPK activation prevents upregulation of NADPH oxidase activity. (A) A confluent monolayer of HeLa cells infected with B. abortus was measured at various time points. NADPH oxidase activity was assayed by DHE fluorescence. (B) Cultured HeLa cells were pretreated with compound C (10 μM) for 30 min and then infected with B. abortus for the indicated times. NADPH oxidase activity was assayed by DHE fluorescence. (C) HeLa cells were transfected with IRE1(K907A) and then infected with B. abortus for the indicated times. NADPH oxidase activity was assayed by DHE fluorescence. Data are expressed as means ± SEM (n = 3 in each group). *, P < 0.05 versus control. (D) HeLa cells or cells transfected with IRE1(K907A) or pretreated with compound C (10 μM) were infected with B. abortus for 8 h. Expression of NADPH oxidase subunits (p47phox and p67phox) was measured by Western blotting. Quantitative data for p47phox and p67phox expression are shown under the Western blotting data. Data are expressed as means ± SEM (n = 3).
As NADPH oxidase activity can be mediated by the expression of its cytosolic subunits p47phox and p67phox (29, 30), we determined whether the decreased NADPH oxidase activity caused by B. abortus was due to changes in expression of NADPH oxidase subunits. As expected, B. abortus infection significantly reduced expression of p67phox and p47phox in HeLa cells (Fig. 3D). HeLa cells transfected with RNase-deficient IRE1(K907A) or pretreated with AMPK inhibitor compound C had significant increases in p67phox and p47phox expression. Taken together, these results suggest that B. abortus infection-induced AMPK activation suppresses NADPH oxidase activity via mediating the protein levels of NADPH subunits p67phox and p47phox.
B. abortus infection-induced AMPK activation promotes bacterial intracellular survival via decreasing NADPH-derived ROS production.
Having shown that B. abortus infection activates IRE1, which in turn activates AMPK that suppresses NADPH, we next determined if the NADPH suppression leads to reductions in ROS. B. abortus infection of HeLa cells induced a significant decrease in ROS over time (Fig. 4A). Pretreatment with an AMPK inhibitor, compound C, led to significantly more B. abortus-stimulated ROS (Fig. 4B). Likewise, HeLa cells transfected with RNase-deficient IRE1(K907A) had substantially more ROS production than control cells (Fig. 4C).
FIG 4.
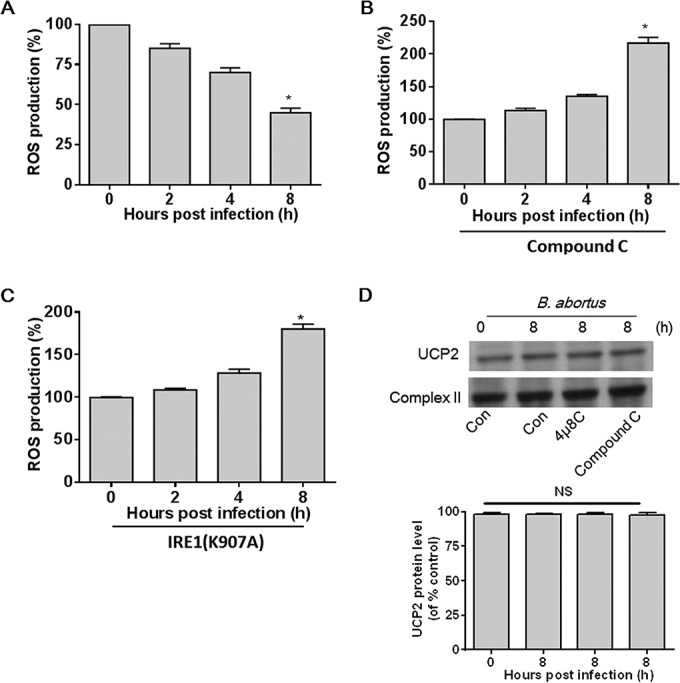
B. abortus infection-induced AMPK activation reduces NADPH-derived ROS production. (A) A confluent monolayer of HeLa cells infected with B. abortus was measured at various time points. ROS production was assayed by DHE fluorescence. (B) Cultured HeLa cells were pretreated with compound C (10 μM) for 30 min and then infected with B. abortus for the indicated times. ROS production was assayed by DHE fluorescence. (C) HeLa cells were transfected with IRE1(K907A) and then infected with B. abortus for the indicated times. ROS production was assayed by DHE fluorescence. (D) HeLa cells were pretreated with DMSO, 4μ8C (30 nM), or compound C (10 μM) for 30 min and then infected with B. abortus for 8 h, and UCP2 in mitochondria was examined by Western blotting. Complex II was used for normalization. Quantitative data for UCP2 expression are shown under the Western blot. Results are representative of three independent experiments. NS, no significance. Data from the ROS assay are expressed as means ± SEM (n = 3 in each group). *, P < 0.05 versus control.
Bronner et al. recently reported that B. abortus (strain RB51) infection initiates NLRP3- and caspase-2-mediated mitochondrial damage, which leads to enhanced ROS production (31). Given that the mitochondrial anion carrier protein uncoupling protein 2 (UCP2) is crucial for modulating mitochondrial ROS and that the generation of mitochondrial ROS is regulated by UCP2 expression (32–34), we examined whether ROS production is also from mitochondria in B. abortus (2308)-infected HeLa cells via checking UCP2 expression. As shown in Fig. 4D, inhibition of AMPK or IRE1 activation did not significantly elicit a change in UCP2 expression in mitochondria, suggesting that mitochondria do not participate in ROS production in B. abortus-infected cells. Taken together, these data indicate that B. abortus infection can prevent NADPH-derived ROS induction in an IRE1- and AMPK-dependent manner.
Finally we asked whether the mechanism is specific for HeLa cells. To address this question, mouse peritoneal macrophages which were pretreated with compound C or 4μ8C were infected with B. abortus, and AMPK activation and ROS production were measured, respectively. Consistent with the results in HeLa cells, AMPK activation caused by B. abortus infection was mediated by IRE1 RNase activity, which suppresses ROS production in macrophages (Fig. 5A and B). As ROS play an essential role in killing intracellular bacteria (3, 35), we also investigated the role of AMPK activation in bacterial intracellular survival at different times by counting B. abortus CFU. As shown in Fig. 5C, the number of intracellular live bacterial decreased significantly in macrophages pretreated with compound C or 4μ8C compared to control cells, suggesting that the suppression of ROS via IRE1 and AMPK activation is essential for intracellular survival.
FIG 5.

IRE1-dependent activation of AMPK promotes B. abortus growth within macrophages. (A) Mouse peritoneal macrophage cells were pretreated with DMSO, 4μ8C (30 nM), or compound C (10 μM) for 30 min and then infected with B. abortus for 8 h, and AMPK phosphorylation in macrophages was examined by Western blotting. (B) Mouse peritoneal macrophage cells were pretreated with DMSO, 4μ8C (30 nM), or compound C (10 μM) for 30 min and then infected with B. abortus for 8 h, and ROS production was assayed by DHE fluorescence. (C) Mouse peritoneal macrophage cells pretreated with DMSO, 4μ8C (30 nM), or compound C (10 μM) were infected with B. abortus for the indicated times. Bacterial intracellular survival within macrophage cells was determined by counting the viable intracellular bacteria (CFU). *, P < 0.05 versus control. Data are expressed as means ± SEM (n = 3 in each group). (D) Proposed molecular mechanism for protective effect of AMPK activation on B. abortus intracellular growth.
DISCUSSION
This study demonstrates that B. abortus infection alters host cellular proteins that are essential for intracellular survival. Our data show that B. abortus infection induces AMPK activation in HeLa cells or peritoneal macrophages that are essential for the suppression of NADPH and ROS production required for intracellular survival of the bacterium. Mechanistically, we discovered a novel role for IRE1 RNase activity in this process whereby RNase-deficient IRE1 is unable to activate AMPK, leading to failure to suppress ROS and suppression of bacterial survival.
One of the most important findings of this study is that B. abortus infection induces AMPK activation. To explore the mechanisms by which B. abortus mediates AMPK activation, we initially evaluated the effects of siRNA knockdown for each of the known AMPK kinases, LKB1, CAMKKβ, and TAK1. Surprisingly, siRNA knockdown of these AMPK kinases did not inhibit B. abortus infection-induced AMPK activation, indicating that each of these known AMPK kinases is dispensable for AMPK activation in B. abortus-infected HeLa cells. IRE1 has been reported to participate in mediating AMPK activation in response to nitric oxide stimulation (18), and recent studies showed that Brucella infection of macrophages or HeLa cells induces activation of IRE1 (13–15), suggesting that IRE1 may influence the activation of AMPK during the process of B. abortus infection. In testing this hypothesis, we made the similar observation that infection with B. abortus activates IRE1. More importantly, our results demonstrated that IRE1 RNase mutation inhibits AMPK activation in response to B. abortus infection. However, the direct effects of B. abortus on IRE1 RNase activity are currently unknown. Quercetin has been reported to stimulate IRE1 RNase activity and activate AMPK as an IRE1 ligand (36, 37). Brucella can deliver different effector proteins across the Brucella-containing vacuole (BCV) membrane into the host cell via the VirB type IV secretion system (T4SS) (1, 38). Therefore, we speculate that B. abortus may generate an effector acting as an IRE1 ligand for activation of IRE1 RNase. Indeed, the effector VceC has been shown to participate in modulating IRE1 RNase activity via binding of the ER chaperone BiP (15). In future experiments, we will further investigate whether VceC or other effectors function as IRE1 ligands to activate AMPK.
Another important finding of this study is that AMPK activation promotes B. abortus intracellular growth via suppressing ROS production in host cells. Previous studies showed that Brucella lipopolysaccharide induces low ROS formation mediated by NADPH oxidase in PMNs (5). However, the underlying mechanism remains elusive. To our knowledge, our study is the first to report that AMPK activation plays an important role in NADPH oxidase-derived ROS production downstream of Brucella infection. NADPH oxidase, which is inhibited by activity of AMPK, is identified as the main source of ROS (6). Our data show that expression of NADPH oxidase subunits was downregulated in the presence of B. abortus infection. Accordingly, decreased activity of NADPH oxidase abolishes ROS production and, consequently, significantly promotes bacterial replication. Importantly, inhibition of AMPK significantly increased the production of NADPH cytosolic oxidase subunits, suggesting that the effects of AMPK on B. abortus infection are directly mediated by inhibition of NADPH oxidase-derived ROS (Fig. 5D).
In this study, pretreatment with AMPK inhibitor compound C exerted a dramatic effect leading to decreases in B. abortus intracellular growth. These data suggest that B. abortus will trigger increased ROS production of the host in the absence of AMPK activation (Fig. 5C). Although compound C is widely utilized to assess the role of AMPK in vitro, the drug may affect other cellular processes besides AMPK (39). The nonspecificity of compound C is one limitation of this study. However, these results supply a rationale to further investigate specific inhibitors of AMPK that might be utilized to suppress Brucella survival. Also, despite nonspecificity, compound C may be useful therapeutically in view of inhibition of B. abortus intracellular growth. Successful inhibition of Brucella virulence in vivo by compound C or other, more selective ROS modulation could open a new avenue of drug development.
In conclusion, the results demonstrate that B. abortus infection-induced AMPK activation promotes bacterial growth within host cells via inhibiting production of ROS by suppression of NADPH oxidase. It is tempting to speculate that targeted intervention with AMPK activation could provide an attractive therapy for management of brucellosis.
ACKNOWLEDGMENTS
We thank Mary Beth Humphrey (Department of Medicine, University of Oklahoma Health Sciences Center, Oklahoma City, OK) for critically reading the manuscript.
This work was funded by National Natural Science Foundation of China grant 31372409 to Qisheng Peng. This work was also supported by Natural Science Foundation of China grants 81472030 and 21175055, Jilin Province Science and Technology Department grants 20110739 and 20150204001YY, and Jilin University Bethune Project B grant 2012210 to Ning Liu.
We declare that we have no competing interests.
REFERENCES
- 1.de Jong MF, Tsolis RM. 2012. Brucellosis and type IV secretion. Future Microbiol 7:47–58. doi: 10.2217/fmb.11.136. [DOI] [PubMed] [Google Scholar]
- 2.Barquero-Calvo E, Chaves-Olarte E, Weiss DS, Guzman-Verri C, Chacon-Diaz C, Rucavado A, Moriyon I, Moreno E. 2007. Brucella abortus uses a stealthy strategy to avoid activation of the innate immune system during the onset of infection. PLoS One 2:e631. doi: 10.1371/journal.pone.0000631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang X, Leonard B, Benson R, Baldwin CL. 1993. Macrophage control of Brucella abortus: role of reactive oxygen intermediates and nitric oxide. Cell Immunol 151:309–319. doi: 10.1006/cimm.1993.1241. [DOI] [PubMed] [Google Scholar]
- 4.Sun YH, den Hartigh AB, Santos RD, Adams LG, Tsolis RM. 2002. virB-mediated survival of Brucella abortus in mice and macrophages is independent of a functional inducible nitric oxide synthase or NADPH oxidase in macrophages. Infect Immun 70:4826–4832. doi: 10.1128/IAI.70.9.4826-4832.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barquero-Calvo E, Mora-Cartin R, Arce-Gorvel V, de Diego JL, Chacon-Diaz C, Chaves-Olarte E, Guzman-Verri C, Buret AG, Gorvel JP, Moreno E. 2015. Brucella abortus induces the premature death of human neutrophils through the action of its lipopolysaccharide. PLoS Pathog 11:e1004853. doi: 10.1371/journal.ppat.1004853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Song P, Zou MH. 2012. Regulation of NAD(P)H oxidases by AMPK in cardiovascular systems. Free Radic Biol Med 52:1607–1619. doi: 10.1016/j.freeradbiomed.2012.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gay B, Sanchez-Teff S, Caravano R. 1984. Ultrastructural localization of NADPH-oxidase activity in murine peritoneal macrophages during phagocytosis of Brucella. Correlation with the production of superoxide anions. Virchows Arch B Cell Pathol Incl Mol Pathol 45:147–155. doi: 10.1007/BF02889861. [DOI] [PubMed] [Google Scholar]
- 8.Lage R, Dieguez C, Vidal-Puig A, Lopez M. 2008. AMPK: a metabolic gauge regulating whole-body energy homeostasis. Trends Mol Med 14:539–549. doi: 10.1016/j.molmed.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 9.Fisslthaler B, Fleming I. 2009. Activation and signaling by the AMP-activated protein kinase in endothelial cells. Circ Res 105:114–127. doi: 10.1161/CIRCRESAHA.109.201590. [DOI] [PubMed] [Google Scholar]
- 10.Song P, Wang SX, He CY, Wang SB, Liang B, Viollet BN, Zou MH. 2011. AMPK alpha 2 deletion exacerbates neointima formation by upregulating Skp2 in vascular smooth muscle cells. Circ Res 109:1230-U1291. doi: 10.1161/CIRCRESAHA.111.250423. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Wang SX, Zhang M, Liang B, Xu J, Xie ZL, Liu C, Viollet B, Yan DG, Zou MH. 2010. AMPK alpha 2 deletion causes aberrant expression and activation of NAD(P)H oxidase and consequent endothelial dysfunction in vivo role of 26S proteasomes. Circ Res 106:1117–1128. doi: 10.1161/CIRCRESAHA.109.212530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei P, Lu Q, Cui GM, Guan ZH, Yang L, Sun CJ, Sun WC, Peng QS. 2015. The role of TREM-2 in internalization and intracellular survival of Brucella abortus in murine macrophages. Vet Immunol Immunopathol 163:194–201. doi: 10.1016/j.vetimm.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 13.Smith JA, Khan M, Magnani DD, Harms JS, Durward M, Radhakrishnan GK, Liu YP, Splitter GA. 2013. Brucella induces an unfolded protein response via TcpB that supports intracellular replication in macrophages. PLoS Pathog 9:e1003785. doi: 10.1371/journal.ppat.1003785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taguchi Y, Imaoka K, Kataoka M, Uda A, Nakatsu D, Horii-Okazaki S, Kunishige R, Kano F, Murata M. 2015. Yip1A, a novel host factor for the activation of the IRE1 pathway of the unfolded protein response during Brucella infection. PLoS Pathog 11:e1004747. doi: 10.1371/journal.ppat.1004747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Jong MF, Starr T, Winter MG, den Hartigh AB, Child R, Knodler LA, van Dijl JM, Celli J, Tsolis RM. 2013. Sensing of bacterial type IV secretion via the unfolded protein response. mBio 4:e00418-12. doi: 10.1128/mBio.00418-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ron D, Walter P. 2007. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 17.Qin QM, Pei J, Ancona V, Shaw BD, Ficht TA, de Figueiredo P. 2008. RNAi screen of endoplasmic reticulum-associated host factors reveals a role for IRE1alpha in supporting Brucella replication. PLoS Pathog 4:e1000110. doi: 10.1371/journal.ppat.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meares GP, Hughes KJ, Naatz A, Papa FR, Urano F, Hansen PA, Benveniste EN, Corbett JA. 2011. IRE1-dependent activation of AMPK in response to nitric oxide. Mol Cell Biol 31:4286–4297. doi: 10.1128/MCB.05668-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen F, He Y. 2009. Caspase-2 mediated apoptotic and necrotic murine macrophage cell death induced by rough Brucella abortus. PLoS One 4:e6830. doi: 10.1371/journal.pone.0006830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Celli J, de Chastellier C, Franchini DM, Pizarro-Cerda J, Moreno E, Gorvel JP. 2003. Brucella evades macrophage killing via VirB-dependent sustained interactions with the endoplasmic reticulum. J Exp Med 198:545–556. doi: 10.1084/jem.20030088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He Y, Reichow S, Ramamoorthy S, Ding X, Lathigra R, Craig JC, Sobral BW, Schurig GG, Sriranganathan N, Boyle SM. 2006. Brucella melitensis triggers time-dependent modulation of apoptosis and down-regulation of mitochondrion-associated gene expression in mouse macrophages. Infect Immun 74:5035–5046. doi: 10.1128/IAI.01998-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang SX, Xu J, Song P, Wu Y, Zhang JH, Choi HC, Zou MH. 2008. Acute inhibition of guanosine triphosphate cyclohydrolase 1 uncouples endothelial nitric oxide synthase and elevates blood pressure. Hypertension 52:484–490. doi: 10.1161/HYPERTENSIONAHA.108.112094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu C, Wu JL, Zou MH. 2012. Activation of AMP-activated protein kinase alleviates high-glucose-induced dysfunction of brain microvascular endothelial cell tight-junction dynamics. Free Radic Biol Med 53:1213–1221. doi: 10.1016/j.freeradbiomed.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao Y, Cui G, Zhang N, Liu Z, Sun W, Peng Q. 2012. Lipopolysaccharide induces endothelial cell apoptosis via activation of Na(+)/H(+) exchanger 1 and calpain-dependent degradation of Bcl-2. Biochem Biophys Res Commun 427:125–132. doi: 10.1016/j.bbrc.2012.09.023. [DOI] [PubMed] [Google Scholar]
- 25.Cui G, Wei P, Zhao Y, Guan Z, Yang L, Sun W, Wang S, Peng Q. 2014. Brucella infection inhibits macrophages apoptosis via Nedd4-dependent degradation of calpain2. Vet Microbiol 174:195–205. doi: 10.1016/j.vetmic.2014.08.033. [DOI] [PubMed] [Google Scholar]
- 26.Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LGD, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. 2003. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 27.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. 2005. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab 2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 28.Herrero-Martin G, Hoyer-Hansen M, Garcia-Garcia C, Fumarola C, Farkas T, Lopez-Rivas A, Jaattela M. 2009. TAK1 activates AMPK-dependent cytoprotective autophagy in TRAIL-treated epithelial cells. EMBO J 28:677–685. doi: 10.1038/emboj.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pasquier F, Boulogne A, Leys D, Fontaine P. 2006. Diabetes mellitus and dementia. Diabetes Metab 32:403–414. doi: 10.1016/S1262-3636(07)70298-7. [DOI] [PubMed] [Google Scholar]
- 30.Ristow M. 2004. Neurodegenerative disorders associated with diabetes mellitus. J Mol Med 82:510–529. [DOI] [PubMed] [Google Scholar]
- 31.Bronner DN, Abuaita BH, Chen XY, Fitzgerald KA, Nunez G, He YQ, Yin XM, O'Riordan MXD. 2015. Endoplasmic reticulum stress activates the inflammasome via NLRP3- and caspase-2-driven mitochondrial damage. Immunity 43:451–565. doi: 10.1016/j.immuni.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mailloux RJ, Harper ME. 2011. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic Biol Med 51:1106–1115. doi: 10.1016/j.freeradbiomed.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 33.Negre-Salvayre A, Hirtz C, Carrera G, Cazenave R, Troly M, Salvayre R, Penicaud L, Casteilla L. 1997. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J 11:809–815. [PubMed] [Google Scholar]
- 34.Xie Z, Zhang J, Wu J, Viollet B, Zou MH. 2008. Upregulation of mitochondrial uncoupling protein-2 by the AMP-activated protein kinase in endothelial cells attenuates oxidative stress in diabetes. Diabetes 57:3222–3230. doi: 10.2337/db08-0610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Deffert C, Cachat J, Krause KH. 2014. Phagocyte NADPH oxidase, chronic granulomatous disease and mycobacterial infections. Cell Microbiol 16:1168–1178. doi: 10.1111/cmi.12322. [DOI] [PubMed] [Google Scholar]
- 36.Ahn J, Lee H, Kim S, Park J, Ha T. 2008. The anti-obesity effect of quercetin is mediated by the AMPK and MAPK signaling pathways. Biochem Biophys Res Commun 373:545–549. doi: 10.1016/j.bbrc.2008.06.077. [DOI] [PubMed] [Google Scholar]
- 37.Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, Towler MC, Brown LJ, Ogunbayo OA, Evans AM, Hardie DG. 2010. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab 11:554–565. doi: 10.1016/j.cmet.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marchesini MI, Herrmann CK, Salcedo SP, Gorvel JP, Comerci DJ. 2011. In search of Brucella abortus type IV secretion substrates: screening and identification of four proteins translocated into host cells through VirB system. Cell Microbiol 13:1261–1274. doi: 10.1111/j.1462-5822.2011.01618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saito S, Furuno A, Sakurai J, Park HR, Shin-ya K, Tomida A. 2012. Compound C prevents the unfolded protein response during glucose deprivation through a mechanism independent of AMPK and BMP signaling. PLoS One 7:e45845. doi: 10.1371/journal.pone.0045845. [DOI] [PMC free article] [PubMed] [Google Scholar]