

Abstract
The 15q13.3 microdeletion is a recurrent CNV, presumably mediated by NAHR between segmental duplications in chromosome 15. The 15q13.3 deletion and duplication are associated with a wide range of clinical manifestations, such as intellectual deficits, seizures, autism, language and developmental delay, neuropsychiatric impairments, and behavioral problems illustrating incomplete penetrance and expressivity. This study comprises an evaluation of 106 symptomatic patients carrying the heterozygous deletion, as well as of 21 patients carrying the duplication, who have been described in previous studies. The analysis shows considerable heterogeneity for the manifestation of different key symptoms and familiar occurrence. Furthermore, 8 new patients are introduced. Convoluted familiar connections give new insights into the complexity of symptomatic manifestation. In previous studies, different opinions have been expressed as to the nature and precise location of the deletion breakpoints. Here, we show that not CHRNA7 and CHRFAM7A, but rather FAM7A or GOLGA8, serve as breakpoint regions concerning our patients. The deletion is described as heterogeneous in size. However, we assume that not only different breakpoints but also the imprecision of aCGH analysis on chromosome 15 due to segmental duplications accounts for the variability in size.
Key Words: Copy number variations, Microdeletion 15q13.3, Segmental duplications
The 15q13.3 microdeletion syndrome (OMIM 612002), which has been first described by Sharp et al. [2008], is a recurrent CNV, presumably mediated by NAHR between segmental duplications (BP3-BP5, BP4-BP5) in chromosome 15. The 15q13.3 deletion and duplication are associated with a wide range of neurodevelopmental disorders, such as intellectual deficits, seizures, autism, language and developmental delay, neuropsychiatric impairments, and behavioral problems showing incomplete penetrance and variable expressivity. The typical 1.6-Mb deletion harbors 7 genes: ARHGAP11B, MTMR10, MTMR 15, TRPM1, KLF13, OTUD7A, and CHRNA7. CHRNA7 encodes for the neuronal alpha7 nicotinic acetylcholine receptor and, therefore, is regarded as the major candidate gene responsible for the clinical features expressed.
In this study, we describe 6 so far unpublished patients carrying the typical 15q13.3 microdeletion between BP4 and BP5, including a family with 4 affected members who are described further below. In addition, 1 patient with a 15q13.3 duplication and 1 patient carrying a 3.4-Mb deletion between BP3 and BP5 are described.
Moreover, in order to explain the variety of clinical manifestations and the severity of symptoms, we reviewed the data of 106 symptomatic patients with heterozygous 15q13.3 deletion and 21 patients with duplications which have been reported.
In recent studies, different opinions have arisen as to the nature and precise location of the deletion breakpoints. We investigated the breakpoint location using FISH in order to confirm in how far these options prove true for our patients.
Methods
DNA Extraction and Array CGH
Peripheral blood leukocytes were used as a source of DNA. Array-CGH analysis was performed using an array slide Sure Print 4x180K (Agilent Technologies, Santa Clara, Calif., USA). Patient DNA was labeled with Cy3, reference DNA with Cy5. Hybridization was carried out using Cot-1 DNA (1.0 µg/ml). Test DNA samples were hybridized with gender-matched reference DNA (Agilent). Purification, hybridization and washing steps were performed according to the manufacturer's instructions. Sure Scan microarray Scanner G2600D (Agilent), Feature Extraction software (Agilent) and Agilent Cytogenomics Software Edition 2.0.6.0 were used.
FISH and Breakpoint Analysis
Slides with metaphases of cultivated peripheral lymphocytes had been stored at −70°C. Before hybridization, slides were processed in an alcohol series (70, 80 and 100%), followed by pepsin treatment (15 min, 37°C). Fluorescence-labeled BACs (Illumina®, BlueFish) were used as FISH probes. Probe preparation, hybridization and washing steps were performed according to Illumina's instructions.
BAC probes used were RP11-11H9, 22,067,176-22,300,706, chr.15q11.2; RP11-40J8, 30,546,758-30,724,265, chr.15q13.2; RP11-348B17, 31,281,641-31,502,115, chr.15q.13.3; RP11-265I17, 32,293,149-32,457,541, chr.15q13.3; RP11-280K19, 32,654,212-32,821,799, chr.15q13.3, and RP11-232J12, 53,792,861-53,948,902, chr.15q21.3 (see fig. 1, hg19).
Fig. 1.

Schematic overview of the region 15q13.3. Location of FISH probes is given by colored numbers.
Results
Clinical Assessment
The patients were initially observed on the occasion of neuropediatric examination because of intellectual deficits and/or developmental delay. Table 1 summarizes the cytogenetic and clinical findings.
Table 1.
Overview of newly introduced patients
| Patient |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1, female | 2, female | 3, female | 4, male | 5, male | 6, male | 7, male | 8, male | ||
| Abberation size | 1.97 Mb | 1.85 Mb | 1.78 Mb | 1.47 Mb | 1.85 Mb | 3.47 Mb | 2.26 Mb | 1.97 Mb | |
| Intellectual deficits | + | IQ 63 | + | IQ 56 | IQ 78 | + | + | + | |
| Development delay | + | + | NA | + | – | + | + | + | |
| Parental phenotype noticeable | + | + | + | + | – | – | – | – | |
| OFC | P>97 | P3–10 | P50 | P50 | NA | P50 | P10 | P50 | |
| Weight | P10 | P50 | P>97 | P97 | NA | P50 | P3–10 | P10 | |
| Language problems | – | + | – | + | NA | + | + | + | |
| Behavioral problems | ADHD | ADHD | Stereotypies | ADHD | ADHD | ADHD | – | + | |
| Epilepsy | – | – | – | – | + | – | – | – | |
| Hypotonia | NA | NA | NA | NA | – | + | NA | + | |
For aCGH results see online supplementary material 2 and figure 2 for the pedigree of patients 1 −4. NA = No data available; + = feature expressed; – = feature not expressed.
Description of Patients
Patient 1 (III/1)
The 13-year-old girl presented with attention deficit hyperactivity disorder (ADHD), intellectual deficits, developmental delay, recurrent respiratory infections, and scoliosis. Her weight and head circumference (OFC) vary around the 10th percentile (P), respectively P90. She attends a special school. Her mother (II/2, in fig. 2) also attended a school for special education and completed it successfully. Both parents have not been available for further investigation. Patient 1 is a half sister of patients 2 and 3, and the cousin of patient 4.
Fig. 2.

Pedigree of patients 1-4. Filled circles and squares indicate tested symptomatic deletion carriers. Circle with vertical line indicates a deletion carrier by evidence, who was not tested. P1-P4 were tested. aCGH results are shown in online supplementary material 2. Patients III/4 and III/5 were tested, but clinical evaluation was not possible. Patient III/9 was tested (no carrier), and patients III/7 and III/8 were not tested.
Patient 2 (III/2)
The female patient was introduced at the age of 9 and showed developmental and language delay as well as intellectual deficits (K-ABC-Test: IQ 63). A waking EEG showed general alterations, but so far, no seizures have occurred. Her OFC is between P3 and P10, weight and height are in normal ranges. ADHD is treated with methylphenidate. She is described as a cheerful but also aggressive and impulsive person and attends a special school. Both parents have not been available for further investigation.
Patient 3 (III/3)
The 10-year-old patient was diagnosed with a learning disorder, which was not further defined. The girl's body measurements show a high variation in time. Until the age of 2, her OFC was P<3; currently, her weight is P97. She is obese, shows stereotyped behavior and attends a special school.
Patient 4 (III/6)
The examination of the patient (13 years old) revealed language and psychomotor delay as well as mild intellectual deficits (HAWIK-IV: SW 56). Noticeable features are multiple dyslalia and ADHD, which is treated with methylphenidate (10-15 mg/day). The boy's EEG is unremarkable, his OFC is normal, height P90, and his weight P97. The patient's character is described as friendly but at times aggressive and impulsive. He attends a school for the handicapped. Both parents attended a special school. His father (II/5) is imprisoned for assault. His 2 sisters (III/7, III/8) also are visiting a special school, and his half brother (III/9) shows a delay in development and language, but does not carry the deletion. The patient is the cousin of patients 1, 2 and 3. The parents have not been available for investigation.
Patient 5
The patient (14 years old) presented with abnormal social behavior and ADHD. Due to his poor school performance and the discrepancy between his intellectual performance (HAMIK-IV, IQ 78) and the family's mean, a medical examination was initiated. After an epileptic seizure, he has been treated successfully with Orfiril®. ADHD is treated with methylphenidate (5-10 mg/day). His parents have not been available for investigation.
Patient 6
As an infant, patient 6 manifested developmental and language delay. He showed signs of hypotonia. At the time of examination, the 19-year-old patient presented with intellectual deficits and ADHD. The treatment with methylphenidate was stopped after the patient started high-performance sport, which proved to be an effective treatment of the ADHD symptoms. His mother is not a deletion carrier. His father has not been available for investigation.
Patient 7
The patient was examined at the age of 7 and revealed mild intellectual deficits (K-ABC-Test, SW: 63). His body measurements are all within P10. He is described as restless and inattentive. He attends a special school. His parents have been unavailable for investigation.
Patient 8
The patient was born with a heart defect (ventricular septal defect and atrial septal defect). He shows dysmorphic features such as low-set ears, hypertelorism, strabism, and hypotonia. His psychomotor development is delayed. The patient's birth length and OFC were P50, his weight was P10. He inherited the duplication from his father who shows an unremarkable phenotype. An uncle (on the maternal site) with epilepsy and malignant hyperthermia has been reported.
Breakpoint Analysis
In previous studies, the heterogeneity of the deletion size has been a topic of interest. Shinawi et al. [2009] assumed FAM7A1/2 to function as a breakpoint, whereas Szafranski et al. [2010] suspected CHRNA7 and CHRFAM7A to be responsible for the recurrent deletion in the 15q13.3 locus. Antonacci et al. [2014] suggested the gene family GOLGA8 as possible candidates. An attempt was made to validate these theories by using FISH. Probe 4 marks CHRNA7, probe 5 marks FAM7A, probe 2 marks CHRFAM7A, and probe 3 marks a region distally to CHRFAM7A (fig. 1). In our patients, FAM7A is detectable in both chromosomes 15. This finding indicates that the distal breakpoint is located between CHRNA7 and FAM7A. The analysis of patients 7 and 4 show that the proximal breakpoint is located distally to CHRFAM7A (figs. 3, 4). In patient 7, probe 3 bridges the breakpoint. Therefore, CHRNA7 and CHRFAM7A do not mediate the recurrent deletion regarding our patients.
Fig. 3.
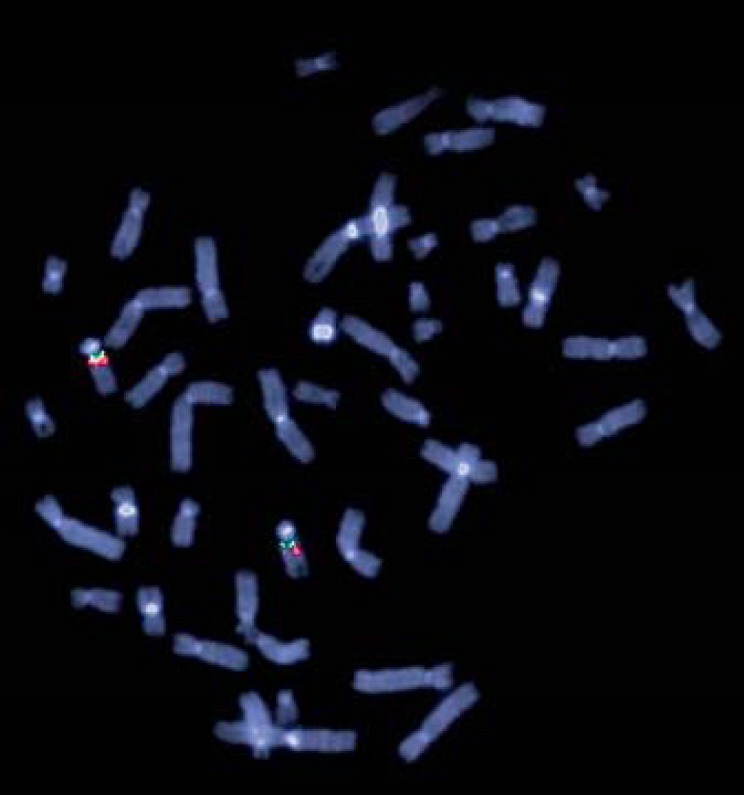
Metaphase FISH using BAC RP11-40J8 (probe 2, labeled in green) and RP11-348B17 (probe 3, labeled in red) showing the proximal breakpoint of patient 7. Probe 3 bridges the breakpoint (weak red signal).
Fig. 4.
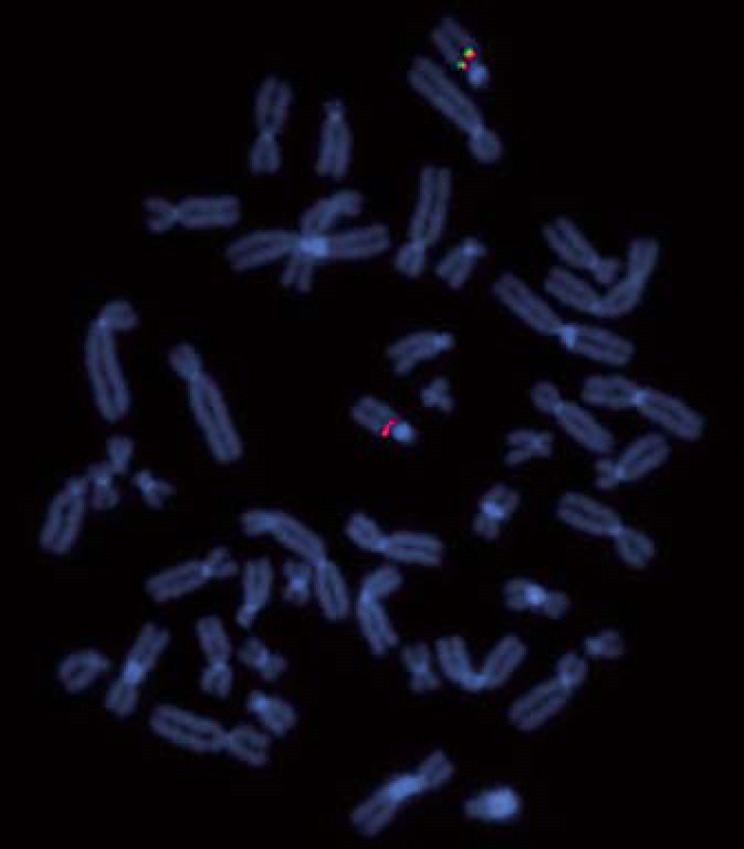
Metaphase FISH using BAC RP11-265I17 (probe 4, labeled in green) and RP11-280K19 (probe5, labeled in red) showing the distal breakpoint of patient 4.
Evaluation of Symptomatic Patients Described in Previous Studies
For patients and studies see table 2 and online supplementary material 1 (for all online suppl. material, see www.karger.com/doi/10.1159/000443343). Statistical analysis was performed by Fisher's exact test, using software ‘R’. Results with p < 0.05 were assessed as significant.
Table 2.
Summary of features of patients described in previous studies
| Feature | Percentage | n/N |
|---|---|---|
| Size | ||
| <1.0 Mb | 24.53 | 26/106 |
| 1–1.6 Mb | 63.21 | 67/106 |
| >1.6 Mb | 12.26 | 13/106 |
| Intellectual deficits | ||
| No | 20.22 | 18/89 |
| Undefined | 11.24 | 10/89 |
| Mild | 40.45 | 36/89 |
| Severe | 28.09 | 25/89 |
| Developmental delay | 41.77 | 33/79 |
| Epilepsy | 30.61 | 30/98 |
| Autism | 27.71 | 23/83 |
| Paternal | 26.47 | 18/68 |
| Maternal | 54.41 | 37/68 |
| De novo | 19.12 | 13/68 |
| Parental phenotype noticeable | 66.67 | 34/51 |
| Male | 55.56 | 45/81 |
| Female | 44.44 | 36/81 |
| OFC | ||
| P<25 | 26.09 | 18/69 |
| P25–75 | 53.62 | 37/69 |
| P>75 | 20.29 | 14/69 |
| Weight | ||
| P<25 | 14.29 | 8/56 |
| P25–75 | 64.29 | 36/56 |
| P>75 | 21.42 | 12/56 |
| Language problems | 75.34 | 55/73 |
| Hypotonia | 39.02 | 16/41 |
| Behavioral problems | 63.64 | 49/77 |
For a list of patients see online supplementary material 1. n/N = Affected/total patients.
Discussion
The family we present shows interesting features. On the one hand, the degree of relationship varies; on the other hand, the symptoms differ as well (fig. 2). However, there is no demonstrable connection between the degree of relationship and symptoms. Patients 2 and 3, who are sisters, are remarkably different. They vary considerably in body measurements, character and the severity of symptoms, although they are exposed to similar environmental factors and carry the same deletion size. Therefore, other genetic factors are bound to influence the degree of clinical manifestations. A possible explanation could be an altered expression of her genes in the genome, which may have diverse effects [Le Pichon et al., 2013]. This approach has been further confirmed by Henrichsen et al. [2009], who have shown that CNV regions are expressed at lower and more variable levels and modify the expression of neighboring genes. Chaignat et al. [2011] stated that CNVs alter the expression timing of genes during development in mice. Additionally, different temporal patterns of expression can be found in different individuals. Furthermore, altered expression or mutations of different genes coding for different steps of the same neurobiological pathway could intensify neurological symptoms. Poot et al. [2011] suggest different mechanisms which can lead to phenotypic pleiotropy of CNVs, for example, the interaction of CNVs, genetic epistasis or allelic exclusion.
Three out of 4 family members manifest ADHD; all show developmental delay. As the origin of ADHD apparently is multifactorial, it is not surprising to find an accumulation in one family. Remarkably, all children are affected by the deletion. Particular familial genetic constellations possibly increase the probability to manifest symptoms. Furthermore, the intrauterine surroundings of the deletion-carrying mother may have affected the phenotype of her children. However, no shared CNVs correlating with the occurrence of symptoms could be identified, and no second pathogenic CNV could be identified in any of our patients.
The breakpoint analysis shows that the distal breakpoint is positioned distally to CHRNA7. Possible locations would be within the gene families FAM7A or GOLGA8, as has already been suggested, or within the locus control regions [Shinawi et al., 2009; Anttonaci et al., 2014]. The proximal breakpoint is located distally to CHRFAM7A. In patients 1-4, who belong to one family, the deletion sizes detected by the aCGH-analysis software differ. These differences are brought about by gaps in the distribution pattern of oligonucleotides in the aCGH within and around the breakpoint regions. Slight changes in the log ratio of marginally located oligonucleotides result in a considerable shift of deletion size detected by the analysis software. The obvious difference in size of the recurrent deletion 15q13.3 is most likely an artifact of the processing software. We assume the deletion to be identical in size for all family members (see online suppl. material 2).
Different CNV studies have revealed an association of the 15q13.3 microdeletion with schizophrenia, epilepsy and autism. Several publications have been issued presenting new cases and theories about breakpoints and factors which alter symptoms [e.g. Freedman et al., 2001; Dibbens et al., 2009; Mikhail et al., 2011]. Efforts were made to merge this information in order to find new connections.
The results leave room for speculations. Deletions are inherited in 80.88%. Maternal decent is described disproportionately more often for deletions (54.41%, p = 0.001) and duplications (53.33%, p = 0.027).
An explanation might be given by Sinkus et al. [2011]. They studied the impact of CHRNA7 promoter polymorphisms, which decrease the level of transcription by ∼25% and influence the cortisol level in mother and child. The cortisol level of the child is lowered further, if the mother as well as the child are polymorphism carriers. The impact of the intrauterine surrounding may also have an influence on other factors. The OFC is reduced in patients if the deletion is inherited by the mother (P<25 in 50%, p = 0.002). Therefore, we assume that if the aberration is inherited from the mother, the intrauterine surrounding will intensify the symptoms. As we only included symptomatic patients in this assessment, this would explain why the majority of aberrations are apparently of maternal origin (54%, p = 0.001). In this context, it is striking that the gender distribution of the deletion is equal. Our data confirms the findings of Lowther et al. [2015]. In their comprehensive review of the 15q13.3 deletion, they combined symptomatic and asymptomatic patients. As we only included symptomatic patients, the proportions of distributional patterns of features are similar but the relative numbers differ. Regarding the increased maternal decent of deletions, they state a reduced reproductive fitness in males as a possible explanation.
The size of the deletion does not consistently correlate with the degree of symptoms. For example, comparing the degree of intellectual deficits with the size of the deletion shows no linear connection. Patients with the typical 1.6-Mb deletion show severe deficits more frequently than patients with smaller or larger deletions (41.07%, p = 0.00045). Intellectual deficits are reported significantly more often in patients with deletions than duplications (79.78 and 55.56%, p = 0.037). As expected, normal intelligence is described most often in patients with deletions <1 Mb (45%, p = 0.0037).
Deletions with a size of 1-1.6 Mb are most frequent (63.21%, p = 0.0002). Duplications <1 Mb (66.67%, p = 0.0004) are most commonly described. Regarding deletions, intellectual deficits (79.78%), language disorder (75.34%), a noticeable phenotype of the parents (66.67%), and behavioral problems (63.64%) are described much more often than epilepsy (30.61%) or autism (27.71%) (p = 0.02-3.931E-09). Comparing duplications and deletions, duplications are associated more often with autism (p = 0.039) and the phenotype of the parents is noticeably less frequent (p = 0.027).
Epilepsy is significantly associated with mild intellectual deficits (59.09%, p = 0.0029). Language disorder is associated with intellectual deficits in 79.24% of the cases (p = 0.003).
To conclude, it can be said that the clinical phenotype of the microdeletion 15q13.3 is of multifactorial origin. Even close family members exposed to similar environmental factors differ significantly in their clinical manifestations. Genetic counseling in families with 15q13.3 microdeletions or notably microduplications remains thus complex and occasionally inadequate. Further investigations in genetic alterations regarding the nondeleted allele or promotor regions of affected genes may be more promising when it comes to discovering influencing factors.
Statement of Ethics
The authors have no ethical conflicts to disclose.
Disclosure Statement
The authors have no conflicts of interest to declare.
Supplementary Material
Supplementary data
Supplementary data
Acknowledgements
The authors thank Margot Fliegauf, Monika Heinkelein, Helga Heitzler, and Claudia Ladwig in the cytogenetic group of the Institute of Human Genetics in Freiburg for their experimental advice and assistance. We also thank Ekkehart Lausch, Elke Botzenhart, Susanne Munk-Schulenburg, and Andreas Busche for patient care. We are grateful to the patients and their families for their kind cooperation.
References
- 1.Antonacci F, Dennis MY, Huddleston J, Sudmant PH, Steinberg KM, et al. Palindromic GOLGA8 core duplicons promote chromosome 15q13.3 microdeletion and evolutionary instability. Nat Genet. 2014;46:1293–1302. doi: 10.1038/ng.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaignat E, Yahya-Graison EA, Henrichsen CN, Chrast J, Schütz F, et al. Copy number variation modifies expression time courses. Genome Res. 2011;21:106–113. doi: 10.1101/gr.112748.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dibbens LM, Mullen S, Helbig I, Mefford HC, Bayly MA, et al. Familial and sporadic 15q13.3 microdeletions in idiopathic generalized epilepsy: precedent for disorders with complex inheritance. Hum Mol Genet. 2009;18:3626–3631. doi: 10.1093/hmg/ddp311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Freedman R, Leonard S, Gault JM, Hopkins J, Cloninger CR, et al. Linkage disequilibrium for schizophrenia at the chromosome 15q13-14 locus of the alpha7-nicotinic acetylcholine receptor subunit gene (CHRNA7) Am J Med Genet. 2001;105:20–22. [PubMed] [Google Scholar]
- 5.Henrichsen CN, Chaignat E, Reymond A. Copy number variants, diseases and gene expression. Hum Mol Genet. 2009;18:R1–R8. doi: 10.1093/hmg/ddp011. [DOI] [PubMed] [Google Scholar]
- 6.Le Pichon JB, Yu S, Kibiryeva N, Graf WD, Bittel DC. Genome-wide gene expression in a patient with 15q13.3 homozygous microdeletion syndrome. Eur J Hum Genet. 2013;21:1093–1099. doi: 10.1038/ejhg.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lowther C, Costain G, Stavropoulos DJ, Melvin R, Silversides CK, et al. Delineating the 15q13.3 microdeletion phenotype: a case series and comprehensive review of the literature. Genet Med. 2015;17:149–157. doi: 10.1038/gim.2014.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mikhail FM, Lose EJ, Robin NH, Descartes MD, Rutledge KD, et al. Clinically relevant single gene or intragenic deletion encompassing critical neurodevelopmental genes in patients with developmental delay, mental retardation, and/or autism spectrum disorder. Am J Med Genet A. 2011;155A:2386–2396. doi: 10.1002/ajmg.a.34177. [DOI] [PubMed] [Google Scholar]
- 9.Poot M, van der Smagt JJ, Brilstra EH, Bourgeron T. Disentangling the myriad genomics of complex disorders, specifically focusing on autism, epilepsy, and schizophrenia. Cytogenet Genome Res. 2011;135:228–240. doi: 10.1159/000334064. [DOI] [PubMed] [Google Scholar]
- 10.Sharp AJ, Mefford HC, Li K, Baker C, Skinner C, et al. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat Genet. 2008;40:322–328. doi: 10.1038/ng.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shinawi M, Schaaf CP, Bhatt SS, Xia Z, Patel A, et al. A small recurrent deletion within 15q13.3 is associated with a range of neurodevelopmental phenotypes. Nat Genet. 2009;41:1269–1271. doi: 10.1038/ng.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sinkus ML, Wamboldt MZ, Barton A, Fingerlin TE, Laudenslager ML, Leonard S. The α7 nicotinic acetylcholin receptor and the acute stress response: maternal genotype determines offspring phenotype. Physiol Behav. 2011;104:321–326. doi: 10.1016/j.physbeh.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szafranski P, Schaaf CP, Person RE, Gibson IB, Xia Z, et al. Structures and molecular mechanisms for common 15q13.3 microduplications involving CHRNA7: benign or pathological? Hum Mutat. 2010;31:840–850. doi: 10.1002/humu.21284. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data
Supplementary data