

Background: Several E3 ligases regulate cytosolic β-catenin during Wnt-off phase. The fate of critical form active β-catenin in Wnt-on phase remains poorly defined.
Results: Casitas B-lineage lymphoma (c-Cbl) ubiquitinates cytosolic β-catenin and translocates to the nucleus with Wnt induction to also ubiquitinate active nuclear β-catenin.
Conclusion: c-Cbl is a unique E3 ligase targeting active nuclear β-catenin.
Significance: This study uncovers a novel layer of Wnt regulation.
Keywords: Angiogenesis, Cancer Biology, Gene Transcription, Ubiquitin Ligase, Wnt Signaling, Cbl, Dimerization
Abstract
Regulation of transcriptionally active nuclear β-catenin during the Wnt-on phase is crucial to ensure controlled induction of Wnt target genes. Several ubiquitin E3 ligases are known to regulate cytosolic β-catenin during the Wnt-off phase, but little is known about the fate of active nuclear β-catenin in the Wnt-on phase. We now describe ubiquitination of active β-catenin in the Wnt-on phase by a RING finger ubiquitin E3 ligase, Casitas B-lineage lymphoma (c-Cbl) in endothelial cells. c-Cbl binds preferentially to nuclearly active β-catenin in the Wnt-on phase via the armadillo repeat region. Wild-type c-Cbl suppresses and E3 ligase-deficient c-Cbl-70Z increases Wnt signaling. Wnt induces nuclear translocation of c-Cbl where it ubiquitinates nuclear β-catenin. Deletion of the c-Cbl UBA domain abrogates its dimerization, binding to β-catenin, Wnt-induced c-Cbl nuclear translocation, and ubiquitination of nuclear β-catenin. c-Cbl activity inhibits pro-angiogenic Wnt targets IL-8 and VEGF levels and angiogenesis in a β-catenin-dependent manner. This study defines for the first time c-Cbl as a ubiquitin E3 ligase that targets nuclearly active β-catenin in the Wnt-on phase and uncovers a novel layer of regulation of Wnt signaling.
Introduction
The Wnt/β-catenin axis is a highly conserved signaling pathway, which is activated by Wnt ligand and plays key roles in several cellular functions. In the absence of Wnt ligand (Wnt-off phase), β-catenin localizes in the cytosol at the core of the destruction complex consisting of adenomatous polyposis coli and axin and undergoes serine and threonine phosphorylation by glycogen synthase kinase-3β (GSK-3β) at the N terminus that dictates its stability (1, 2). Phosphorylated β-catenin is specifically recognized by the E3 ligase β-TrCP and Jade-1, which induce β-catenin ubiquitination and subsequent 26 S proteasomal degradation (3–5). In response to Wnt ligand (Wnt-on phase), β-catenin in its hypophosphorylated form escapes degradation, undergoes nuclear translocation, and activates several Wnt target genes, including IL-8 and VEGF (active β-catenin). The current model supports the notion that β-catenin activity is regulated mainly by protein degradation primarily in cytosol during Wnt-off phase (1, 2). Because β-catenin undergoes free nucleo-cytoplasmic shuttling, it is thought that E3 ligases that target the cytosolic β-catenin for degradation could indirectly reduce nuclear β-catenin levels and hence inhibit the Wnt nuclear activity in Wnt-off phase. The molecular regulation of nuclearly active β-catenin specifically during Wnt-on phase remains largely elusive.
Wnt signaling plays prominent roles in fundamental cellular functions, including cell fate, survival and motility, and angiogenesis (6–8). Targeted disruption of Wnt/frizzled genes such as Wnt2, -4, and -7b and Frizzled 5 leads to vascular defects (8). The Frizzled 4 gene is linked to familial exudative vitreoretinopathy, a hereditary disorder characterized by peripheral retinal vascularization failure (9), further underscoring the importance of Wnt signaling in angiogenesis-associated diseases. At the molecular level, Wnt signaling regulates angiogenesis through the transcriptional activity of nuclear β-catenin in endothelial cells (ECs)3 by inducing expression of key pro-angiogenic factors, including VEGF-A and IL-8 (7, 10).
c-Cbl is originally identified as a cytosolic RING finger domain ubiquitin E3 ligase that ubiquitinates various receptor tyrosine kinases (RTKs) and RTK substrates and regulates cell proliferation, survival, and movement (11–13). Recently, c-Cbl has emerged as a negative regulator of angiogenesis (14–19) with a poorly defined mechanism. Expression of c-Cbl in EC inhibits proliferation, tube formation, and sprouting, whereas c-Cbl-70Z, an E3 ligase-deficient variant of c-Cbl, or silencing c-Cbl enhances angiogenesis by increasing EC proliferation and sprouting (17). c-Cbl activity also has been linked to pathological angiogenesis such as laser- and tumor-induced angiogenesis (17, 19). Given the prominent importance of Wnt/β-catenin signaling in angiogenesis and the emerging anti-angiogenesis function of c-Cbl, in this study we demonstrate that c-Cbl is distinctly involved in the regulation of Wnt signaling and angiogenesis. c-Cbl undergoes Wnt-induced nuclear translocation and serves as a specific ubiquitin E3 ligase for nuclearly active β-catenin highlighting the potential therapeutic value of targeting c-Cbl in angiogenesis-associated diseases such as cancer and ocular neovascularization.
EXPERIMENTAL PROCEDURES
Cell Culture, Transfections, and Chemical Treatment
The HEK293T cells were grown as described previously (5). Human aortic endothelial cells and umbilical vein endothelial cells (HUVECs) (Promocell, Germany) pooled from three donors were grown in endothelial growth medium-2 (EGM-2) (Promocell, Germany). EGM-2 was prepared by supplementing endothelial basal medium (EBM-2) with fetal bovine serum (2%), hydrocortisone (1 μg/ml), fibroblast growth factor-1 (10 ng/ml), epidermal growth factor (5 ng/ml), insulin-like growth factor (20 ng/ml), ascorbic acid (1 μg/ml), and heparin (90 μg/ml). Porcine aortic endothelial cells (PAECs) and ECs KO and Ki for c-Cbl were grown in DMEM + 10% FBS and 5% penicillin and streptomycin. Lithium (Sigma) and BIO (Calbiochem) suspended in water and MG132 (Calbiochem) suspended in DMSO were used in cell culture medium. Thrombin 1 unit (New England Biolab) was used in PBS at 4 °C for 3 h to cleave β-catenin from purified recombinant GST-tagged β-catenin.
Cultured cells plated overnight were transiently transfected using Lipofectamine 2000 (Invitrogen) per the manufacturer's instructions. Human recombinant Wnt3a and DKK1 (obtained from R&D Systems) dissolved in PBS + 0.1% bovine serum albumin was obtained from R&D Systems.
Antibodies
Monoclonal β-catenin antibody, polyclonal β-catenin, and active β-catenin (recognizes active form of β-catenin, dephosphorylated on Ser-37 or Thr-41) were from BD Biosciences, Santa Cruz Biotechnology, and Upstate (Millipore), respectively. Polyclonal c-Cbl, monoclonal tubulin, fibrillarin, VE cadherin, HA tag, and Myc tag antibodies were purchased from Cell Signaling. FLAG tag antibody was from Stratagene. Monoclonal actin and ubiquitin antibody were obtained from Santa Cruz Biotechnology. Goat anti-mouse and anti-rabbit HRP-conjugated secondary antibodies were from Bio-Rad for immunofluorescence. Alexa 647 goat anti-mouse and Alexa 488 goat anti-rabbit used as secondary antibodies (Invitrogen).
Constructs
HA-tagged c-Cbl and c-Cbl-70Z, 70ZG306E, and silencing c-Cbl retroviral vectors have been described previously (14, 17). All of the β-catenin constructs have been previously described (5). Promoter-reporter constructs pBARLS and pfuBARLS were obtained from the Randal T. Moon laboratory (University of Washington, Seattle) (20). pBAR (β-catenin-activated reporters) contain 12 transcription factor 4 (TCF)-binding sites separated by distinct five-base linkers, which are directly upstream of a minimal TK promoter that then drives the expression of firefly luciferase. The pfuBAR reporter (found unresponsive β-catenin-activated reporter) has a two-base substitution in each TCF-binding site making it unresponsive to β-catenin. Reporters contain a separate PGK promoter that constitutively drives the expression of a puromycin resistance gene. FLAG-tagged c-Cbl WT, delUBA, and Dimer were generated using site-directed mutagenesis using the following primers sense and antisense: delUBA antisense: UBA region from 856–909, GAGCTCGGATCCCTAAGGTGAGGCGGTGGCAGCAGA; artificial dimerization motif, LLLLLLLLLQLISGSL (21); Dimer antisense, GAGCTCGGATCCCTAAAGGCTTCCGCTAATAAGTTGAAGAAGAAGAAGAAGAAGAAGAAGAAGAGGTGAGGCGGTGGCAGCAGA.
The PCR products digested by NotI and BamHI in pQCXIP retroviral vector and viral particles were generated in HEK293T cells as described below. The target cells transduced with viral particles were selected using puromycin.
Immunoblotting and Immunoprecipitation
Cells were lysed in 50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 30 mm EDTA, 0.5% Triton X-100 with complete protease inhibitor (Roche Applied Science). Immunoblotting and immunoprecipitation were performed as described previously (5).
Immunofluorescence
Cells were grown in Chamber slides (Lab-Tek) and fixed and processed as described previously. Alexa 488 goat anti-rabbit and Alexa 647 goat anti-mouse were used as secondary antibodies. ImageJ was used to generate profile and scatter plots as described previously (5).
Cellular Fractionation
Subcellular fractionation was performed using Dounce homogenization as described previously (5).
Digitonin Extraction
Cells washed with ice-cold PBS were covered with buffer containing 120 mm KCl, 5 mm KH2PO4, 10 mm HEPES, pH 7.4, 2 mm EGTA, 0.15 mg/ml digitonin (Sigma) and gently rocked on ice for 15 min as described previously (5).
Generation of Viral Particles
Retroviral constructs with HA-tagged c-Cbl or c-Cbl-70Z and c-Cbl silencing constructs were cotransfected in HEK293T packaging cells along with packaging, envelope, and reverse transcriptase vectors using Lipofectamine 2000 per the manufacturer's instructions. Medium containing active viral particles collected after 48 h was centrifuged and stored at −80 °C. Lentiviral particles of TOP- and FOP-Flash were generated similarly by cotransfecting the lentiviral constructs with packaging, envelope, and reverse transcriptase vectors using Lipofectamine 2000 per the manufacturer's instructions. For viral transduction, the cells were seeded at 50–60% confluence. The cells were treated overnight with the medium containing active viral particles along with hexadimethrine bromide (Sigma), a cationic polymer, to increase the efficiency of infection. Puromycin (Sigma) selection was initiated after 24 h. The cells were harvested after 4 days to examine the effect on protein levels. The retroviral system was employed to knock-in (KI) c-Cbl or c-Cbl-70Z in c-Cbl KO ECs.
GST-tagged Protein Purification
GST purification of pGEX-2T c-Cbl(1–358), c-Cbl(359–909), and β-catenin constructs was performed as described previously (5).
GST Pulldown Assay
c-Cbl(1–358) or -(359–909) tethered to glutathione-SepharoseTM beads was incubated with cell lysates of HEK293T cells for 2 h at 4 °C in 50 mm Tris-HCl, pH 7.6, 250 mm NaCl, 30 mm EDTA, 0.5% Triton X-100. Beads extensively washed with the same buffer containing 300 mm NaCl were boiled in Laemmli buffer (Boston Bioproducts).
Tcf/β-Catenin-responsive Luciferase Reporter Assay
The cells seeded in 6-well plates stably expressing HA-tagged c-Cbl or c-Cbl-70Z were cotransduced with lentiviral particles of pBARLS or pfuBARLS. After 48 h of transfection, luciferase assays were performed using the Dual-Luciferase® kit (Promega) and normalized using protein content determined by the Bradford assay (Bio-Rad).
In Vivo Ubiquitination Assay
The HEK293T cells were treated with 10 μm proteasome inhibitor MG132 (Boston Biochem) for 8 h before harvesting in lysis buffer containing 50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 30 mm EDTA, 0.5% Triton X-100 with complete protease inhibitor (Roche Applied Science). For immunoprecipitation, cell lysates were mixed with 1 μg of β-catenin antibody overnight at 4 °C and further processed as described previously (5).
Ex Vivo Ubiquitination of β-Catenin Using HeLa Cell S100 Fraction
HeLa S100 conjugation kit (Boston Biochem) was used per the manufacturer's instructions. Briefly, 2.8 μm GST-β-catenin on glutathione-SepharoseTM beads was incubated with 200 μg of HeLa S100 fraction (pretreated with 200 μm MG132 and 100 μm ubiquitin aldehyde for 15 min at RT), 750 nm c-Cbl(359–909), 600 μm Myc-tagged ubiquitin, and 5 μl of energy-regenerating solution in 50 mm HEPES, pH 7.6, for 90 min at 37 °C. β-Catenin ubiquitination has been described previously (5).
In Vitro Ubiquitination Reaction
Ubiquitination reactions were reconstituted in 30 μl with ubiquitination buffer (50 mm Tris-HCl, pH 7.5, 0.5 mm DTT) containing 225 nm E1-activating enzyme (Boston Biochem), 500 nm E2 conjugase, 600 μm Myc-tagged ubiquitin (Boston Biochem), 1 mm MgCl2-ATP, and 2.8 μm GST-β-catenin on glutathione beads with IPed c-Cbl or c-Cbl-70Z and incubated at 37 °C for 60 min.
ELISA
The media were harvested and concentrated using Amicon filters 3KD (Millipore) and subjected to ELISAs for IL-8 and VEGF (R&D Systems) per the manufacturer's instructions.
In Vitro Angiogenesis Tube Formation Assay
Early passage pooled HUVECs pretransduced with HA-tagged c-Cbl or c-Cbl-70Z, 70ZG306E were transiently transfected with control or β-catenin siRNA oligos (SMART-pool siRNA oligos, Dharmacon) using Dharmafect-1 per the manufacturer's instructions. The cells were then divided into two sets. In the first set, 15,000 HUVECs were seeded in 96-well plates precoated with 106 μl of growth factor Matrigel (BD Biosciences) (10 mg/ml) and incubated at 37 °C for 24 h. Tube formation was imaged using phase contrast microscopy, and tube lengths were analyzed using ImageJ software. The second set of cells was seeded for 24 h on a 6-well plate and harvested for protein expression.
Statistical Analysis
In all figures, data are expressed as average ± S.E. Student's t test followed by Bonferroni's correction was conducted to determine statistical differences between groups. Values of p < 0.05 were considered significant.
RESULTS
c-Cbl Inhibits Wnt Activity in Endothelial Cells
To test the hypothesis that Wnt signaling is regulated by c-Cbl activity, we initially examined the β-catenin in porcine aortic endothelial cells stably expressing HA-tagged c-Cbl or c-Cbl-70Z, an E3 ligase-deficient mutant c-Cbl. Our observation showed that Wnt activity was significantly inhibited in porcine aortic endothelial cells stably expressing c-Cbl and increased in c-Cbl-70Z, an E3 ligase-deficient mutant (Fig. 1A). The levels of β-catenin also reduced in c-Cbl and conversely increased in c-Cbl-70Z expressing ECs (Fig. 1A), suggesting a possible link between c-Cbl activity and Wnt signaling. Because c-Cbl has an E3 ligase activity (11–13) and proteasome inhibition with MG132 abrogated c-Cbl-mediated β-catenin down-regulation (supplemental Fig. S1A), we further probed c-Cbl regulation of β-catenin at the post-translational level.
FIGURE 1.

Direct interaction of c-Cbl and β-catenin in ECs independent of Wnt status. A, differential Wnt activity in c-Cbl and c-Cbl-70Z expressing PAECs. Cell lysates of PAECs stably expressing HA-tagged c-Cbl or c-Cbl-70Z and TCF-responsive promoter-reporter pBAR and nonresponsive control (Ctr) reporter pfuBAR tethered to the luciferase reporter (20) were analyzed for luciferase assay. Activity of the Wnt signaling pathway is quantified by measuring relative firefly luciferase units normalized to protein concentration. Mean results of three experiments are shown. Student's t test was applied to determine statistical significance, p = 0.015 and 0.004 for c-Cbl and c-Cbl-70Z (70Z) compared with control. Error bars = S.E. Inset, cell lysates of PAECs were probed using β-catenin, HA tag, and actin antibodies. Densitometry was performed to quantitate β-catenin normalized to actin using ImageJ. Representative immunoblot of three experiments is shown. B, endogenous c-Cbl/β-catenin interaction is enhanced with Wnt activation, and c-Cbl binds stronger to nuclear β-catenin in the Wnt-on phase. Immunoprecipitations (IPs) were performed with 500 μg of EC lysates from ECs pretreated with vehicle (Veh), Wnt3a (50 ng/ml), and Wnt3a with DKK1 (250 ng/ml) for 4 h using 1 μg of either rabbit polyclonal c-Cbl or rabbit preimmune serum (Ctr). The co-IPed β-catenin was detected using monoclonal β-catenin antibody. β-Catenin was IPed as described above using monoclonal β-catenin antibody or isotype control (Ctr). The co-IPed c-Cbl was detected using polyclonal c-Cbl antibody. Ten percent of cell lysates are shown as input and normalized to actin (data not shown) using ImageJ. The co-IPed β-catenin and c-Cbl were normalized to the immunoglobulin. Representative immunoblot of three experiments is shown. C, schematic of β-catenin constructs. β-Catenin constructs have an N-terminal Myc tag and include β-catenin wild-type (WT), S33A naturally occurring point mutation (S33A), N-terminal deletion (delN), C-terminal deletion (delC), and deletions of both the C and N termini with only the ARM region (ARM). D, c-Cbl binds to β-catenin ARM. HEK293T cells stably expressing HA tag c-Cbl were transiently transfected with Myc tag β-catenin constructs, and digitonin-extracted cytosolic fractions (5) were used for IP using HA and Myc antibodies, and co-IPed Myc-β-catenin or HA-c-Cbl was detected. Five percent of cell lysates are shown as input. Representative immunoblot from two experiments is shown. E, HA-tagged c-Cbl colocalizes with Myc-tagged β-catenin. HEK293T cells stably expressing HA-tagged c-Cbl and transiently expressing Myc-tagged β-catenin constructs were fixed and stained with polyclonal HA tag and monoclonal Myc tag antibodies. The scatter plots for colocalization were generated using the ImageJ program (National Institutes of Health) in cells having comparable signal levels for both constructs (5). Scatter plot points along the x or y axes represent absence of colocalization, whereas scatter plot points along a diagonal represent evidence of colocalization. F, schematic of c-Cbl constructs. TKB = tyrosine kinase binding domain; Linker region spans between tyrosine kinase binding and the RING finger domain, where the latter possesses E3 ligase activity; UBA = ubiquitin binding domain. c-Cbl-70Z construct lacks 17 amino acids from the linker domain and a part of RING domain. G, Wnt signaling enhances c-Cbl/β-catenin interaction and c-Cbl binds stronger to the nuclear active β-catenin in the Wnt-on phase. Purified recombinant GST-tagged c-Cbl(359–909) bound to glutathione-SepharoseTM beads was incubated with 500 μg of EC lysates pretreated with vehicle or Wnt-3a 50 ng/ml for 3 h prior to harvest. The eluent was immunoblotted using total β-catenin and active β-catenin antibodies. Five percent of cell lysates or GST-tagged c-Cbl are shown as input. GST-tagged proteins are shown as an input using Coomassie stain. Representative immunoblot from two experiments is shown. H, bacterially purified recombinant GST-tagged c-Cbl interacts with GST-tagged β-catenin in vitro. Purified recombinant GST-β-catenin was cleaved using thrombin, and the supernatant was treated with the purified recombinant GST or GST-tagged c-Cbl(359–909) bound to glutathione-SepharoseTM beads for 4 h. The beads were washed with buffer containing 0.25 m sodium chloride, and eluent was immunoblotted with β-catenin antibody. GST-tagged proteins are shown as an input using Coomassie stain. Representative immunoblot are of two experiments. WB, Western blot.
c-Cbl Interacts with β-Catenin in ECs in Both the Phases of Wnt Signaling
Because c-Cbl expression alters β-catenin abundance, we examined if the effect of c-Cbl on β-catenin is due to its direct physical interaction with β-catenin in different phases of Wnt signaling. The data demonstrated that endogenous c-Cbl interacts with endogenous β-catenin in ECs stimulated with the vehicle (Wnt-off state), Wnt3a (Wnt-on state), or Wnt3a plus DKK-1 (Wnt-inhibited state) (Fig. 1B). Similarly, porcine aortic endothelial cells overexpressing c-Cbl or c-Cbl-70Z reciprocally coimmunoprecipitated endogenous β-catenin (supplemental Fig. S1B) in both phases of Wnt signaling.
c-Cbl Interacts with the Armadillo Repeat Domain of β-Catenin
Wnt activity regulates β-catenin interactions depending on the region of β-catenin to which each partner binds. Jade-1 and β-TrCP bind at the N terminus of β-catenin, a region where multiple serine and threonine residues are phosphorylated by GSK-3β upon Wnt induction creating a docking site for Jade-1 and β-TrCP (1, 2, 5) (Fig. 1C). Because c-Cbl binding with β-catenin persists during both Wnt phases, we hypothesized that c-Cbl would bind at a region other than the N terminus and does not require phosphorylation of β-catenin. The data demonstrate that the armadillo (ARM) domain of β-catenin alone is sufficient for c-Cbl binding with β-catenin (Fig. 1, D and E). A naturally occurring N terminus oncogenic mutant β-catenin S33A lacking a GSK-3β phosphorylation site, which fails to bind β-TrCP and Jade-1 (1, 2,5,), also retained c-Cbl interaction (Fig. 1, D and E). β-Catenin crystal structure reveals that the ARM region is composed of 13 compactly organized repetitive units to form a tight hydrophobic domain suggesting that deletion of any of these ARM repeats could introduce a structural impairment in β-catenin protein making the entire region unstable (22, 23). Therefore, further mapping of c-Cbl binding within the ARM region was not pursued. Taken together, c-Cbl, unlike β-TrCP and Jade-1, distinctly recognizes β-catenin through its ARM domain, and this interaction is independent of Wnt activity or phosphorylation of β-catenin.
β-Catenin Interacts with C Terminus of c-Cbl, a Site Distinct from the Interacting Domain with RTKs
The Src homology 2 domain of c-Cbl (also known as tyrosine kinase (TK domain)) is responsible for its recognition of RTKs (Fig. 1F). To determine whether the Src homology 2/TK domain of c-Cbl is also involved in the recognition of β-catenin, we generated a panel of GST-tagged truncated c-Cbl constructs and tested for their potential to bind with β-catenin. The data showed that endogenous or GST-tagged β-catenin specifically and strongly interacts with the C terminus of c-Cbl encompassing 359–909 amino acids, which lacks the Src homology 2/TK domain retaining its binding even in the presence of 250 mm sodium chloride wash buffer indicating strong interaction of both the proteins (Fig. 1G). Purified recombinant GST-tagged β-catenin interacted with purified recombinant c-Cbl(359–909), indicating a direct interaction between two proteins (Fig. 1H). These data also indicate that c-Cbl employs a domain different from that used for its interaction with RTKs. Interestingly, although c-Cbl(309–909) binds both to β-catenin (transcriptionally inactive) in Wnt-off and to transcriptionally active (active) β-catenin in the Wnt-on phase (Fig. 1G), the binding of c-Cbl with the nuclearly active β-catenin in the Wnt-on phase was stronger compared with the Wnt-off phase (Fig. 1, B and G). In this regard c-Cbl binding with β-catenin is distinct from Jade-1 or β-TrCP, which exhibits predominant binding with cytosolic β-catenin in Wnt-off phase (3–5).
Wnt-induced Nuclear Translocation of c-Cbl Depends on β-Catenin
c-Cbl is thought to be a cytosolic protein (11–13), whereas β-catenin predominantly resides in the cytosol during Wnt-off and in the nucleus in the Wnt-on phase (1, 2). We hypothesized that c-Cbl/β-catenin interaction during the Wnt-on phase would be feasible if c-Cbl translocates also in the nucleus, the same compartment as β-catenin upon Wnt activation. Our immunofluorescence microscopy analysis showed that indeed Wnt activation results in the nuclear translocation of c-Cbl, where it colocalized with the nuclear β-catenin (Fig. 2A). These data indicate that although c-Cbl interaction with β-catenin is a Wnt-independent event, c-Cbl nuclear localization distinctly is controlled by Wnt activation. Of note, β-catenin does not require c-Cbl for nuclear translocation, and in c-Cbl knock-out (KO) ECs, β-catenin still undergoes Wnt-induced nuclear translocation (supplemental Fig. S2A). In contrast, Wnt-induced nuclear translocation of c-Cbl was substantially abrogated, although not completely abolished, with β-catenin silencing (Fig. 2B and supplemental Fig. S2B). These results suggest that c-Cbl requires β-catenin for nuclear translocation.
FIGURE 2.

Wnt-induced nuclear translocation of c-Cbl depends on its dimerization. A, upper panel, Wnt-induced nuclear translocation of endogenous c-Cbl. Primary HUVEC serum-starved for 16 h followed by treatment with vehicle (Veh) or Wnt3a 50 ng/ml for 3 h were fixed and stained with monoclonal β-catenin and polyclonal c-Cbl antibodies. Lower panel, Wnt-induced nuclear translocation of overexpressed c-Cbl. HUVEC stably expressing HA-tagged c-Cbl (upper panel) or c-Cbl-70Z (lower panel) were stimulated with Wnt3a and processed using polyclonal β-catenin and monoclonal HA tag antibodies. Confocal microscopy was performed. Images are representative of 150 randomly analyzed subconfluent ECs. Profile plots were generated using ImageJ to demonstrate quantitative distribution of c-Cbl and β-catenin (5). Student's t test was performed to compare number of ECs with nuclear c-Cbl. p = 0.001 for ECs with nuclear c-Cbl in Wnt-on and Wnt-off phase. B, β-catenin is required for the nuclear translocation of c-Cbl. ECs silenced using control (Csi) or β-catenin siRNA oligos (βsi) were serum-starved and stimulated by Wnt3a (50 ng/ml) for 3 h. The cytosol and nuclear fractions were probed for β-catenin and c-Cbl. Tubulin and fibrillarin served as markers for the cytosol and nuclear fractions, respectively, and loading controls. Representative immunoblot from two experiments is shown. C, schematic representation of various c-Cbl constructs. FLAG-tagged c-Cbl constructs were generated lacking UBA domain (dUBA) or with an artificial dimerization motif (Dim) mutant. D, c-Cbl dimerizes through UBA domain, and Wnt enhances c-Cbl dimerization. EC stably coexpressing HA-tagged c-Cbl wild-type with FLAG-tagged c-Cbl wild-type or delUBA (dU) or c-Cbl with artificial dimerization motif (Dim) were serum-starved, stimulated with Wnt3a (50 ng/ml), fractionated followed by immunoprecipitation using FLAG, and immunoblotted using HA antibody. Five percent of lysates are shown as inputs. Representative immunoblot of two experiments is shown. E, binding to β-catenin depends on c-Cbl dimerization. Lysates of ECs stably expressing FLAG c-Cbl constructs were serum-starved, stimulated with Wnt3a, fractionated followed by immunoprecipitation using FLAG, and immunoblotted using β-catenin. Five percent of lysates are used as inputs. Representative immunoblot of two experiments is shown. F, c-Cbl dimerization mediates c-Cbl nuclear translocation. ECs stably expressing FLAG-tagged c-Cbl or dUBA or Dimer constructs were stimulated with Wnt and were fixed for immunofluorescence using FLAG tag monoclonal antibody and DAPI for nuclear staining. Microscopy and statistical analysis were performed, as described above. Images are representative of 150 randomly analyzed subconfluent ECs. p = 0.13 for delUBA and p = 0.02 for Dimer for number of ECs with nuclear c-Cbl in Wnt-on and Wnt-off phase. WB, Western blot.
c-Cbl Dimerization Regulates Binding to β-Catenin and c-Cbl Nuclear Translocation
c-Cbl exists as both monomers and dimers, and its dimerization regulates binding to different substrates (12, 24, 25). Hence, we examined contribution of c-Cbl dimerization to β-catenin binding and Wnt-induced c-Cbl nuclear translocation. c-Cbl dimerizes through the C-terminal UBA domain (Fig. 2C) (12, 24). To perturb c-Cbl dimerization, we utilized c-Cbl constructs lacking the UBA domain (c-Cbl dUBA) or rescued dimerization with the addition of an artificial dimerization motif (c-Cbl dimer) (Fig. 2C) (21). In concurrence with others (26), we observed that wild-type c-Cbl exists in part as dimerized species at base line. Although deletion of the UBA domain abrogates c-Cbl dimerization, this is rescued by the artificial dimerization motif (Fig. 2D). Wnt activation further increased its dimerization.
In addition to c-Cbl dimerization, we assayed c-Cbl/β-catenin binding. In the Wnt-off phase, deletion of the UBA domain substantially abolished β-catenin binding, whereas c-Cbl with the artificial dimerization motif exhibited increased binding to β-catenin (Fig. 2E). Wnt activation enhanced the binding of c-Cbl dimers with β-catenin (Fig. 2E). We also tested the ability of c-Cbl dimerization mutants to translocate to the nucleus in response to Wnt induction. Interestingly, c-Cbl dUBA failed to undergo nuclear translocation, whereas c-Cbl Dimer retained its capacity (Fig. 2F), suggesting that c-Cbl's ability to dimerize and bind to β-catenin regulates its Wnt-induced nuclear translocation. All these data are consistent with the notion that c-Cbl exhibits base-line dimerization, which mediates binding to β-catenin in both phases of Wnt signaling and Wnt-induced c-Cbl nuclear translocation.
c-Cbl Down-regulates β-Catenin Independent of Wnt Status
Because c-Cbl interacts with β-catenin independent of Wnt activity, this suggest that c-Cbl could regulate β-catenin during both phases of Wnt signaling. To this end, we analyzed the effect of c-Cbl in the down-regulation of β-catenin. c-Cbl down-regulated wild-type and the phosphorylation-resistant oncogenic S33A β-catenin (Fig. 3A), and treatment with the proteasome inhibitor MG132 abrogated β-catenin down-regulation (supplemental Fig. 3A). Moreover, c-Cbl down-regulated both the cytosolic and nuclear pools of endogenous β-catenin but not the membrane pool of β-catenin (supplemental Fig. 3B). Silencing of c-Cbl by shRNA increased the endogenous β-catenin in four different cell lines by 2–4-fold (Fig. 3B) and re-expression of c-Cbl rescued the β-catenin regulation (supplemental Fig. 3C). c-Cbl KO ECs revealed increased levels of endogenous β-catenin in both the cytosol and in the nuclear compartments compared with c-Cbl knock-in (KI-c) ECs during both the phases of Wnt signaling (Fig. 3C). c-Cbl-70Z (Ki-7), although it binds to β-catenin (Fig. 2A), was unable to down-regulate β-catenin. c-Cbl-70Z rather up-regulated endogenous β-catenin, thus serving as a dominant negative. We used lithium and BIO, a specific GSK-3β inhibitor, which stabilize β-catenin from Jade-1- or β-TrCP-mediated degradation (1, 2, 5). Yet c-Cbl was able to down-regulate stabilized species of β-catenin (Fig. 3D) indicating that c-Cbl regulates β-catenin independent of Wnt activity and GSK-3β activity.
FIGURE 3.

c-Cbl destabilizes β-catenin during both the phases of Wnt signaling. A, c-Cbl down-regulates β-catenin. HUVECs stably expressing control vector (Ctr), HA-tagged c-Cbl (c), or c-Cbl-70Z (70Z) were transiently transfected with various constructs of Myc-tagged β-catenin. Digitonin-extracted cytosol fractions were immunoblotted using HA, Myc, and actin antibodies. Representative immunoblot from two experiments is shown. B, c-Cbl silencing in ECs up-regulates β-catenin. Lysates of cells stably silenced with control (pSup) or c-Cbl (sil) retrovirus were probed with β-catenin, c-Cbl, and actin antibodies. Representative immunoblot from three experiments is shown. C, increased β-catenin levels in c-Cbl knock out (KO) ECs. Lysates from ECs from c-Cbl KO or with knock-in (Ki) with HA-tagged c-Cbl (Ki-c) or c-Cbl-70Z (Ki-7) and pretreated with vehicle (veh) or Wnt3a 100 ng/ml were fractionated and immunoblotted using β-catenin, HA, and actin antibodies. Representative immunoblot from three experiments is shown. D, c-Cbl regulates β-catenin stabilized by inhibition of GSK-3β. ECs stably expressing control (Ctr) or FLAG-c-Cbl were serum-starved and treated with vehicle (Veh) or lithium (10 mm) and BIO (100 nm) for 12 h. Whole cell lysates were probed for β-catenin, FLAG, and actin antibodies. β-Catenin bands were normalized to actin using ImageJ. Representative immunoblot from two experiments is shown. E, c-Cbl dimerization-dependent regulation of β-catenin. ECs stably expressing FLAG-tagged c-Cbl constructs were serum-starved and stimulated with Wnt3a (50 ng/ml) followed by subcellular fractionation. The lysates were probed for β-catenin. Tubulin and fibrillarin served as markers of cytosolic and nuclear fractions and as loading controls, respectively. Representative immunoblot from two experiments is shown. F, c-Cbl destabilizes endogenous β-catenin in both the phases of Wnt signaling. Lysates from c-Cbl KO or KI ECs serum-starved for 16 h and pretreated with Wnt3a (100 ng/ml) for 3 h and 20 μm emetine were fractionated at different time intervals and immunoblotted for endogenous β-catenin. Tubulin and fibrillarin served as cytosolic and nuclear markers, respectively, and loading controls. Densitometric values are normalized loading controls. Representative immunoblot of three experiments is shown. G, percent β-catenin remaining was analyzed by densitometry after normalizing to tubulin and fibrillarin, respectively. Graph shows mean result of three experiments. Error bars, S.E. WB, Western blot.
Furthermore, dimerization mutant c-Cbl (dUBA) failed to down-regulate β-catenin (Fig. 3E), whereas the c-Cbl mutant bearing an artificial dimerization motif down-regulated β-catenin in both Wnt phases (Fig. 3E). Collectively, these data indicate that β-catenin down-regulation is dictated by c-Cbl's ability to dimerize and depends on its E3 ligase activity.
c-Cbl Destabilizes β-Catenin during Both Phases of Wnt Signaling
c-Cbl ubiquitinates its substrates for proteasomal degradation (11–13). Both c-Cbl silencing and expression of c-Cbl-70Z in ECs increased the half-life of the cytosolic β-catenin extracted with digitonin-containing lysis buffer (5) by 3.25-fold (supplemental Fig. 3, D–F), indicating that the c-Cbl destabilized β-catenin and that its ubiquitin E3 ligase activity is required for destabilization of β-catenin.
Compared with c-Cbl KO ECs, c-Cbl KI shortened the half-life of cytosolic β-catenin by 40% in the Wnt-off phase and the nuclear β-catenin half-life by 70% in the Wnt-on phase (Fig. 3, F and G). c-Cbl expression significantly shortened the half-life of both the cytosolic and nuclear β-catenin (supplemental Fig. 3, G and H). These data indicate that similar to the Wnt-off phase, the nuclear β-catenin also undergoes degradation in the Wnt-on phase. Furthermore, the data demonstrate that c-Cbl exerts higher destabilizing activity on nuclear β-catenin in the Wnt-on phase, which is in contrast to Jade-1 and β-TrCP, which destabilize cytosolic β-catenin in the Wnt-off phase (3, 5, 27).
c-Cbl Ubiquitinates Nuclearly Active β-Catenin
The cytosolic β-catenin undergoes ubiquitination followed by 26 S proteasome-mediated degradation during the Wnt-off phase (28). Because c-Cbl is an E3 ligase and destabilizes β-catenin, we examined c-Cbl-dependent ubiquitination of β-catenin. In both an in vitro ubiquitination assay and ex vivo HeLa S100 fraction, c-Cbl ubiquitinates GST-tagged β-catenin (Fig. 4, A and B). We examined β-catenin ubiquitination by c-Cbl in denaturing conditions to minimize the contribution of the ubiquitination signal by other potential interactors (Fig. 4C). ECs stably expressing c-Cbl, but not 70Z, enhanced β-catenin ubiquitination (Fig. 4C and supplemental Fig. 4, A and B). Silencing c-Cbl by shRNA increased the endogenous β-catenin in four different cell lines by 2–4-fold (Fig. 3B) and re-expression of c-Cbl rescued the β-catenin regulation (supplemental Fig. 3C). c-Cbl-70Z that binds and fails to ubiquitinate β-catenin inhibited c-Cbl-mediated β-catenin ubiquitination, thus serving as a dominant negative (supplemental Fig. 4D). Given that c-Cbl destabilizes nuclear β-catenin, we specifically examined ubiquitination of nuclear β-catenin in the Wnt-on phase. The data revealed that c-Cbl expression increased (Fig. 4D) and c-Cbl silencing reduced the ubiquitination of endogenous β-catenin during Wnt-off and also of the nuclear β-catenin in the Wnt-on phase (Fig. 4E). In line with the binding ability, c-Cbl could also ubiquitinate β-catenin S33A (Fig. 4F). The dimerization-impaired mutant c-Cbl (dUBA) failed, whereas c-Cbl Dimer retained the capacity to ubiquitinate β-catenin in both phases of Wnt signaling (supplemental Fig. 4E), in line with the inability to bind β-catenin and undergo nuclear translocation in response to Wnt signaling. These data indicate that c-Cbl is a unique E3 ligase for the cytosolic and nuclear β-catenin in both phases of Wnt signaling.
FIGURE 4.

c-Cbl ubiquitinates β-catenin in both the phases of Wnt signaling. A, in vitro ubiquitination of β-catenin by c-Cbl. Ubiquitination reaction mixture was reconstituted in vitro using GST or GST-β-catenin and IPed FLAG-c-Cbl or c-Cbl-70Z from ECs along with 225 nm E1-activating enzyme, 500 nm E2 conjugase, 600 μm Myc-ubiquitin, 1 mm MgCl2-ATP and incubated at 37 °C for 60 min. Reactions without E1, E2, c-Cbl or ubiquitin served as negative control. β-Catenin cleaved from GST beads using thrombin and then eluent were immunoblotted using Myc antibody. The blot was stripped and reprobed using β-catenin and FLAG antibodies. Representative immunoblot of two experiments is shown. B, ex vivo ubiquitination of purified β-catenin using the HeLa cell cytosolic S100 fraction. Purified recombinant GST-β-catenin on glutathione-SepharoseTM beads was incubated with HeLa cell S100 fraction (pretreated with ubiquitin aldehyde and MG132), Myc-tagged human recombinant ubiquitin (Ub), recombinant GST-tagged c-Cbl(359–909), and energy regeneration solution. Eluent was resolved with SDS-polyacrylamide gel. The nitrocellulose membrane was stained with Ponceau stain for c-Cbl input and immunoblotted using monoclonal ubiquitin and reprobed using β-catenin antibodies. Lanes designated as -ERS or -UB were reactions without energy-regenerating solution (ERS) or ubiquitin only, respectively. Representative immunoblot was from two experiments. C, c-Cbl, but not 70Z enhances β-catenin ubiquitination. HEK293T cells cotransfected with HA-Ub and Myc empty vector or c-Cbl or 70Z were treated with 10 μm of MG132 for 16 h. The cells were then lysed in RIPA buffer and immunoprecipitated with HA antibody and immunoblotted using β-catenin antibody. Five percent of cell lysates were probed as input. Representative image of two experiments is shown. D, c-Cbl ubiquitinates cytosolic β-catenin in Wnt-off phase and nuclear β-catenin in Wnt-on phase. Human aortic endothelial cells stably expressing control (Ctr), HA-tagged c-Cbl (cbl), or c-Cbl-70Z pretreated with MG132 at 10 μm for 12 h were subjected to subcellular fractionation and IP as above. The membrane was stripped and reprobed with β-catenin antibody. α-Tubulin and fibrillarin served as markers of cytosolic and nuclear fractions, respectively. Representative immunoblot from two experiments is shown. E, c-Cbl silencing reduces β-catenin ubiquitination in both the phases of Wnt signaling. 500 μg of cytosolic or nuclear lysates of ECs cells transduced with control (pSup) or c-Cbl silencing (sil) and serum-starved and treated with vehicle or Wnt3a were processed as above. Because both β-catenin and c-Cbl are predominantly located in the cytosol in Wnt-off and in the nucleus in Wnt-on phase (Fig. 2A), the respective fractions are shown. The blot was stripped and reprobed for β-catenin. Ten percent of cell lysates were probed for endogenous c-Cbl. Representative immunoblot from three experiments is shown. F, c-Cbl ubiquitinates naturally occurring oncogenic mutant β-catenin S33A. Lysates of HEK293T cells stably expressing HA-tagged c-Cbl and transiently transfected with Myc-tagged β-catenin and treated with MG132 at 10 μm for 12 h were subjected to IP using 0.5 μg of Myc antibody. The eluents were probed with ubiquitin antibody and reprobed with Myc antibody. Representative immunoblot of two experiments. G, both c-Cbl and β-TrCP bind β-catenin in Wnt-off, whereas only c-Cbl binds to β-catenin in Wnt-on phase. HEK293T cells stably expressing HA c-Cbl (c) or c-Cbl-70Z (7) and transiently coexpressing Myc-β-TrCP were serum-starved for 16 h, treated with vehicle or Wnt3a (50 ng/ml) for 3 h, and IPed using β-catenin antibody and immunoblotted with HA and Myc antibodies. The co-IPed c-Cbl and β-TrCP were normalized to immunoglobulin. Five percent of lysates are shown as inputs. Representative immunoblot of three experiments is shown. H, β-TrCP regulates β-catenin in Wnt-off, whereas c-Cbl regulates β-catenin in both the phases of Wnt signaling. HEK293T cells stably expressing HA c-Cbl (c) or c-Cbl-70Z (7) and transiently coexpressing Myc-dominant negative β-TrCP (DN) were serum-starved for 16 h and treated with vehicle or Wnt3a (50 ng/ml) for 3 h and fractionated and probed with endogenous β-catenin; tubulin and fibrillarin served as cytosolic and nuclear markers, respectively, and as loading controls. Representative immunoblot of three experiments is shown.
c-Cbl Is a Unique E3 Ligase for β-Catenin in Wnt-on Phase
β-TrCP and Jade-1 specifically bind and regulate the phosphorylated β-catenin in the Wnt-off phase (3, 5, 27). Because β-TrCP also modulates angiogenesis (15–19), we directly compared c-Cbl with β-TrCP in regulating β-catenin during different phases of Wnt signaling. In the Wnt-off phase, both β-TrCP and c-Cbl bind to β-catenin. However, in the Wnt-on phase β-TrCP-β-catenin complex binding decreased despite an increase in the expression of endogenous β-TrCP, whereas c-Cbl/β-catenin binding persisted and increased in the Wnt-on phase (Fig. 4G). Because expression of c-Cbl reduced β-TrCP interaction with β-catenin, these findings may also suggest that c-Cbl influences β-TrCP-β-catenin binding. In the same vein, β-TrCP lacking the F box E3 ligase domain serving as a dominant negative stabilized β-catenin in Wnt-off phase, but much less in the Wnt-on phase (Fig. 4H). In contrast, c-Cbl-mediated down-regulation and c-Cbl-70Z-mediated up-regulation of β-catenin persisted during both phases of Wnt signaling and also in the presence of dominant negative β-TrCP. These data indicate that like β-TrCP, c-Cbl down-regulates β-catenin in Wnt-off phase, but unlike β-TrCP, c-Cbl continues to bind and down-regulate the active β-catenin in the Wnt-on phase. These data indicate that c-Cbl is a unique E3 ligase for β-catenin in Wnt-on phase.
c-Cbl Regulates Wnt Target Genes and Mediates Angiogenesis through Wnt Signaling
To determine the biological significance of the β-catenin/c-Cbl interaction, we evaluated TCF/β-catenin-mediated transcription. c-Cbl KO ECs exhibited significantly higher spontaneous Wnt activity compared with c-Cbl KI cells (Fig. 5A). Similarly, c-Cbl-silenced ECs had significantly higher Wnt activity that could be rescued by c-Cbl but not 70Z expression (supplemental Fig. 5). Pro-angiogenic mediators such as IL-8 and VEGF are known Wnt targets, whose promoters contain β-catenin response elements (10, 29). ECs expressing c-Cbl reduced and c-Cbl-70Z increased amounts of IL-8 and VEGF levels (Fig. 5, B and C).
FIGURE 5.

c-Cbl inhibits Wnt activity and Wnt targets in ECs and regulates angiogenesis through Wnt signaling. A, spontaneous Wnt activity in c-Cbl KO ECs. ECs from c-Cbl KO animals and KI ECs were transfected with TCF-responsive promoter-reporter pBARLS and nonresponsive control reporter pfuBARLS tethered to luciferase reporter. Activity of the Wnt signaling pathway quantified by measuring relative firefly luciferase units normalized to protein concentration. Mean results of three experiments are shown. Student's t test was performed to determine statistical significance, p = 0.022 for KO and KI. Error bars, S.E. The immunoblot depicting the expression of c-Cbl these lysates is shown. B, β-catenin silencing reduces c-Cbl-70Z-induced proangiogenic Wnt target IL-8 levels. ECs stably expressing control vector (Ctr) or c-Cbl or c-Cbl-70Z (70Z) retrovirus were transfected with control (Con si) or β-catenin (β si) siRNA oligonucleotides. Media harvested after 24 h were analyzed for IL-8 by ELISA and normalized to number of cells. The mean of triplicates samples is shown. Student's t test was performed to determine statistical significance. # compares control and c-Cbl p = 0.03, and p = 0.001 c-Cbl-70Z. * compares control and βsi siRNA. p = 0.001 for control, p = 0.01 for c-Cbl, and 0.005 for c-Cbl-70Z. Error bars, S.E. The input is shown in D. C, β-catenin silencing significantly reduces c-Cbl-70Z-induced proangiogenic Wnt target VEGF levels. ECs stably expressing control (Ctr) or c-Cbl or c-Cbl-70Z (70Z) retrovirus were transfected with control (Con) or β-catenin siRNA oligos (β si). Media concentrated after 24 h were analyzed for VEGF by ELISA and normalized to number of cells. The mean of triplicates samples is shown. Student's t test was performed to determine statistical significance; # compares empty vector and c-Cbl p = 0.04, and c-Cbl-70Z p = 0.003. * compares control and β si siRNA. Empty vector p = 0.05, c-Cbl p = 0.01, and c-Cbl-70Z p = 0.001. Error bars, S.E. The input is shown in D. D, β-catenin silencing significantly abrogates c-Cbl- or c-Cbl-70Z-mediated angiogenesis. Left panel, HUVECs stably expressing control (Ctr), HA-tagged c-Cbl (cbl), or c-Cbl-70Z were transiently transfected with control (Con si) or β-catenin siRNA oligonucleotides (β si). Cells were divided in two sets. One set of 15,000 ECs were seeded per well of a 96-well plate coated with Matrigel and were analyzed for tube formation within 24 h after serum starvation and treating with Wnt3a (50 ng/ml). Middle panel, tube lengths were measured with ImageJ. Mean of six images are shown. Mean of tube length from two separate experiments performed in triplicate is shown. Student's t test was performed to determine statistical significance, p = 0.015 for c-Cbl and 0.001 for c-Cbl-70Z compared with control. Error bars = S.E. Right panel, other set of cells seeded in 6-well plate was harvested after 24 h and probed for input. E, c-Cbl-70ZG306E up-regulates β-catenin and angiogenesis but not PDGFα. Left panel, HUVECs stably expressing control (Ctr), HA-tagged c-Cbl (cbl), or c-Cbl-70Z or 70ZG306E were then divided into two sets. One set of 15,000 ECs were seeded per well of a 96-well plate coated with Matrigel and were analyzed for tube formation within 24 h after serum starvation and treating with Wnt3a (50 ng/ml). Middle panel, tube lengths were measured with ImageJ. Mean of six images shown. Mean of tube length from two separate experiments performed in triplicate is shown. Student's t test was performed to determine statistical significance; * compares the control and c-Cbl p = 0.029, c-Cbl-70Z p = 0.008, and 70ZG306E p = 0.001. However, we compared c-Cbl-70Z with 70ZG306E, p = 0.13. Error bars, S.E. Right panel, other set of cells was harvested after 24 h and probed using β-catenin, PDGFα, and HA antibodies. F, c-Cbl regulates proangiogenic Wnt target genes dependent on its ability to undergo dimerization. Media harvested from ECs stably expressing control (Ctr) or various FLAG-c-Cbl constructs and serum-starved and pretreated with Wnt3a (50 ng/ml) were analyzed as above. The mean of triplicates samples is shown. Student's t test was performed to determine statistical significance for IL-8 compared with Wnt-treated control medium, p = 0.026 for c-Cbl wild type, 0.021 for dUBA = 0.003, and 0.001 for Dimer; VEGF p = 0.031 for c-Cbl wild type, 0.026 for dUBA, and Dimer, 0.001. Error bars, S.E. G, c-Cbl regulates angiogenesis depending on its ability to undergo dimerization. Upper panel, HUVECs stably expressing control (Ctr) or various FLAG-c-Cbl constructs seeded in a 96-well plate coated with Matrigel were analyzed for tube formation within 24 h after serum starvation and treating with Wnt3a (50 ng/ml). Lower panel, tube lengths were measured with ImageJ. Mean of six images is shown. Mean of tube length from two separate experiments performed in triplicate is shown. Student's t test was performed to determine statistical significance; * compares wild-type c-Cbl with dUBA, p = 0.029, and Dimer, p = 0.001. Error bars, S.E.
c-Cbl regulates angiogenesis through multiple mediators such as RTKs. Specific contribution of Wnt signaling in c-Cbl-regulated angiogenesis was examined using β-catenin silencing and c-Cbl-70ZG306E, a specific mutant unable to bind and regulate some of the angiogenic mediators ZAP70/Syk, PDGF receptor α, and EGF receptor (30). β-Catenin silencing significantly reduced IL-8 and VEGF secretion (Fig. 5, B and C) and the tube formation in ECs expressing c-Cbl or c-Cbl-70Z (Fig. 5D). Mutation of G306E in c-Cbl-70Z abrogated up-regulation of PDGFRα but retained up-regulation of β-catenin and angiogenesis (Fig. 5E). Both these approaches indicate that c-Cbl regulates angiogenesis through Wnt signaling.
Consistent with the c-Cbl binding pattern with β-catenin, deletion of UBA abrogated c-Cbl's inhibitory activity on IL-8 and VEGF levels and tube formation with Wnt induction (Fig. 5, F and G). These observations indicate that Wnt signaling is a critical mediator of c-Cbl-induced angiogenesis and that c-Cbl's regulation of Wnt target genes and angiogenesis corresponds to its ability to bind to β-catenin and undergo Wnt-mediated nuclear translocation to regulate active nuclear β-catenin.
DISCUSSION
Wnt stimulation regulates various cellular functions mainly through the transcriptional coactivator activity of nuclear β-catenin that induces different Wnt target genes. Rigorous control of β-catenin-mediated transcription is necessary to prevent relentless activation of Wnt target genes, thus underscoring a critical need for regulating transcriptionally active nuclear β-catenin during the Wnt-on phase (1, 2, 31). Ubiquitination and proteasomal degradation by a small set of E3 ligases constitute the cornerstone of β-catenin regulation. However, all of the known ubiquitin E3 ligases of β-catenin, including Jade-1 and β-TrCP, degrade β-catenin predominantly in the cytosol in Wnt-off phase or in specific cell types (Ozz in developing myocytes) or in response to unique stimulus (Siah1 with DNA damage pathway) (32, 33, 34). Even though both Jade-1 and β-TrCP are present in the nucleus, their interaction and regulation of active β-catenin in Wnt-on is substantially abrogated (5, 35). Thus, the regulation of active β-catenin remains unclear, despite its critical importance in Wnt signaling.
This study describes ubiquitin-proteasomal degradation of nuclear β-catenin through c-Cbl as a unique E3 ligase of active β-catenin in the Wnt-on phase. Two aspects bestow c-Cbl with this ability as follows: the domain of interaction on β-catenin, and the nuclear translocation of c-Cbl. The armadillo repeat region of β-catenin has not been shown to be modified by the Wnt status, thus allowing Wnt-independent binding to c-Cbl. Wnt-induced nuclear translocation of c-Cbl ensures continual interaction and ubiquitination of nuclear β-catenin in the Wnt-on phase.
Both c-Cbl-mediated β-catenin regulation and Wnt-induced nuclear translocation of c-Cbl require direct c-Cbl/β-catenin interaction. These data suggest that c-Cbl dimerization is a critical requirement for binding and regulating β-catenin, because deletion of the UBA domain abrogates while artificial dimerization motif recapitulates binding in both the phases of Wnt signaling, Wnt-induced c-Cbl nuclear translocation and ubiquitination of nuclear β-catenin in Wnt-on phase (Figs. 2, D and E, and 3E). Wnt-enhanced c-Cbl dimerization is intriguing. It is plausible that Wnt activation through yet an undefined signal results in a conformational change to enhance c-Cbl dimers. Thus, Wnt induction shifts the c-Cbl pool to predominantly dimers, which may impart a stoichiometric advantage to either bind to more numbers of β-catenin molecules or accelerates c-Cbl-β-catenin binding kinetics to keep up with the rapidly expanding pool of active β-catenin during Wnt-on phase. That c-Cbl binding with β-catenin is critical for its Wnt-induced nuclear translocation (Fig. 2, E and F) raises an interesting possibility that c-Cbl may cotranslocate with β-catenin into the nucleus. This is especially plausible as c-Cbl has no putative nuclear localizing signal and is likely to take advantage of β-catenin nucleo-cytoplasmic shuttling. Furthermore, Wnt induction up-regulates c-Cbl (Figs. 1B and 2D), which may also contribute to β-catenin regulation in Wnt-on phase. Although more work is needed to further understand the above findings, it remains novel in Wnt signaling that an E3 ligase translocates into the same compartment as β-catenin to continue exerting its regulation on β-catenin.
Both Wnt signaling and c-Cbl play roles in physiological and pathological angiogenesis (6, 8). Loss- and gain-of-function mutation studies of various Wnt signaling components upstream of β-catenin have shown effects in physiological angiogenesis as evident by defects of vascular organization, length, and branching patterns of intersomitic vessels (36). Similarly, genetic evidence from both the c-Cbl KO animal (15–19) and ECs obtained from these animals strongly supports the critical role of c-Cbl in angiogenesis. Wnt signaling is critical in hypoxic or oxidative stress-induced angiogenesis, too (37, 38). c-Cbl regulates VEGF-, tumor-, and laser-induced choroidal angiogenesis (17, 19). Although we examined the c-Cbl/β-catenin relationship in physiological angiogenesis, it raises a tantalizing possibility that Wnt signaling may be a crucial mediator of c-Cbl-regulated proliferative retinal vascular diseases or tumor-induced angiogenesis.
c-Cbl regulates angiogenesis by other substrates, including RTKs (15–18). In in vitro angiogenesis models, using overexpression and silencing β-catenin with siRNA oligos exhibit significant suppression of c-Cbl-70Z mediated up-regulation of IL-8 and VEGF and angiogenesis (Fig. 5, B–D). The mutation of G306E in c-Cbl, which abrogates binding with other angiogenic mediators such as EGF receptor, PDGF, etc., continued to up-regulate β-catenin and angiogenesis (Fig. 5E). The fact that c-Cbl directly interacts with β-catenin through a domain distinct from that of the RTK binding domain (Fig. 1, G and H) indicates that c-Cbl directly and independently regulates angiogenesis through Wnt/β-catenin signaling. Overall, these approaches indicate that Wnt signaling is a crucial mediator of c-Cbl-regulated angiogenesis.
The above findings along with the present understanding of Wnt signaling can be explained based on the following model (Fig. 6). Wnt signaling is a critical mediator of angiogenesis and stimulates angiogenesis with Wnt induction in ECs. The transcriptional activity of nuclear β-catenin (by up-regulating pro-angiogenic Wnt target genes such as IL-8 and VEGF) mediates the bulk of this function (6–8). In the Wnt-off phase, angiogenesis is suppressed by down-regulation of cytosolic β-catenin by E3 ligases interacting via multiple regions such as the N terminus by β-TrCP in GSK-3β-dependent and ARM by c-Cbl in a GSK-3β-independent manner. Dimerized species of c-Cbl mediates β-catenin binding and its regulation. Wnt stimulation allows accumulation of hypophosphorylated β-catenin. Stabilized β-catenin “escapes” into the nucleus to activate transcription of Wnt target genes (active β-catenin). β-TrCP interaction with active β-catenin reduces in this phase of Wnt signaling. In parallel, Wnt activation increases c-Cbl dimerization, binding with β-catenin, and induces c-Cbl nuclear translocation. c-Cbl “chases” and destabilizes the active β-catenin in the nucleus, suppresses pro-angiogenic IL-8 and VEGF Wnt targets, and inhibits angiogenesis induced by Wnt signaling. Thus, c-Cbl is a unique E3 ligase targeting active β-catenin in the nucleus, a species of β-catenin that remains immune to degradation by known E3 ligases. Thus, these data uncover a novel layer of Wnt regulation.
FIGURE 6.
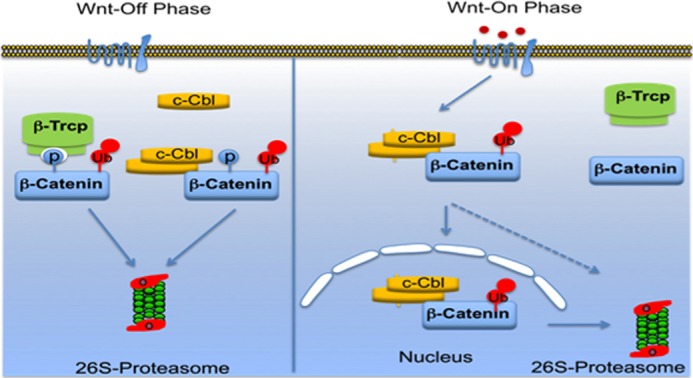
Proposed model of c-Cbl in regulating β-catenin in Wnt signaling. In Wnt-off phase, cytosolic phosphorylated β-catenin is targeted by E3 ligases, including c-Cbl and β-TrCP. Dimerized species of c-Cbl mediates β-catenin binding and its down-regulation. Wnt stimulation allows accumulation of hypophosphorylated β-catenin, which exhibits reduced binding and regulation by E3 ligases such as Jade-1 and β-TrCP. Stabilized β-catenin undergoes nuclear translocation to activate transcription of Wnt target genes (active β-catenin). In parallel, Wnt activation increases c-Cbl dimerization, binding with β-catenin, and induces c-Cbl nuclear translocation. c-Cbl destabilizes the active β-catenin in nucleus. Thus, c-Cbl is a unique E3 ligase targeting active β-catenin in nucleus.
These observations ask a fundamental question about the role of c-Cbl in β-catenin ubiquitination in both the compartments under physiological conditions. In the Wnt-off phase, c-Cbl, in addition to other E3 ligases, ensures effective degradation of phosphorylated cytosolic β-catenin to maintain the Wnt inactive status. As c-Cbl degrades hypophosphorylated cytosolic species of β-catenin, it too may facilitate controlled increase in β-catenin during the Wnt-on phase. Because with Wnt induction, c-Cbl translocates to the nucleus to degrade active nuclear β-catenin, it may also ensure “switching-off” of β-catenin-mediated transcription. Thus, c-Cbl may serve as a dynamic “break” to exert controlled physiological angiogenesis. It is also plausible that reduced c-Cbl activity may contribute to pathological angiogenesis. Under pathological conditions driven by hyperactive Wnt signaling due to stabilized β-catenin, c-Cbl may represent a new layer of regulation, whose loss of function results in excess angiogenesis or cancer induction or progression. This model also illustrates a critical paradigm that cells employ multiple “guards” for a potent oncoprotein such as β-catenin in every compartment and with different Wnt activity status.
c-Cbl as an E3 ligase for nuclear β-catenin regulating Wnt signaling may have implications beyond angiogenesis in areas such as tumor cell growth. A tumor suppressor function of c-Cbl for example is linked to its ubiquitin ligase activity in hematopoietic cells resulting in a myeloproliferative disorder (39, 40). Although this study establishes relationship of c-Cbl and Wnt signaling in ECs, it is conceivable that the c-Cbl also down-regulates Wnt signaling in other cell types, including hematopoietic cells or colonic epithelial cells. Thus, inhibition of Wnt signaling may be an important mechanism of c-Cbl tumor suppressor function. Further investigation of c-Cbl in hyperactive Wnt status and Wnt signaling in the diseases linked to defective E3 ligase activity of c-Cbl will provide a deeper understanding into the c-Cbl/β-catenin relationship.
Supplementary Material
Acknowledgments
We thank R. Kemler (Max-Planck Institute for Immunobiology, Germany) for providing the β-catenin S33A construct., and W. Birchmeier (Max-Delbruck-Center for Molecular Medicine, Germany) for β-catenin C and N termini and CR deletion constructs. We thank Randal Moon (University of Washington, Seattle) for BAR and fuBAR constructs in lentiviral plasmids. We appreciate John Schwartz (Boston University School of Medicine) and Ann Fiegen (Harvard University) for the critical review of the manuscript and insightful suggestions.
This work was supported, in whole or in part, by National Institutes of Health Grants K08 DK080946, R01 GM-49039 (to E. E.), and R01 EY017955 (to N. R.) from NIDDK. This work was also supported by a Young Investigator Award from the National Kidney Foundation (to V. C.) and Grant FI-DGR 2011 from Generalitat de Catalunya, Spain (to J. M.).

This article contains supplemental Figs. 1–5.
- EC
- endothelial cell
- RTK
- receptor tyrosine kinase
- HUVEC
- human umbilical vein endothelial cell
- PAEC
- porcine aortic endothelial cell
- TK
- tyrosine kinase
- KI
- knock-in
- IPed
- immunoprecipitated
- ARM
- armadillo
- oligos
- oligonucleotides
- UBA
- ubiquitin binding domain
- GSK-3β
- glycogen synthase kinase-3β
- β-TrCP
- β-transducin repeat containing protein
- TCF
- transcription factor 4.
REFERENCES
- 1. MacDonald B. T., Tamai K., He X. (2009) Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nusse R. (2005) Wnt signaling in disease and in development. Cell Res. 15, 28–32 [DOI] [PubMed] [Google Scholar]
- 3. Kitagawa M., Hatakeyama S., Shirane M., Matsumoto M., Ishida N., Hattori K., Nakamichi I., Kikuchi A., Nakayama K. (1999) An F-box protein, FWD1, mediates ubiquitin-dependent proteolysis of β-catenin. EMBO J. 18, 2401–2410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Winston J. T., Strack P., Beer-Romero P., Chu C. Y., Elledge S. J., Harper J. W. (1999) The SCFβ-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IκBα and β-catenin and stimulates IκBα ubiquitination in vitro. Genes Dev. 13, 270–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chitalia V. C., Foy R. L., Bachschmid M. M., Zeng L., Panchenko M. V., Zhou M. I., Bharti A., Seldin D. C., Lecker S. H., Dominguez I., Cohen H. T. (2008) Jade-1 inhibits Wnt signalling by ubiquitylating β-catenin and mediates Wnt pathway inhibition by pVHL. Nat. Cell Biol. 10, 1208–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dejana E. (2010) The role of Wnt signaling in physiological and pathological angiogenesis. Circ. Res. 107, 943–952 [DOI] [PubMed] [Google Scholar]
- 7. Zerlin M., Julius M. A., Kitajewski J. (2008) Wnt/Frizzled signaling in angiogenesis. Angiogenesis 11, 63–69 [DOI] [PubMed] [Google Scholar]
- 8. van Amerongen R., Berns A. (2006) Knockout mouse models to study Wnt signal transduction. Trends Genet. 22, 678–689 [DOI] [PubMed] [Google Scholar]
- 9. Robitaille J., MacDonald M. L., Kaykas A., Sheldahl L. C., Zeisler J., Dubé M. P., Zhang L. H., Singaraja R. R., Guernsey D. L., Zheng B., Siebert L. F., Hoskin-Mott A., Trese M. T., Pimstone S. N., Shastry B. S., Moon R. T., Hayden M. R., Goldberg Y. P., Samuels M. E. (2002) Mutant frizzled-4 disrupts retinal angiogenesis in familial exudative vitreoretinopathy. Nat. Genet. 32, 326–330 [DOI] [PubMed] [Google Scholar]
- 10. Masckauchán T. N., Shawber C. J., Funahashi Y., Li C. M., Kitajewski J. (2005) Wnt/β-catenin signaling induces proliferation, survival and interleukin-8 in human endothelial cells. Angiogenesis 8, 43–51 [DOI] [PubMed] [Google Scholar]
- 11. Blake T. J., Heath K. G., Langdon W. Y. (1993) The truncation that generated the v-cbl oncogene reveals an ability for nuclear transport, DNA binding and acute transformation. EMBO J. 12, 2017–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schmidt M. H., Dikic I. (2005) The Cbl interactome and its functions. Nat. Rev. Mol. Cell Biol. 6, 907–918 [DOI] [PubMed] [Google Scholar]
- 13. Thien C. B., Langdon W. Y. (2005) Negative regulation of PTK signalling by Cbl proteins. Growth Factors 23, 161–167 [DOI] [PubMed] [Google Scholar]
- 14. Singh A. J., Meyer R. D., Navruzbekov G., Shelke R., Duan L., Band H., Leeman S. E., Rahimi N. (2007) A critical role for the E3-ligase activity of c-Cbl in VEGFR-2-mediated PLCγ1 activation and angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 104, 5413–5418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kobayashi S., Sawano A., Nojima Y., Shibuya M., Maru Y. (2004) The c-Cbl/CD2AP complex regulates VEGF-induced endocytosis and degradation of Flt-1 (VEGFR-1). FASEB J. 18, 929–931 [DOI] [PubMed] [Google Scholar]
- 16. Rahimi N. (2009) A role for protein ubiquitination in VEGFR-2 signalling and angiogenesis. Biochem. Soc. Trans. 37, 1189–1192 [DOI] [PubMed] [Google Scholar]
- 17. Meyer R. D., Husain D., Rahimi N. (2011) c-Cbl inhibits angiogenesis and tumor growth by suppressing activation of PLCγ1. Oncogene 30, 2198–2206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kassenbrock C. K., Hunter S., Garl P., Johnson G. L., Anderson S. M. (2002) Inhibition of Src family kinases blocks epidermal growth factor (EGF)-induced activation of Akt, phosphorylation of c-Cbl, and ubiquitination of the EGF receptor. J. Biol. Chem. 277, 24967–24975 [DOI] [PubMed] [Google Scholar]
- 19. Husain D., Meyer R. D., Mehta M., Pfeifer W. M., Chou E., Navruzbekov G., Ahmed E., Rahimi N. (2010) Role of c-Cbl-dependent regulation of phospholipase Cγ1 activation in experimental choroidal neovascularization. Invest. Ophthalmol. Vis. Sci. 51, 6803–6809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Biechele T. L., Moon R. T. (2008) Assaying β-catenin/TCF transcription with β-catenin/TCF transcription-based reporter constructs. Methods Mol. Biol. 468, 99–110 [DOI] [PubMed] [Google Scholar]
- 21. Sal-Man N., Gerber D., Shai Y. (2005) The identification of a minimal dimerization motif QXXS that enables homo- and hetero-association of transmembrane helices in vivo. J. Biol. Chem. 280, 27449–27457 [DOI] [PubMed] [Google Scholar]
- 22. Graham T. A., Weaver C., Mao F., Kimelman D., Xu W. (2000) Crystal structure of a β-catenin/Tcf complex. Cell 103, 885–896 [DOI] [PubMed] [Google Scholar]
- 23. Xing Y., Takemaru K., Liu J., Berndt J. D., Zheng J. J., Moon R. T., Xu W. (2008) Crystal structure of a full-length β-catenin. Structure 16, 478–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Swaminathan G., Tsygankov A. Y. (2006) The Cbl family proteins: ring leaders in regulation of cell signaling. J. Cell. Physiol. 209, 21–43 [DOI] [PubMed] [Google Scholar]
- 25. Liu J., Kimura A., Baumann C. A., Saltiel A. R. (2002) APS facilitates c-Cbl tyrosine phosphorylation and GLUT4 translocation in response to insulin in 3T3-L1 adipocytes. Mol. Cell. Biol. 22, 3599–3609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bartkiewicz M., Houghton A., Baron R. (1999) Leucine zipper-mediated homodimerization of the adaptor protein c-Cbl. A role in c-Cbl's tyrosine phosphorylation and its association with epidermal growth factor receptor. J. Biol. Chem. 274, 30887–30895 [DOI] [PubMed] [Google Scholar]
- 27. Latres E., Chiaur D. S., Pagano M. (1999) The human F box protein β-Trcp associates with the Cul1/Skp1 complex and regulates the stability of β-catenin. Oncogene 18, 849–854 [DOI] [PubMed] [Google Scholar]
- 28. Aberle H., Bauer A., Stappert J., Kispert A., Kemler R. (1997) β-Catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 16, 3797–3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang X., Gaspard J. P., Chung D. C. (2001) Regulation of vascular endothelial growth factor by the Wnt and K-ras pathways in colonic neoplasia. Cancer Res. 61, 6050–6054 [PubMed] [Google Scholar]
- 30. Lupher M. L., Jr., Reedquist K. A., Miyake S., Langdon W. Y., Band H. (1996) A novel phosphotyrosine-binding domain in the N-terminal transforming region of Cbl interacts directly and selectively with ZAP-70 in T cells. J. Biol. Chem. 271, 24063–24068 [DOI] [PubMed] [Google Scholar]
- 31. Mosimann C., Hausmann G., Basler K. (2009) β-Catenin hits chromatin: regulation of Wnt target gene activation. Nat. Rev. Mol. Cell Biol. 10, 276–286 [DOI] [PubMed] [Google Scholar]
- 32. Nastasi T., Bongiovanni A., Campos Y., Mann L., Toy J. N., Bostrom J., Rottier R., Hahn C., Conaway J. W., Harris A. J., D'Azzo A. (2004) Ozz-E3, a muscle-specific ubiquitin ligase, regulates β-catenin degradation during myogenesis. Dev. Cell 6, 269–282 [DOI] [PubMed] [Google Scholar]
- 33. Matsuzawa S. I., Reed J. C. (2001) Siah-1, SIP, and Ebi collaborate in a novel pathway for β-catenin degradation linked to p53 responses. Mol. Cell 7, 915–926 [DOI] [PubMed] [Google Scholar]
- 34. Liu J., Stevens J., Rote C. A., Yost H. J., Hu Y., Neufeld K. L., White R. L., Matsunami N. (2001) Siah-1 mediates a novel β-catenin degradation pathway linking p53 to the adenomatous polyposis coli protein. Mol. Cell 7, 927–936 [DOI] [PubMed] [Google Scholar]
- 35. Sadot E., Simcha I., Iwai K., Ciechanover A., Geiger B., Ben-Ze'ev A. (2000) Differential interaction of plakoglobin and β-catenin with the ubiquitin-proteasome system. Oncogene 19, 1992–2001 [DOI] [PubMed] [Google Scholar]
- 36. Ye X., Wang Y., Cahill H., Yu M., Badea T. C., Smallwood P. M., Peachey N. S., Nathans J. (2009) Norrin, frizzled-4, and Lrp5 signaling in endothelial cells controls a genetic program for retinal vascularization. Cell 139, 285–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kaidi A., Williams A. C., Paraskeva C. (2007) Interaction between β-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat. Cell Biol. 9, 210–217 [DOI] [PubMed] [Google Scholar]
- 38. Essers M. A., de Vries-Smits L. M., Barker N., Polderman P. E., Burgering B. M., Korswagen H. C. (2005) Functional interaction between β-catenin and FOXO in oxidative stress signaling. Science 308, 1181–1184 [DOI] [PubMed] [Google Scholar]
- 39. Niemeyer C. M., Kang M. W., Shin D. H., Furlan I., Erlacher M., Bunin N. J., Bunda S., Finklestein J. Z., Sakamoto K. M., Gorr T. A., Mehta P., Schmid I., Kropshofer G., Corbacioglu S., Lang P. J., Klein C., Schlegel P. G., Heinzmann A., Schneider M., Starý J., van den Heuvel-Eibrink M. M., Hasle H., Locatelli F., Sakai D., Archambeault S., Chen L., Russell R. C., Sybingco S. S., Ohh M., Braun B. S., Flotho C., Loh M. L. (2010) Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat. Genet. 42, 794–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Martinelli S., De Luca A., Stellacci E., Rossi C., Checquolo S., Lepri F., Caputo V., Silvano M., Buscherini F., Consoli F., Ferrara G., Digilio M. C., Cavaliere M. L., van Hagen J. M., Zampino G., van der Burgt I., Ferrero G. B., Mazzanti L., Screpanti I., Yntema H. G., Nillesen W. M., Savarirayan R., Zenker M., Dallapiccola B., Gelb B. D., Tartaglia M. (2010) Heterozygous germline mutations in the CBL tumor-suppressor gene cause a Noonan syndrome-like phenotype. Am. J. Hum. Genet. 87, 250–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.