

Abstract
Expression of indoleamine-2,3-dioxygenase (IDO), an immunosuppressive enzyme in human tumors, leads to immune evasion and tumor tolerance. IDO is therefore a tumor immunotherapeutic target, and several IDO inhibitors are currently undergoing clinical trials. IDO inhibitors can enhance the efficacy of common cancer chemotherapeutics. Here we investigated Pt(IV) – (D)-1-methyltryptophan conjugates 1 and 2 for combined immunomodulation and DNA cross-link-triggered apoptosis for cancer ‘immuno-chemotherapy’. Compound 2 effectively kills hormone-dependent, cisplatin-resistant human ovarian cancer cells, inhibiting IDO by transcriptional deregulation of the auto-crine-signaling loop IDO-AHR-IL6, which blocks kynurenine production and promotes T-cell proliferation. Additionally, 1 and 2 display low toxicity in mice and are stable in blood. To our knowledge, this construct is the first Pt drug candidate with immune checkpoint blockade properties.
Graphical Abstract
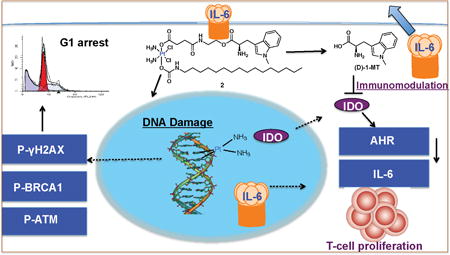
Attractive immunotherapy approaches have included chimeric antigen receptor (CAR) T-cell therapies, cancer vaccines, dendritic cell therapies, and immune checkpoint inhibitors.1 Immune checkpoint therapy has become a clinically viable treatment alternative to conventional chemotherapy for cancer following the FDA approval of ipilumumab, pembrolizumab, and nivolumab.2 Several immune checkpoints are involved in tumor immune escape with varied biological functions, signaling pathways, and expression levels in tumors.3 The programmed death (PD-1), cy-totoxic T-lymphocyte antigen CTLA, T-cell immunoglobulin and mucin 3 domain (TIM3),4 and IDO are common inhibitory immune checkpoint targets under investigation. Immune checkpoint therapy targets regulatory pathways that affect T-cells to enhance antitumor immune responses.5 Combining this therapy, by using small molecule immune checkpoint inhibitors, with standard chemotherapy is likely to provide survival benefit to patients.
IDO is a heme-containing oxidoreductase encoded by the INDO gene. IDO catalyzes the degradation of the essential amino acid tryptophan to kynurenine with the exception of dietary tryptophan, which is catabolized by the liver enzyme tryptophan dioxygenase (TDO).6 The depletion of tryptophan mediates immune tolerance by suppressing effector T-cell function through G1 arrest and subsequent inactivation.7 In a variety human tumors and host antigen-presenting cells, elevated levels of IDO are characteristic of poor prognosis.8 Small molecule inhibitors of IDO that stimulate antitumor immunity have emerged with (D)-1 methyltryptophan ((D)-1-MT)9 and INCB-243671c in Phase I/II clinical trials for the treatment of breast, brain, melanoma, and pancreatic cancers. Promising IDO inhibitors with unique chemical scaffolds continue to attract attention, among which include brassinins, quinones, phenylimidazoles, and hydroxy–amidines.10 These small molecules have the advantage of being easy to produce and deliver, low cost, and compatible with conventional cancer therapies.
IDO inhibitors enhance the efficacy of common chemotherapeutics11 and are synergistic with radiation therapy.12 The IDO inhibitor methylthiohydantoin-tryptophan (MTH-Trp) in combination with cisplatin regresses autochthonous murine breast tumors.11 Induction of IDO-blockade using (D)-1-MT and NLG919 works synergistically with temozolomide (TMZ), cyclophosphamide, and radiotherapy to treat GL261 tumors (glioblastoma)13 in a syngeneic mouse model. Combination chemotherapy incorporating IDO inhibitors holds promise for cancer therapy. A dual – threat construct14 having a potent chemotherapeutic and immune checkpoint inhibitor has thus far not been reported.
Platinum-based chemotherapy is first line treatment for many cancers in the clinic.15 The FDA-approved Pt agents include cisplatin, carboplatin, and oxaliplatin. They induce apoptosis in cancer cells, primarily through DNA damage.16 Despite the efficacy of Pt drugs, toxicity, tumor recurrence, acquired and inherent resistance, and deactivation are associated drawbacks that remain problematic.17 To overcome these problems, one chemical strategy that we and others have employed has been to design an inert Pt(IV) prodrug that can be activated by intracellular reduction following cellular uptake. Given the aforementioned limitations of conventional chemotherapy and immunotherapy, and taking advantage of the potential synergy between platinum drugs and immune checkpoint inhibitors, we employed the Pt(IV) prodrug strategy to combine immunomodulation with Pt-DNA cross-linking-induced apoptosis, affording the first effective chemo-immunotherapeutic.
A symmetric manifestation of our design attaches two (D)-1-MT units at the axial positions of a cisplatin pro-drug (1, Fig. 1). An asymmetric construct having a hexadecyl hydrophobic chain at one axial position and (D)-1-MT at the other was also prepared (2, Fig. 1). The latter synthetic strategy provides a unique double prodrug, activated both by intracellular reduction and by esterase activity. The use of the long hydrophobic chain was motivated by our earlier report showing Pt(IV) binding to human serum albumin for drug delivery.18 Detailed chemical studies revealed that 2 binds HSA, as evidenced in Fig. S1 by FPLC, graphite furnace atomic absorption spectrophotometry (GFAAS), and ESI mass-spectrometric analysis. Furthermore, the degree of lipophilicity of (D)-1-MT, 1 and 2 was determined by measuring the extent of compound partition between octanol and water, Po/w. Experimentally determined Log P values, shown in Table S1, increase from (D)-1-MT (Log P = −2.98±0.15), to 1 (Log P = −0.21±0.08) to 2 (Log P = 1.35±0.26), which determines their cellular differential uptake. We hypothesized that release of (D)-1-MT from 1 or 2 inside cancer cells would block IDO to prevent T-cell degradation, while generation of cisplatin would concomitantly induce DNA damage-induced cell death. A thorough investigation of this novel, dual-threat prodrug approach provided the results presented here, including the synthesis and characterization, cellular mechanism, functional IDO inhibition, and cellular immunomodulation of 1 and 2. The potent anti-proliferative ability of the constructs, nanoparticle formulation, and stability in mice were also investigated.
Figure 1.

Structures of key compounds under investigation
Compounds 1 and 2 were prepared and characterized as outlined in Scheme S1 (Supporting Information, Fig. S2 – S9) and tested against a panel of human ovarian cancer cells with varying sensitivities to cisplatin and constitutive expression of IDO. We found that SKOV3 cells constitutively express robust levels of IDO, A2780 cells express the protein minimally, and NIH:OVCAR3 not at all (Fig. S10). The conversion of tryptophan to kynurenine by IDO suppresses the anti-tumor immune response.7 We therefore investigated the ability of 1 and 2 to block kynurenine production in cells. With the use of HPLC (Fig. S11), we observed that 2 blocks kynurenine release in the medium of SKOV3 cultured cells by a factor of three in comparison to treatment with (D)-1-MT (Fig. 2A). Using immunoblotting and qRT-PCR, we found that 2 selectively targets and effectively blocks IDO protein expression (Fig. 2B), whereas 1 requires a 10-fold higher concentration (100 μM), as shown in Figs. S12 and S13. Previously we demonstracted that cisplatin-DNA adducts block transcription, ultimately triggering apoptosis.19 However, investigation of the control platinum construct C16-Pt-Suc did not show IDO inhibition relative to 2 (Fig. 2B). This result indicates that Pt-DNA adducts do not block IDO transcription or induce IDO mRNA instability, but (D)-1-MT released in the cells does. Collectively, our findings demonstrate the ability of 2 to interrupt the kynurenine pathway by depleting IDO.
Figure 2.

(A) Kynurenine inhibition by 2 (10 μM) in comparison to (D)-1-MT (1 mM) and an untreated control (B) Immunoblotting of the pharmacological inhibition of human IDO in SKOV3 cells. Cells were treated with 10 μM platinum concentrations for 1, 2 and C16-Pt-Suc.
The ability of 1, 2, and cisplatin as a control to promote cell death was evaluated by the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl–tetrazolium bromide] assay across a panel of human ovarian cancer cells. The concentration required for inducing 50% cell kill (IC50) ranged from high nanomolar to micromolar, as extrapolated from dose-response curves; the results are summarized in Table S2. Significantly, complex 2 was the most potent with no cross-resistance with cisplatin as typified by its ability to induce cell death in both cisplatin-sensitive and resistant ovarian cancer cell lines A2780 and A2780/CP70, SKOV3 and NIH:OVCAR3. Encouragingly, 2 displayed approximately 2–4000-fold potency in IDO expressing cells, A2780 and SKOV3 over cisplatin, co-incubated [cisplatin+(D)-1MT] and C16-Pt-Suc-NHC2H4OH treated cells (Table S2).
The intracellular behavior of the Pt(IV)-(D)-1-MT construct (2) was analyzed by using an RNA interference (RNAi) methodology and cell cycle analysis.20 This mechanism of action predictive RNAi methodology uses lymphoma cells that are partially infected with one of eight GFP-labelled short hairpin RNAs (shRNAs). Each shRNA has the potential ability to confer resistance or sensitivity to a particular drug depending on its mechanism of action. Thus, after treatment with the drugs of interest, the cells are subjected to flow cytometry to assess GFP percentage. The resulting pattern of resistance and sensitivity to a given drug is then processed by a probabilistic K-nearest neighbors algorithm that assigns novel compounds to a category by comparing their signatures to that of reference set of drugs. The RNAi signatures of cisplatin, cis,cis,trans-[Pt(NH3)2Cl2(OCOCH3)2] (Di-acetate Pt(IV)), and 2 were obtained. Interestingly, although 2 displays features of a DNA damaging agent, such as strong p53 and Chk2 shRNA enrichment, it differs significantly in other shRNAs, as shown in Fig. 3A. In addition, DNA content assessment by flow cytometry revealed that 2 induces a cell cycle block at G1 phase in a time dependent fashion (Fig. S14), which deviates from the behavior of the classical G2/M arrest phase imposed by cisplatin (Fig. S15). The collective evidence from both RNAi signature assays and cell cycle analysis indicates a unique mechanism of action triggered by the activity of (D)-1-MT provided by the construct (Fig. 3B).
Figure 3.

(A) RNAi signatures derived from the treatment of Eμ-Myc lymphoma cells with 2, cisplatin, and diacetate Pt(IV) at LD80–90 for each compound after 72 h. (B) Principal component analysis plot of the RNAi signatures obtained from (A) and reference set of drugs to elucidate the mechanism of action of complex 2.
The ability of 2 to induce DNA damage was investigated by monitoring changes in expression of markers of the damage response pathway by immunoblotting analyses (Fig. S16 and S17). SKOV3 cells incubated with 2 for up to 48 h and 72 h showed a marked time-dependent increase in expression of phosphorylated ATR, ATM, BRCA1, Chk1, Chk2, H2AX, and p53, indicative of DNA damage. Notably, the high phosphoBRCA1 response confirms the DNA damage role of BRCA1 specific to ovarian and breast cancer.
To gain further insight into the mode of action of 1 and 2, we studied the subcellular distribution of platinum. SKOV3 cells were incubated with the drug candidates (5 μM) for 17 h, using cisplatin as a control. The platinum content was measured by using GFAAS. Whereas the platinum content within the nucleus following 2 treatment was comparable to that of cisplatin, it was 7-fold higher in the cytoplasm (Fig. S18). This result indicates that, over the duration of the study, 2 is preferentially taken up over both 1 and cisplatin, possibly due to its lipophilic axial chain. In addition, DNA platination studies shown in Fig. S19 reveal the occurrence of 310 ± 75 Pt adducts/104 nucleotides for 1, 409 ± 54 Pt adducts/104 nucleotides for cisplatin and 970 ±110 adducts/104 nucleotides for 2. These results demonstrate that, like cisplatin, the new platinum constructs target genomic DNA.
Mechanistic studies of the effect of 2 on the IDO pathway were performed by examining the autocrine-signaling loop of IDO-AHR-IL6 (Fig. S20). The activation of AHR, a cytosolic transcription factor that translocates to the nucleus upon binding xenobiotic ligands such as kynurenine, is involved in tumor formation.21 The signal transducer and activator of transcription STAT3 mediates the process of carcinogenesis.22 We hypothesized that 2, after spontaneous intracellular reduction and esterase activation, releases (D)-1MT into the cytosol to block IDO, leading to inactivation of AHR owing to kynurenine inhibition. Recent findings show that interleukin 6 (IL-6) modulates IDO expression via STAT3.23 To support this hypothesis, SKOV3 cells were treated with 2 and the RNA was harvested after 24 h for qPCR. The results show that mRNA expression levels of AHR decreased by 5-fold (Fig. S21) and those of IL-6 were reduced by 10-fold (Fig. S22) relative to untreated controls. These results support the role of 2 to interrupt the IDO pathway by deactivating the downstream AHR and IL-6, thereby destabilizing the auto-crine loop implicated in constitutive IDO expression.
To target a key component of the immunosuppression promoting IDO-AHR-IL6 loop, we investigated the immunomodulatory phenotype of 2. A mixed leukocyte reaction (MLR) was carried out. In this experiment, suspended 2×105 peripheral blood mono-nuclear cells (PBMCs) stimulated with phytohemagglutinin (PHA-P), and 2×105 cells of PBMCs from unrelated healthy donors as responders stained with carboxyfluorescein succin-imidyl ester (CFSE), a cell permeable fluorescent dye that binds amine residues (mostly lysines) covalently, through its succin-imidyl ester group were cocultured with adherent SKOV-3 (2000 cells). After 6 days of culture, PBMCs were harvested by centrifugation of medium, stained with APC-anti CD3 and T-cell proliferation was assessed by flow cytometry. T-cells express CD3, a cell surface multimeric protein complex involved in T-cell development and activation. PBMCs used were CD3high as assessed by flow cytometry using APC labeled anti-CD3, as shown in Fig 4A. Consistent with our results of a decrease in kynurenine production by 2 due to downregulation of IDO and its downstream targets, the presence of 2 in the SKOV3/MLR coculture experiment resulted in the enhancement of alloreactive T-cell proliferation assessed by live-cell tracing for multiple generations using flow cytometry (Fig. 4B). Collectively, our cellular data show that 2 plays a role in immunomodulation, which results in T-cell proliferation.
Figure 4.

(A) Estimation of T-cell population in PBMCs by measuring CD3 cell surface marker using flow cytometry. (B) Mixed leukocyte reaction to assess T-cell proliferation induced by 2 (10 μM) after 6 days of compound incubation with PBMCs and SKOV3 cells. T-cell division accounts for multiple generations (bands) measured by the intensity of CFSE dilution using flow cytometry.
Therapeutic nanoparticles (NPs) have been widely investigated for their potential to enhance cancer treatment.24 The chemical structure of 2 bears both hydrophilic (1-D-MT) and hydrophobic (hexadecyl isocyanate) axial ligands, conferring amphiphilic character to the molecule. Work in our lab has shown that hexadecyl isocyanate axial ligand facilitates non-covalent binding to human serum albumin, protecting the Pt(IV) center from premature reduction in blood.18 In the presence of the biodegradable block copolymer, poly(lactic-co-glycolic acid)-polyethylene glycol (PLGA-PEG), 2 readily self-assembles into NPs suitable for pre-clinical studies (Fig. 5A). Characterization of the NPs using DLS showed sizes of 120 ± 3.50 nm, applicable for preclinical studies. (Fig. S23). Having obtained the nanoparticles we proceeded to investigate the ability of 2 and its NP to induce apoptosis. Translocation of phosphatidylserine residues to the exterior is a characteristic of apoptotic cells, which have a compromised cell membrane, and can be detected by annexin V.25 Using a dual staining annexin V/sytox green apoptosis dead cell assay, we examined the apoptotic behavior of SKOV3 cells after 72 h of treatment with 2 and its NP by flow cytometry. Both 2 and its NP induced large populations of SKOV3 cells to undergo apoptosis, with the NPs doing so severely (Fig. S24).
Figure 5.

A) Schematic representation of PLGA-PEG nanoparticles used to encapsulate 2. B) Time dependent stability of 2 in mice treated with 2-NP (8mg/kg).
In vivo evaluation of 2 revealed that mice are tolerant of the platinum-immune checkpoint inhibitor conjugates. Dynamic blood stability experiments of 2 in six-week old female Balb/c mice revealed a t1/2 of approximately 1 h, as shown in Fig 5B. Mice were intravenously injected with a sterile NP formulation of 2 at a relatively high dose of 8 mg/kg and blood was drawn at different time intervals (10 min, 1h, 3 h, 6 h and 24 h). The hydrophobic complex 2 was extracted with octanol,18 diluted, and the Pt concentration was determined by GFAAS. By 24 h the Pt concentration reached non-detectable levels suggesting complete reduction of hydrophobic Pt(IV) to hydrophilic Pt(II) species. In addition, blood stability in human blood showed that the t1/2 of 2 is 2.2 h in comparison to that reported for cisplatin (t1/2 = 21.6 min)26 and satraplatin (t1/2 = 6 min)27 as seen in Fig. S25.
In conclusion, we present a Pt small molecule immune checkpoint inhibitor platform that preferentially targets the immuno-suppressive enzyme IDO to enhance T-cell proliferation. We investigated the synergy of Pt and checkpoint inhibitor biology as well as the molecular mechanism of (D)-1-MT. Our cellular data provide evidence for DNA damage induced by 2, which leads to G1 arrest and cell death. Furthermore, IDO is inhibited to evoke downregulation of AHR and IL-6, which are key genes involved in the auto-regulation of constitutive IDO expression. This action leads to immunomodulation and enhanced T-cell proliferation. Work is currently underway to evaluate the in vivo tumor efficacy of this and other Pt-immune checkpoint inhibitors in immuno-competent mice. We foresee that this molecular design has the potential to facilitate discovery of metal-based small-molecule immunomodulators in targeting cancer cells by immuno-chemotherapy.
Supplementary Material
Acknowledgments
This work is supported by the NCI under grant CA034992.
Footnotes
Experimental details, characterization of compounds 1 and 2, and data regarding all molecular biology, cellular, mice, and blood stability studies. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Couzin-Frankel J. Science. 2013;342:1432. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]; (b) Kim MS, Ma JS, Yun H, Cao Y, Kim JY, Chi V, Wang D, Woods A, Sherwood L, Caballero D, Gonzalez J, Schultz PG, Young TS, Kim CH. J Am Chem Soc. 2015;137:2832. doi: 10.1021/jacs.5b00106. [DOI] [PubMed] [Google Scholar]; (c) BioMedTracker. Cancer Immunotherapies. 2014 [Google Scholar]
- 2.Sharma P, Allison JP. Science. 2015;348:56. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 3.Muller AJ, Scherle PA. Nat Rev Cancer. 2006;6:613. doi: 10.1038/nrc1929. [DOI] [PubMed] [Google Scholar]
- 4.Kane LP. J Immunol. 2010;184:2743. doi: 10.4049/jimmunol.0902937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma P, Allison JP. Cell. 2015;161:205. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mellor AL, Munn DH. Nat Rev Immunol. 2004;4:762. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- 7.Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. J Exp Med. 1999;189:1363. doi: 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T, Van den Eynde BJ. Nat Med. 2003;9:1269. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]; (b) Okamoto A, Nikaido T, Ochiai K, Takakura S, Saito M, Aoki Y, Ishii N, Yanaihara N, Yamada K, Takikawa O, Kawaguchi R, Isonishi S, Tanaka T, Urashima M. Clin Cancer Res. 2005;11:6030. doi: 10.1158/1078-0432.CCR-04-2671. [DOI] [PubMed] [Google Scholar]; (c) Brandacher G, Perathoner A, Ladurner R, Schneeberger S, Obrist P, Winkler C, Werner ER, Werner-Felmayer G, Weiss HG, Gobel G, Margreiter R, Konigsrainer A, Fuchs D, Amberger A. Clin Cancer Res. 2006;12:1144. doi: 10.1158/1078-0432.CCR-05-1966. [DOI] [PubMed] [Google Scholar]
- 9.(a) Munn DH, Sharma MD, Lee JR, Jhaver KG, Johnson TS, Keskin DB, Marshall B, Chandler P, Antonia SJ, Burgess R, Slingluff CL, Jr, Mellor AL. Science. 2002;297:1867. doi: 10.1126/science.1073514. [DOI] [PubMed] [Google Scholar]; (b) Godin-Ethier J, Hanafi LA, Piccirillo CA, Lapointe R. Clin Cancer Res. 2011;17:6985. doi: 10.1158/1078-0432.CCR-11-1331. [DOI] [PubMed] [Google Scholar]
- 10.Jiang T, Sun Y, Yin Z, Feng S, Sun L, Li Z. Future Med Chem. 2015;7:185. doi: 10.4155/fmc.14.151. [DOI] [PubMed] [Google Scholar]
- 11.Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Nat Med. 2005;11:312. doi: 10.1038/nm1196. [DOI] [PubMed] [Google Scholar]
- 12.Hou DY, Muller AJ, Sharma MD, DuHadaway J, Banerjee T, Johnson M, Mellor AL, Prendergast GC, Munn DH. Cancer Res. 2007;67:792. doi: 10.1158/0008-5472.CAN-06-2925. [DOI] [PubMed] [Google Scholar]
- 13.Li M, Bolduc AR, Hoda MN, Gamble DN, Dolisca SB, Bolduc AK, Hoang K, Ashley C, McCall D, Rojiani AM, Maria BL, Rixe O, MacDonald TJ, Heeger PS, Mellor AL, Munn DH, Johnson TS. J Immunother Cancer. 2014;2:21. doi: 10.1186/2051-1426-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dhar S, Lippard SJ. Proc Natl Acad Sci U S A. 2009;106:22199. doi: 10.1073/pnas.0912276106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galanski M, Jakupec MA, Keppler BK. Curr Med Chem. 2005;12:2075. doi: 10.2174/0929867054637626. [DOI] [PubMed] [Google Scholar]
- 16.Wang D, Lippard SJ. Nat Rev Drug Discov. 2005;4:307. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 17.Siddik ZH. Oncogene. 2003;22:7265. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 18.Zheng YR, Suntharalingam K, Johnstone TC, Yoo H, Lin W, Brooks JG, Lippard SJ. J Am Chem Soc. 2014;136:8790. doi: 10.1021/ja5038269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Todd RC, Lippard SJ. Metallomics. 2009;1:280. doi: 10.1039/b907567d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Jiang H, Pritchard JR, Williams RT, Lauffenburger DA, Hemann MT. Nat Chem Biol. 2011;7:92. doi: 10.1038/nchembio.503. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pritchard JR, Bruno PM, Gilbert LA, Capron KL, Lauffenburger DA, Hemann MT. Proc Natl Acad Sci U S A. 2013;110:E170. doi: 10.1073/pnas.1210419110. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Pritchard JR, Bruno PM, Hemann MT, Lauffenburger DA. Mol Biosyst. 2013;9:1604. doi: 10.1039/c2mb25459j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller M, Jugold M, Guillemin GJ, Miller CL, Lutz C, Radlwimmer B, Lehmann I, von Deimling A, Wick W, Platten M. Nature. 2011;478:197. doi: 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- 22.Yu H, Pardoll D, Jove R. Nat Rev Cancer. 2009;9:798. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Litzenburger UM, Opitz CA, Sahm F, Rauschenbach KJ, Trump S, Winter M, Ott M, Ochs K, Lutz C, Liu X, Anastasov N, Lehmann I, Hofer T, von Deimling A, Wick W, Platten M. Oncotarget. 2014;5:1038. doi: 10.18632/oncotarget.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nat Nanotechnol. 2007;2:751. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 25.Quinn PJ. Subcell Biochem. 2002;36:39. doi: 10.1007/0-306-47931-1_3. [DOI] [PubMed] [Google Scholar]
- 26.Erdlenbruch B, Nier M, Kern W, Hiddemann W, Pekrun A, Lakomek M. Eur J Clin Pharmacol. 2001;57:393. doi: 10.1007/s002280100319. [DOI] [PubMed] [Google Scholar]
- 27.Carr JL, Tingle MD, McKeage MJ. Cancer Chemother Pharmacol. 2002;50:9. doi: 10.1007/s00280-002-0462-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.