

Abstract
Background
High-fat diet (HFD) promotes endothelial dysfunction and proinflammatory monocyte activation, which contribute to atherosclerosis in obesity. We investigated whether HFD also induces the dysfunction of red blood cells (RBCs), which serve as a reservoir for chemokines via binding to Duffy antigen receptor for chemokines (DARC).
Methods and Results
A 60% HFD for 12 weeks, which produced only minor changes in lipid profile in C57/BL6 mice, markedly augmented the levels of monocyte chemoattractant protein-1 bound to RBCs, which in turn stimulated macrophage migration through an endothelial monolayer. Levels of RBC-bound KC were also increased by HFD. These effects of HFD were abolished in DARC−/− mice. In RBCs from HFD-fed wild-type and DARC−/− mice, levels of membrane cholesterol and phosphatidylserine externalization were increased, fostering RBC-macrophage inflammatory interactions and promoting macrophage phagocytosis in vitro. When labeled ex vivo and injected into wild-type mice, RBCs from HFD-fed mice exhibited ≈3-fold increase in splenic uptake. Finally, RBCs from HFD-fed mice induced increased macrophage adhesion to the endothelium when they were incubated with isolated aortic segments, indicating endothelial activation.
Conclusions
RBC dysfunction, analogous to endothelial dysfunction, occurs early during diet-induced obesity and may serve as a mediator of atherosclerosis. These findings may have implications for the pathogenesis of atherosclerosis in obesity, a worldwide epidemic.
Keywords: atherosclerosis, erythrocytes, leukocytes, obesity
Obesity caused by chronic consumption of a hypercaloric, high-fat diet (HFD) is a worldwide epidemic, representing one of the greatest threats to global health.1,2 Leukocytes play a key role in fueling adipose tissue inflammation and insulin resistance in obesity, and also promote atherosclerotic lesion formation and myocardial infarction, the leading cause of death in these patients.1,2 Although the proatherogenic effects of HFD and hyperlipidemia on monocytes have been described in mice and humans,3–5 the impact of HFD on other marrow-derived cells that regulate atherosclerosis is ill-defined.
Evidence is emerging that erythrocytes (red blood cells, RBCs) play an important modulatory role in the development of atherosclerosis. RBCs avidly bind proinflammatory chemokines such as monocyte chemoattractant protein-1 (MCP-1), KC/interleukin-8, and RANTES via the Duffy antigen receptor for chemokines (DARC).6 Reversible binding to DARC may alter chemokine levels in the atherosclerotic milieu, depending on factors altering the balance of uptake versus release in RBCs traversing through plaque microvessels. Importantly, RBCs can become entrapped within atherosclerotic lesions at sites of intraplaque hemorrhage, where they are taken up by macrophages. RBC membranes are enriched in cholesterol, and, in human coronary plaques, cholesterol colocalizes with glycophorin-A, a membrane marker of RBCs.7 Intraplaque hemorrhage positively correlates with advanced atherosclerosis, with the extent of RBC extravasation proportional to plaque development.8 Nevertheless, the impact of chronic HFD on RBC function is largely unexplored.
Here, we examined the effects of HFD on RBC function in mice. We observed that HFD dramatically enhances chemokine release from RBC-DARC, triggering proinflammatory responses in macrophages and increased macrophage binding to the vascular endothelium. Moreover, RBCs from HFD-fed mice exhibit increased levels of reactive oxygen species and membrane phosphatidylserine (PS) externalization, as well as enhanced phagocytosis by macrophages in vitro. In vivo, these RBCs are more avidly taken up by the spleen, the major organ responsible for the clearance of senescent/ apoptotic RBCs. These findings have important implications regarding the potential role of RBCs in the pathogenesis of obesity-related atherosclerosis.
Materials and Methods
Animals and Whole Blood Collection
Please see the online-only Data Supplement for details on mice and blood harvesting procedures.
Enzyme-Linked Immunosorbent Assay and Chemokine Array Assays
Specimens of EDTA anticoagulated blood were divided in half, treated with either 50 U/mL of heparin in phosphate-buffered saline (PBS) or vehicle control (PBS), and incubated at room temperature for 30 minutes on a rocking platform.6 Platelet-poor plasma was harvested by centrifugation at 1500g for 10 minutes at 25°C and used to measure MCP-1 and KC protein levels with and without heparin treatment by commercial enzyme-linked immunosorbent assay kits according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN). Mouse chemokine array (R&D Systems, ARY020) was performed according to the manufacturer’s protocol. In brief, array membranes were blocked and incubated with 100 μg of RBC membranes and array panel detection antibody cocktail overnight at 4°C. After washing, membranes were incubated with streptavidin-horseradish peroxidase, washed, and visualized by using chemiluminescence; mean pixel density was measured using the Bio-Rad Gel Doc XR+ imaging system.
Monocyte Transmigration Assay
bEnd.3 cells (murine endothelial cells, ATCC) were grown to confluence on 3.0-μm pore size 24-well inserts (BD Biosciences); 1×107 packed RBCs from chow diet (CD)–fed (CD-RBC) or HFD-fed (HFD-RBC) mice in defined medium were added to the bottom of the wells. Then, J744.1 cells (murine monocyte/macrophage cell line, ATCC) were added to the inserts and allowed to adhere for 1 hour at 37°C, 5% CO2. Nonadherent cells were removed by aspiration, and the inserts were filled with fresh defined medium. Migration proceeded for 16 hours at 37°C, 5% CO2. Inserts were excised and fixed in ice-cold methanol, after which nonmigrated macrophages on the luminal side of the inserts were removed by using a cotton swab, whereas transmigrated macrophages on the abluminal side were preserved by placing the inserts on slides and mounted by using Vectashield/DAPI (Vector Labs). Images were captured using fluorescence (Nikon Microphot-FXA, 10×, n=5 representative fields per insert), and results were analyzed using Image J (National Institutes of Health).
Flow Cytometry
PS externalization, reactive oxygen species (ROS), and DARC protein levels were measured by flow cytometry after labeling RBCs with Annexin V (Molecular Probes), 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA, Invitrogen), and anti-DARC antibody (R&D Systems, catalog no. AF6695), respectively, using standard techniques as described previously.9
Assessment of RBC Deformability
RBCs were subjected to uniform shear stress ranging from 0.3 to 100.0 Pa and the elongation index was determined by using an automated optical rotational analyzer (LoRRca Maxsis, Mechatronics, The Netherlands) and the manufacturer’s protocol. The elongation index is calculated by dividing the difference between the major and minor RBC axes by the sum of the axes.
Determination of RBC Membrane Cholesterol Content
For all experiments, RBC membranes (pink ghosts) were prepared according to Hanahan et al10; in brief, blood was centrifuged at 1000g for 30 minutes at 4°C, plasma and buffy coat were removed, and RBCs were suspended in 1 mL of 310 mOsm/L (0.172 mol/L) Tris-HCl buffer (pH 7.6). Samples were washed twice with 310 mOsm/L Tris-HCl buffer and resuspended to a final hematocrit of 50%. One mL of RBC suspension was pelleted; RBCs were resuspended in 1 mL of hypotonic (20 mOsm/L) Tris-HCl buffer (pH 7.6) and lysed on ice for 5 minutes. RBC membranes were centrifuged at 20 000g for 40 minutes at 4°C, washed 4 times, and resuspended in 20 mOsm/L Tris-HCl buffer to a final volume of 1 mL. Colorimetric assay was performed in 96-well plates; samples were prepared by combining 36 μL of RBC membrane suspensions with 324 μL of Infinity cholesterol reagent (Thermo Scientific catalog no. TR13521), and incubating the mixtures for 1 hour at 37°C. Plates were read at 500 nm; commercial cholesterol preparation (Pointe Scientific, Core Laboratory Supplies catalog no. C7510) was used to derive a standard curve.
Electron Paramagnetic Resonance Analysis of SNO-Hb
Six-week-old C57BL/6 mice were either continued on CD or switched to HFD for 12 weeks. Mice were anesthetized by using isoflurane and blood collected via retro-orbital bleed using EDTA capillary tubes and heparin-coated microfuge tubes. Whole blood was then drawn into a 1-mL syringe, snap-frozen to make a blood core, transferred to a 15-mL conical tube, and stored in liquid nitrogen. S-nitrosohemoglobin (SNO-Hb) measurements were performed by using a Bruker E-scan SC0420 and an electron paramagnetic resonance (EPR) finger dewar filled with liquid nitrogen; blood cores were transferred to the finger dewar to keep them frozen during measurements and placed in e-Scan sample cavity. All e-scan spectra were collected as an average of 30 scans with the following settings: sweep time of 5.243 seconds, modulation amplitude of 3.0906 Gauss, modulation frequency of 86.000 kHz, microwave power of 0.00219 mW, and microwave frequency of 9.742222 GHz. To compare SNO-Hb content in RBCs collected from mice on CD and HFD, spectra for each group were constructed and the area under the curve was calculated from the double integral by using WinEPR processing software.
Biotin Switch Assay
Six-week-old C57BL/6 mice were either continued on CD or switched to HFD for 12 weeks. Mice were anesthetized by using isoflurane, and blood was collected via retro-orbital bleed using EDTA capillary tubes and heparin-coated microfuge tubes. Whole blood was separated by centrifugation and washed twice with PBS. Packed RBCs (300 μL) were transferred to a new microfuge tube and lysed with 800 μL of 100 μM HEPES, 1 mM EDTA, and 1 mM Neocuproine; pH 7.7 buffer containing 1% NP40 and protease inhibitors. Samples were spun at 12 000g for 10 minutes at 4°C. Fifty μL of RBC lysate were then transferred to a new tube and treated with 5 mL of 100% acetone for 20 minutes at −20°C to precipitate proteins. Lysates were pelleted by spinning at 4000g for 5 minutes at 4°C and then washed 3 times with 70% acetone. Protein pellets were then dried and resuspended in HEN buffer+1% sodium dodecyl sulfate (SDS). A positive control was generated by treating one of the samples with 100 μmol/L of Cysnitric oxide (NO) and incubating in the dark for 30 minutes. After another round of acetone precipitation and wash, proteins were resuspended in HEN buffer+2.5% SDS with 20 mmol/L s-methyl methanethiosufonate and incubated at 50°C with vortexing every 4 minutes. Proteins were then precipitated in acetone and washed, and pellets were resuspended in 1 mL of PBS containing 1% SDS, 10 nmol/L copper, 10 mmol/L ascorbate, and 200 μmol/L HPDP-biotin, and incubated in the dark for 1 hour. Proteins were precipitated and washed, and pellets were resuspended in 250 μL of HEN+1% SDS and 200 μmol/L dl-dithiothreitol, and aliquots of total protein were stored at −20°C. Neutralization buffer (750 μL) was added to the remaining protein suspension, samples were transferred to a microfuge tube containing 80 μL of washed streptavidin beads, and the samples were incubated overnight at 4°C with gentle rotation. The following day, streptavidin complexes were washed 3 times with neutralization buffer and eluted at 95°C with 100 μL of HEN+2.5% SDS and 3 mmol/L biotin. Eluted samples and total protein aliquots were normalized to total protein content and the degree of S-nitrosylation determined by Western blotting and quantified by densitometry.
In Vitro Erythrophagocytosis Assay
RBCs from EDTA-anticoagulated whole blood were washed 3 times (PBS, 1000g, 5 minutes, room temperature) and buffy coat was removed. Packed washed RBCs were fluorescently labeled by using 1 μmol/L calcein-AM (BD Biosciences) for 30 minutes at 37°C, washed 3 times with PBS, and resuspended in defined medium. Either CD-RBC or HFD-RBC at 2% hematocrit were added to 1×105 thioglycolate-elicited peritoneal macrophages obtained from chow or HFD mice in a 24 well plate. After 4 hours of incubation, the medium was aspirated, nonphagocytosed RBCs were lysed by using RBC lysis buffer, and the macrophages were washed 3 times with PBS. Macrophages were then lysed using 0.1% Triton-X in PBS, and fluorescence was measured using Omega Polaris plate reader (excitation 485 nm, emission 525 nm).
In Vivo Splenic Uptake Assay
One hundred μL of packed CD-RBC or HFD-RBC were labeled with 15 μmol/L Cell-Tracker Red CMTPX fluorescent dye (Invitrogen) for 30 minutes at 37°C, washed with PBS, and suspended in defined medium at 50% hematocrit. Two hundred μL of RBCs were infused via retro-orbital route into CD-fed mice, and after 24 hours the animals were anesthetized and euthanized by cardiac puncture/perfusion with PBS. Spleens were then isolated, rinsed in PBS, embedded in optimum cutting temperature medium crosswise that included 4 to 5 cut segments per spleen, snap-frozen, and stored at −80°C. With the use of a cryotome, 10-μm sections were cut (≈50 sections per each spleen), fixed with 10% paraformaldehyde, and visualized under fluorescence (Nikon Microphot-FXA). Sections were first scanned at low power (1.5× magnification), and then images were captured for analysis at 100× magnification. At least 3 representative fields for each section of each spleen were examined such that images were obtained to cover the entire cross section of the spleen. Images were uniformly contrast adjusted to reduce background (autofluorescence), converted to binary, and analyzed with Image J software to determine cell number (fluorescent signal density over surface area). Events with pixel sizes >100 were deemed true cell events.
Microarray Analysis of Gene Expression
Peritoneal macrophages were obtained by thioglycolate induction of C57BL/6 mice fed either CD or HFD. Erythrophagocytosis was performed as described above by using CD-RBC or HFD-RBC. A third group (no RBCs) served as control. At the end of incubation, macrophages were washed with PBS to remove free RBCs, and total RNA was extracted by using RNeasy kit (Qiagen, Valencia, CA). RNA was then converted to cDNA, amplified, fragmented, and labeled for microarray analysis by using WTA-Ovation FFPE V2 kit and Encore biotin module (Nugen, San Carlos, CA) according to the manufacturer’s instructions. Affymetrix Human Gene 1.0 ST microarray chips were used to assess the gene expression profile (Microarray Core Facility, Cincinnati Children’s Hospital and Medical Center). Transcripts that were differentially expressed as a result of either CD-RBC or HFD-RBC phagocytosis were identified based on filtering for probe sets with Robust Multichip Average–normalized raw expression of >6.0 in either of the 3 pairs of replicates, that differed between treated and untreated macrophages by at least 1.5-fold with P<0.05 using a Welch t test. A total of 642 genes were identified that exhibited significant expression responses as a function of exposure to CD-RBC/HFD-RBC, HFD, or both; 41 genes of the 642 were >1.6-fold upregulated or downregulated in macrophages isolated from HFD versus CD-fed animals. Primary .cel files have been uploaded into GEO database (accession no. GSE50240). The relative levels of expression of mRNA encoding CCL3, interleukin-1β, and chemokine C-X-C motif ligand (CXCL) 2 were determined by using the corresponding Taqman probe/primer sets (all from Roche Diagnostics, Indianapolis, IN).
Aortic Ring Assay
Abdominal aortas were isolated from CD-fed C57/BL6 mice; aortic rings (≈1 mm) were prepared, and 2 rings were used per each experimental condition. Citrated blood from either CD- or HFD-fed mice was centrifuged, RBCs were washed 2 times with PBS, and ≈100 μL of packed RBC (hematocrit-normalized) were added and incubated with aortic rings in defined media for 4 hours at 37°C (90 rpm). A third group (no RBCs) served as control. The aortic rings were then washed to remove RBCs, and calcein-AM–labeled mouse peritoneal macrophages from mice on CD were added and incubated for 2 hours at 37°C (90 rpm). Subsequently, the aortic rings were washed with PBS and embedded in optimum cutting temperature medium crosswise, snap-frozen, and stored at −80°C. Multiple 10-μm sections were cut using a cryotome, fixed in formalin, and analyzed under fluorescence. Images were captured, and macrophages that adhered to the luminal endothelium were counted.
Statistical Analyses
Statistical analyses were performed and dot plots were generated using SigmaPlot v12.5. The following tests were used: for data sets with ≥3 groups, Kruskal-Wallis; for 2-group data sets, Wilcoxon rank sum; and for paired data sets (heparin versus no heparin), Wilcoxon signed-rank. The appropriate 2-group comparisons were performed and the Bonferroni-Dunn multiple comparisons procedure was used. In the dot plots, thick bars depict mean values and thin bars show standard deviation. P≤0.05 was deemed significant.
Results
RBCs Exhibit a Proinflammatory Phenotype in Response to HFD
To determine the effects of HFD on RBC, mice were fed an obesogenic, 60% fat diet for 12 weeks. Wild-type (WT) and DARC−/− mice similarly gained weight on this diet (Figure IA in the online-only Data Supplement); the fasting lipid profile showed increased cholesterol and triglyceride levels (not shown). Overt diabetes mellitus did not develop in either HFD group, as evidenced by nonelevated hemoglobin A1C levels, although fasting glucose levels were elevated in HFD-fed DARC−/− mice (Figure IB and IC in the online-only Data Supplement).
To test whether RBCs carry increased levels of MCP-1 as a consequence of high-fat feeding, we measured plasma MCP-1 before and after RBCs were treated with heparin to release DARC-bound MCP-1.6 Plasma MCP-1 levels in HFD-fed WT mice were significantly increased in comparison with those in CD-fed WT mice; heparin treatment caused HFD-RBCs to release MCP-1, whereas there was no release from CD-RBC (Figure 1A). Significantly elevated levels of MCP-1 were also detected on RBC membranes (pink ghosts) of HFD-fed WT mice (Figure 1C). As expected, DARC−/− mice exhibited little or no membrane-associated MCP-1. Plasma MCP-1 levels in CD-fed DARC−/− mice were much lower than in CD-fed WT mice, which is consistent with previous reports,11 and HFD produced only a slight elevation of MCP-1 levels in the plasma of DARC−/− mice (Figure 1A and 1C). To examine whether the decreased circulating MCP-1 in DARC−/− mice may be caused by a decrease in its biosynthesis, we also evaluated MCP-1 expression in adipose tissues, a major source of MCP-1 production in obesity, via real-time PCR and enzyme-linked immunosorbent assay. In subcutaneous adipose tissue, no significant differences were noted in MCP-1 mRNA expression levels (not shown) and secreted protein levels between WT and DARC−/− mice (Figure II in the online-only Data Supplement), whereas, in the epididymal adipose tissues, MCP-1 expression tended to be higher in DARC−/− mice (not shown). These data suggest that alterations in MCP-1 biosynthesis do not account for the reduced RBC-bound MCP-1 in DARC−/− mice.
Figure 1.

HFD-RBCs promote vascular inflammation: increased MCP-1 and KC levels on the surface of the HFD-RBCs in wild-type (WT) mice, but not Duffy antigen receptor for chemokine-deficient (DARC−/−) mice. A and B, ELISA was performed on platelet-poor plasma prepared from whole blood of WT and DARC−/− mice fed CD vs HFD for 12 weeks, with or without heparin treatment. C and D, ELISA was performed on RBC membrane preparations. n=3 to 5 (1 dot = 1 mouse); *P<0.05. CD indicates chow diet; ELISA, enzyme-linked immunosorbent assay; HFD, high-fat diet; MCP-1, monocyte chemoattractant protein-1; n.s., not significant; and RBC, red blood cell.
Evidence is emerging that neutrophils play a major role in atherogenesis12; thus, we examined the levels of KC, a critical neutrophil chemoattractant that binds to DARC,13 in plasma and on RBC membranes. Overall, the results were analogous to those for MCP-1 (Figure 1B and 1D); interestingly, treatment with heparin could not dislodge additional KC protein from RBC membranes, indicating that, in comparison with MCP-1, KC likely binds DARC with a higher affinity (Figure 1B). To examine whether HFD affects the levels of other RBC-bound chemokines, we evaluated pink ghost preparations by using a murine chemokine array. HFD significantly decreased the levels of DARC-bound eosinophil-specific chemokine CCL11 (eotaxin, linked to atherosclerotic progression), and increased the levels of DARC-bound chemokine CXCL5, a chemokine that is protective in the context of atherosclerosis13–15 (Figure III in the online-only Data Supplement). Interestingly, the levels of CXCL12, a chemokine that plays a complex role in atherosclerosis and does not bind to DARC,13,16 were significantly decreased on RBC membranes of HFD-fed WT and DARC−/− mice, suggesting reduced production or binding to an alternative RBC receptor in the setting of HFD (Figure III in the online-only Data Supplement). Importantly, we note that the level of DARC expressed on RBC surfaces was not affected by HFD (Figure IV in the online-only Data Supplement).
Because RBC-bound MCP-1 protein levels are significantly higher in HFD, we reasoned that HFD-RBCs would elicit enhanced monocyte recruitment in comparison with CD-RBCs. To test this hypothesis, we performed monocyte transendothelial migration assays by using WT CD-RBCs and HFD-RBCs as the source of chemoattractant. An increase in monocyte transmigration of ≈1.6-fold (Figure 2) was observed with HFD-RBCs in comparison with CD-RBCs. No significant monocyte transmigration was observed in control experiments in which RBCs were omitted (data not shown). Moreover, CD-RBCs and HFD-RBCs collected from DARC−/− mice did not differ in their ability to elicit monocyte transmigration (Figure 2). These data suggest that HFD can enhance the release of DARC-bound chemokines from RBCs, which in some circumstances may contribute to monocyte migration through the endothelium.
Figure 2.
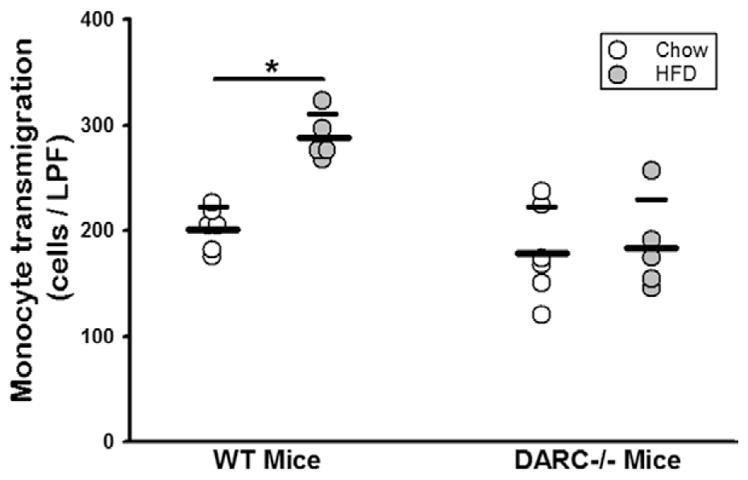
HFD increases DARC-dependent, RBC-induced monocyte transmigration. Monocyte transmigration assay, with packed RBCs from WT and DARC−/− mice fed CD vs HFD for 12 weeks as the chemoattractant source. n=5 (1 dot = 1 mouse); *P<0.05. CD indicates chow diet; DARC, Duffy antigen receptor for chemokines; HFD, high-fat diet; LPF, low-power field; RBC, red blood cell; and WT, wild type.
Production of ROS in WT RBC was studied by flow cytometry by using 2′,7′-dichlorodihydrofluorescein fluorescence, which detects peroxides and other oxidant species. 2′,7′-Dichlorodihydrofluorescein fluorescence was increased ≈1.2-fold in HFD-RBCs in comparison with CD-RBCs (Figure 3A). Damaged and senescent RBCs externalize PS, which is recognized by macrophages via lactadherin-mediated mechanisms,17 resulting in RBC clearance from the circulation through phagocytosis. Staining for Annexin V demonstrated greater externalization of PS in HFD-RBCs than in CD-RBCs (Figure 3B). This difference was not attributable to a higher percentage of reticulocytes (immature RBCs with increased levels of externalized PS) in the HFD mice, suggesting that HFD did not result in a large change in RBC lifespan (data not shown).
Figure 3.

HFD alters the biochemical properties of RBCs. Mice were maintained on CD or HFD for ≥12 weeks. A, Intracellular ROS levels (DCFH fluorescence). B, PS externalization as measured by Annexin V staining. C, Cholesterol content of RBC membranes. D, RBC deformability index as a function of various shear rates (black line, CD RBCs; red line, HFD RBCs), WT mice. E through G, Annexin V staining, cholesterol content, and elongation index, respectively, in DARC−/− mice fed CD vs HFD. A through C, E, F, n=3 to 8 (1 dot = 1 mouse); *P<0.05. H, RBC nitrosohemoglobin levels (EPR assay), n=6; *P<0.05. AUC indicates area under the curve; CD, chow diet; DARC−/−, Duffy antigen receptor for chemokine-deficient; DCFH, 2′,7′-dichlorodihydrofluorescein; EPR, electron paramagnetic resonance; HFD, high-fat diet; PS, phosphatidylserine; RBC, red blood cell; ROS, reactive oxygen species; and WT, wild type.
The rheological properties of human RBCs are altered in a metabolically aberrant hyperoxidative milieu18; moreover, cholesterol is known to influence the flexibility of biological membranes, with higher cholesterol content resulting in a stiffer membrane and thus lower RBC deformability and lower elongation index.19 RBC membrane cholesterol was significantly higher in HFD-RBCs than in CD-RBCs (Figure 3C). Using ektacytometry, we assessed whether HFD caused a change in deformability that may accentuate their clearance from the circulation. In comparison with CD-RBCs, HFD-RBCs had a significantly decreased deformability in the physiological range of shear rates (Figure 3D). Analogous to RBCs from WT mice, RBCs from DARC−/− mice on HFD exhibited significantly enhanced PS externalization, increased membrane cholesterol content, and decreased deformability (Figure 3E through 3G).
Increased ROS could potentially modulate RBC-NO, which has been proposed to regulate RBC deformability and other properties.20 We thus examined the SNO-Hb content in RBCs from mice on CD and HFD by using EPR. As shown in Figure 3H, SNO-Hb levels in HFD-RBCs tended to be lower but did not reach statistical significance. The biotin switch assay yielded analogous findings (Figure V in the online-only Data Supplement); basal levels of SNO-Hb were comparable in RBCs of WT and DARC−/− mice (not shown).
Phagocytosis and Splenic Uptake of RBCs Are Potentiated by HFD
We next sought to determine whether the alterations in RBCs induced by HFD lead to an increase in RBC phagocytosis. In vitro phagocytosis assays performed by using peritoneal macrophages revealed a ≈1.4-fold increase in phagocytosis of HFD-RBCs in comparison with CD-RBCs (Figure 4A). The spleen is a major organ for clearance of senescent RBCs, and splenic monocytes are involved in systemic inflammation. In the mouse, the spleen acts as a depot from which monocytes efflux to sites of inflammation in a triggered manner.21 Hence, we tested whether HFD-RBCs are preferentially taken up by the spleen in comparison with CD-RBCs. We injected labeled packed RBCs retro-orbitally into CD-fed mice, because this is an effective and reliable way to introduce cells into the circulation.22 After euthanization, all mice were perfused with PBS to remove circulating RBCs, and spleens were harvested for analysis. We observed a ≈3-fold increase in splenic uptake of HFD-RBCs in comparison with CD-RBCs (Figure 4B). Judging from the dye pattern, the labeled RBCs were predominately phagocytosed in the marginal zone, which is enriched in monocytes/splenic macrophages.23 It is noteworthy that reduced RBC deformability (Figure 3D and 3G) has been reported to directly promote increased splenic sequestration.24
Figure 4.

Increased phagocytosis and splenic uptake of HFD-RBCs. A, RBC phagocytosis by mouse macrophages. B, Representative images of splenic sections showing uptake of labeled RBCs from CD- or HFD-fed mice in vivo: Top, low-power images of the spleen sections; Bottom, high-power images of the boxed areas. The graph is a quantification of RBC splenic uptake (see text for details). n=3 to 4 (1 dot = 1 mouse); *P<0.05. CD indicates chow diet; HFD, high-fat diet; and RBC, red blood cell.
Phagocytosis of HFD-RBCs Induces a Proinflammatory Phenotype in Macrophages
The increased phagocytosis and splenic uptake of proinflammatory HFD-RBCs prompted us to examine whether HFD-RBCs elicit a proinflammatory shift in the monocytes/macrophages with which they interact. To study the effects on global gene expression in macrophages during RBC phagocytosis, we performed a microarray-based gene expression analysis of the macrophages following in vitro phagocytosis of CD-RBCs and HFD-RBCs. Macrophages elicited from mice on both CD and HFD were used in our assay. As shown in Figure 5A, macrophages from mice on HFD were basally activated toward a proinflammatory phenotype, consistent with previous reports.25 In macrophages isolated from mice on CD, RBC phagocytosis per se yielded a change in the global gene expression pattern, rendering it similar to that in macrophages from mice on HFD: genes involved in leukocyte metabolism/tissue repair were upregulated, whereas the expression of chemoattractants was decreased (Figure 5A, and Figure VI in the online-only Data Supplement). In comparison with phagocytosis of CD-RBCs, phagocytosis of HFD-RBCs by the macrophages from mice on HFD caused a significantly more pronounced upregulation of proinflammatory chemokines known to be involved in atherosclerosis26: microarray results were validated by quantitative real-time polymerase chain reaction to confirm the increased expression of Ccl3, Il1b, and Cxcl2 by ≈2.5 fold, ≈3-fold, and ≈12-fold, respectively (Figure 5A and 5B; Figures VI and VII in the online-only Data Supplement).
Figure 5.

HFD induces a proinflammatory phenotype in RBC-exposed macrophages. A, Heat map, changes in expression of 41 genes in macrophages elicited from chow vs HFD mice and incubated under control conditions (no RBC) or with RBC derived from mice maintained for ≥12 weeks on either CD or HFD; see Methods for details. B through D, qRT-PCR validation of the increased expression of Ccl3, Il1b, and Cxcl2, respectively, in macrophages derived from mice maintained on HFD and exposed to HFD-RBCs vs CD-RBCs. n=3; *P<0.05. CD, chow diet; HFD, high-fat diet; qRT-PCR, quantitative real-time polymerase chain reaction; and RBC, red blood cell.
HFD-RBCs Promote Endothelial–Macrophage Interactions
Activation of the vascular endothelium is one of the earliest events observed in experimental models of atherosclerosis. In humans, endothelial dysfunction is associated with traditional cardiovascular risk factors, including hyperlipidemia, and is a prominent feature of obesity.27 Accordingly, we tested whether exposure of HFD-RBCs to blood vessels leads to endothelial cell activation. We performed an ex vivo experiment wherein we incubated freshly isolated aortic rings with CD-RBCs or HFD-RBCs and then exposed them to fluorescently labeled macrophages. The aortic rings were ≈1 mm thick, and, to ensure the appropriate representation of macrophage adhesion, sections were cut that covered the entire aorta (n=50 per ring). Macrophages that bound to the lumen were counted. Exposure of blood vessels to CD-RBCs had no appreciable effect in comparison with control incubations (in the absence of RBCs); in contrast, HFD-RBCs promoted a ≈3-fold increase in macrophage adhesion to the aortic endothelium (Figure 6).
Figure 6.
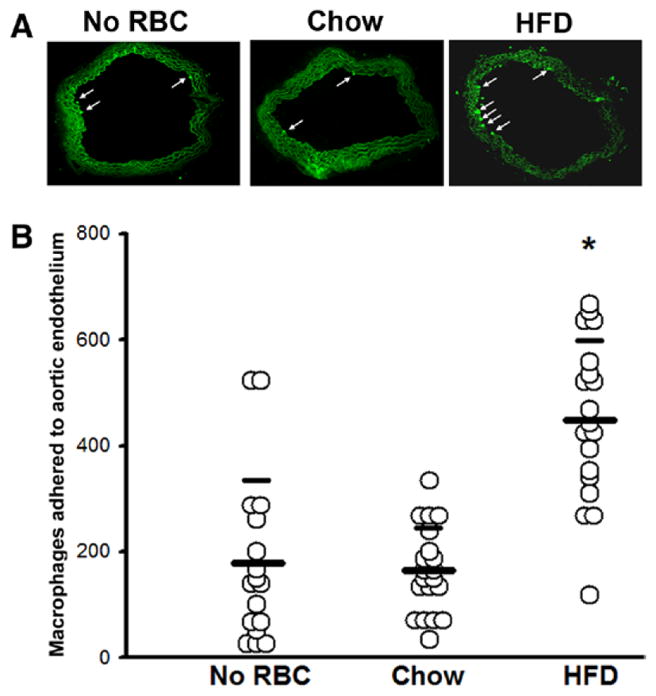
HFD-RBCs increase adhesion of macrophages to the aortic endothelium. Aortic segments were exposed to RBCs from either CD- or HFD-fed mice, washed, and incubated with labeled macrophages; a control group (no RBCs) was also included. The number of macrophages adhered to luminal endothelium was counted in serial sections. A, Representative images (arrows point to macrophages). B, Cumulative data compiled from individual cryosections. *P<0.05. CD indicates chow diet; HFD, high-fat diet; and RBC, red blood cell
Discussion
In this report, we demonstrate that HFD induces dramatic alterations in RBCs which promote inflammatory interactions with macrophages and endothelial cells. These RBC alterations are in part dependent on DARC, because HFD-induced proinflammatory responses in RBCs are abrogated in mice lacking this chemokine receptor. Furthermore, the effects of HFD on RBCs lead to enhanced uptake by the spleen in vivo and by macrophages in vitro. Notably, RBCs become entrapped at sites of intraplaque hemorrhage in advanced atherosclerotic lesions, and uptake of cholesterol from RBC membranes contributes to foam cell formation. This process is likely further accelerated by alterations in RBCs induced by HFD. Thus, our findings may be pertinent to mechanisms of atherosclerosis in the setting of obesity, an emerging worldwide epidemic.
Diets high in saturated fat have long been associated with endothelial dysfunction,28 the precursor to atherosclerosis. To our knowledge, however, the effects of HFD on RBCs have not been previously examined. RBCs can functionally bind to lipoproteins, resulting in lipid transfer and perturbation of membrane fluidity and cell morphology.29 In humans, severe hypercholesterolemia is associated with alterations in RBC cholesterol and membrane fluidity,30 which are improved by treatment with statins.31 However, the majority of obese humans are not severely hypercholesterolemic, as was also the case in the present murine study. The diet we used in this study is rich in saturated fat and sucrose, leading to obesity and glucose intolerance, but the duration was not long enough to induce frank diabetes mellitus. Thus, RBC dysfunction, analogous to endothelial dysfunction, may occur early in the course of diet-induced obesity and function as a marker and, possibly, mediator of atherosclerosis.
HFD caused an increase in MCP-1 and KC levels on the surface of murine RBCs, likely via the increase in binding to DARC, as evidenced by our data with DARC−/− mice (Figure 1). RBC-DARC has been previously shown to bind MCP-1 protein and proposed to regulate its circulating concentration in mice and humans.11,32 The levels of plasma MCP-1 were higher in CD-fed WT mice than in CD-fed DARC−/− mice, suggesting that DARC serves as a buffer-sink for MCP-1 under CD conditions; however, we did not detect increased release of MCP-1 in response to heparin in CD-fed WT mice (Figure 1A). The most likely explanation for this observation is that, at low concentrations, MCP-1 preferentially binds to DARC with high affinity and thus cannot be effectively dislodged by heparin. It is possible that RBC-DARC–bound MCP-1 can elicit vascular inflammation at sites of low flow rates, including atherosclerotic lesions, which contain an extensive microvascular network because of proliferation of vasa vasorum.33 Disruption of the delicate vasa vasorum network is a major cause of intraplaque hemorrhage, which likely contributes to atherosclerosis progression and the development of myocardial infarction.7 MCP-1 released from trapped RBCs may also augment inflammation and thus contribute to this process.
Oxidative stress induces proinflammatory gene expression, antagonizes the beneficial actions of nitric oxide, and precedes the onset of HFD-induced insulin resistance in obesity.34 In this light, we found that HFD induced an increase in the generation of intracellular ROS in murine RBCs, although the mechanism through which this occurs is poorly understood. Increased RBC ROS could potentially diminish NO bound to RBC; however, we were not able to detect a significant reduction in RBC NO/SNO-Hb content in mice fed HFD for 12 weeks, although we cannot exclude the possibility that longer-term HFD feeding would have a greater impact on this parameter. Also, an increase in externalized PS, a marker of senescence in normal RBCs, and a marker for apoptosis in deranged RBCs, as well,35 was noted in HFD-RBCs. Furthermore, as a consequence of increased PS externalization, erythrophagocytosis was increased in HFD-RBCs in an in vitro setting (Figure 4A). These findings may be pertinent with regard to the uptake of extravasated RBCs by macrophages following intraplaque hemorrhage in atherosclerotic lesions.
Swirsky et al21 demonstrated that the spleen acts as a reservoir for undifferentiated monocytes, which are sequestered and subsequently deployed to sites of injury/inflammation. In this context we found that HFD-RBCs are also avidly taken up by the spleen (Figure 4B), where they are phagocytosed by macrophages, especially those surrounding the splenic sinuses. The enhanced phagocytosis of HFD-RBCs by macrophages could potentially act as a trigger for the splenic recruitment and deployment of monocytes, thus influencing atherosclerosis. Considering that splenectomized mice exhibit accelerated atherosclerosis in comparison with sham-operated controls,36 it is tempting to speculate that sequestration of monocytes and RBCs in the spleen in the setting of HFD plays a protective role in atherosclerosis. However, it is important to recognize that the spleen also plays a broad role in regulating immune function, particularly antibody production, and responses to infections. Thus, the role of the spleen in regulating atherosclerosis is likely very complex and merits further investigation.
In macrophages derived from HFD mice, phagocytosis of HFD-RBCs caused a more pronounced upregulation of several proinflammatory genes than phagocytosis of CD-RBC; most notably, Il1b, Ccl3, and Cxcl2 expression was markedly upregulated (Figure 5). Bone marrow from Ccl3(−/−) mice, when transplanted to LDLr(−/−) mice maintained on Western diet, yielded milder atherosclerotic lesions and lower blood cholesterol and triglycerides, implying a role for bone marrow–derived Ccl3 in atherosclerosis; recent studies have implicated Ccl3 (macrophage inflammatory protein-1α) in altered lipid metabolism in hyperlipidemic mice.37 Macrophage secretion of interleukin-1β has likewise been implicated in atherosclerosis.38,39 Because HFD-RBCs carry more cholesterol, phagocytosis of HFD-RBCs by macrophages may trigger the cholesterol-mediated inflammasome pathway,40 thereby upregulating the expression of these proinflammatory molecules.
Finally, we studied the impact of HFD-RBCs on vascular endothelium by incubating the cells with aortic ring segments. We found that HFD-RBCs caused activation of luminal endothelium resulting in increased monocyte binding (Figure 6). Our findings suggest that the erythrocyte dysfunction induced by HFD may be sufficient to fuel endothelial inflammation, thereby contributing to the development of atherosclerosis. Such interactions between RBCs and endothelium are also likely to occur in the microvasculature and may play a role in triggering inflammation in other organs and tissues. The mechanisms whereby HFD-RBCs trigger endothelial activation are likely complex and may involve soluble factors, and cell contact–dependent processes, as well.
In summary, our findings suggest that RBCs play an important role in linking HFD to endothelial dysfunction and activation of macrophages in the pathogenesis of obesity-related atherosclerosis (Figure 7). We propose that erythrocyte dysfunction induced by HFD may be a novel and significant contributor to and mediator of atherogenesis.
Figure 7.

Schematic representation of RBC-mediated processes fueling chronic inflammation in the setting of HFD. RBC dysfunction is promoted by several concomitantly acting mechanisms, both dependent on and independent of DARC on RBC surface (text in purple and black, respectively), leading to an increase in the levels of chemokines in the vasculature, enhanced EC-monocyte interactions, and heightened vascular inflammation. Dashed arrows comprising a possible amplification loop are hypothetical. DARC indicates Duffy antigen receptor for chemokines; EC, endothelial cell; HFD, high-fat diet; PS, phosphatidylserine; and RBC, red blood cell.
Supplementary Material
Suppl. Fig 1: Weight gain (A), fasting glucose (B), and Hb1AC levels (C) in WT and DARC−/− mice maintained on chow and HFD. Animals were fed an obesogenic, 60% fat diet for 12 weeks. n=3–4; *, P<0.05.
Suppl. Fig 2: Levels of MCP-1 protein secreted by subcutaneous adipose tissues are similar in WT and DARC−/− mice maintained on chow and HFD. n=3; *, P<0.05.
Suppl. Fig 3: Levels of CCL11, CXCL5, and CXCL12 on RBC membranes of WT and DARC−/− mice maintained on chow and HFD. n=5; *, P<0.05.
Suppl. Fig 4: Levels of RBC-DARC are unaffected by HFD. A, representative flow cytometry experiment; B, calculated mean fluorescence in graph form; n=3, P<0.05.
Suppl. Fig 5: Levels of SNO-Hb as assessed by biotin switch assay are unaffected by HFD. A, representative western blot; B, calculated mean levels by analysis of relative densitometry in graph form; n=4–5, n.s. chow vs HFD.
Suppl. Fig. 6: Complete unsupervised clustering analysis, changes in gene expression (n=642) observed in macrophages isolated from WT mice maintained on CD or HFD, and exposed to either CD-RBC or HFD-RBC. Note that the bulk of the RBC response signature is recapitulated by HFD-exposed macrophages not exposed to RBC.
Suppl. Fig. 7: Pathway map depicting the major physiological and pathophysiological pathways (group color coded squares: green and brown, respectively) and target genes (red hexagons) affected by the changes in gene expression depicted in Suppl. Fig. 6. The map was compiled using ToppGene Suite (http://toppgene.cchmc.org/). In light green: pathways highly specific for the cells of myeloid lineage as per the compendium maintained by Immunological Genome Project (www.immgen.org).
Clinical Perspective.
High-fat diet (HFD) promotes endothelial dysfunction and proinflammatory monocyte activation, which contribute to atherosclerosis in obesity. Here, we examined the impact of HFD on red blood cells (RBCs), which may play an important modulatory role in atherosclerosis by binding inflammatory chemokines and interacting with macrophages and endothelium within atherosclerotic plaques. We detected a marked increase in the level of chemokines bound to the RBCs of mice fed a 60% HFD for 12 weeks in comparison with mice fed a normal chow diet. Further investigations demonstrated that these chemokines were bound to RBCs via the Duffy antigen receptor for chemokines. Exposure of RBCs from HFD-fed mice to an endothelial monolayer in vitro significantly enhanced macrophage transendothelial migration, confirming the functional importance of RBC-bound chemokines in the setting of HFD. In addition to increasing the level of chemokines bound to RBCs, HFD increased RBC membrane cholesterol content and phosphatidylserine externalization (a marker or RBC damage or senescence), fostering RBC-macrophage inflammatory interactions and promoting the uptake of RBC by macrophages in vitro and by the spleen in vivo. Finally, RBCs from HFD-fed mice augmented macrophage adhesion to the endothelium when incubated with isolated aortic segments, indicating endothelial activation. We propose that RBC dysfunction, analogous to endothelial dysfunction, occurs early during diet-induced obesity and may serve as a mediator of atherosclerosis. These findings may have implications for the pathogenesis of atherosclerosis in obesity, a worldwide epidemic.
Acknowledgments
Authorship: D. Unruh, R. Srinivasan, T. Benson, S. Haigh, D. Coyle, N. Batra, R. Keil, R. Sturm, V. Blanco, M. Palascak, W. Tong, T. Kalfa, and D. Manka designed experiments, performed research, and analyzed data; R.S. Franco, T. Chatterjee, D.H. Hui, W.S. Davidson, B.J. Aronow, A. Peairs, A. Blomkalns, D. Fulton, and J. Brittain participated in designing experiments, analyzed data, and wrote the article; N.L. Weintraub and V.Y. Bogdanov designed research, analyzed data, and wrote the article.
Footnotes
The online-only Data Supplement is available with this article at http://circ.ahajournals.org/lookup/suppl/doi:10.1161/CIRCULATIONAHA.115.017313/-/DC1.
Disclosures
None.
References
- 1.Bornfeldt KE, Tabas I. Insulin resistance, hyperglycemia, and atherosclerosis. Cell Metab. 2011;14:575–585. doi: 10.1016/j.cmet.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med. 2012;18:363–374. doi: 10.1038/nm.2627. [DOI] [PubMed] [Google Scholar]
- 3.Gower RM, Wu H, Foster GA, Devaraj S, Jialal I, Ballantyne CM, Knowlton AA, Simon SI. CD11c/CD18 expression is upregulated on blood monocytes during hypertriglyceridemia and enhances adhesion to vascular cell adhesion molecule-1. Arterioscler Thromb Vasc Biol. 2011;31:160–166. doi: 10.1161/ATVBAHA.110.215434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rothe G, Herr AS, Stöhr J, Abletshauser C, Weidinger G, Schmitz G. A more mature phenotype of blood mononuclear phagocytes is induced by fluvastatin treatment in hypercholesterolemic patients with coronary heart disease. Atherosclerosis. 1999;144:251–261. doi: 10.1016/s0021-9150(99)00061-1. [DOI] [PubMed] [Google Scholar]
- 6.Schnabel RB, Baumert J, Barbalic M, Dupuis J, Ellinor PT, Durda P, Dehghan A, Bis JC, Illig T, Morrison AC, Jenny NS, Keaney JF, Jr, Gieger C, Tilley C, Yamamoto JF, Khuseyinova N, Heiss G, Doyle M, Blankenberg S, Herder C, Walston JD, Zhu Y, Vasan RS, Klopp N, Boerwinkle E, Larson MG, Psaty BM, Peters A, Ballantyne CM, Witteman JC, Hoogeveen RC, Benjamin EJ, Koenig W, Tracy RP. Duffy antigen receptor for chemokines (Darc) polymorphism regulates circulating concentrations of monocyte chemoattractant protein-1 and other inflammatory mediators. Blood. 2010;115:5289–5299. doi: 10.1182/blood-2009-05-221382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kolodgie FD, Gold HK, Burke AP, Fowler DR, Kruth HS, Weber DK, Farb A, Guerrero LJ, Hayase M, Kutys R, Narula J, Finn AV, Virmani R. Intraplaque hemorrhage and progression of coronary atheroma. N Engl J Med. 2003;349:2316–2325. doi: 10.1056/NEJMoa035655. [DOI] [PubMed] [Google Scholar]
- 8.Virmani R, Roberts WC. Extravasated erythrocytes, iron, and fibrin in atherosclerotic plaques of coronary arteries in fatal coronary heart disease and their relation to luminal thrombus: frequency and significance in 57 necropsy patients and in 2958 five mm segments of 224 major epicardial coronary arteries. Am Heart J. 1983;105:788–797. doi: 10.1016/0002-8703(83)90242-9. [DOI] [PubMed] [Google Scholar]
- 9.Barber LA, Palascak MB, Joiner CH, Franco RS. Aminophospholipid translocase and phospholipid scramblase activities in sickle erythrocyte subpopulations. Br J Haematol. 2009;146:447–455. doi: 10.1111/j.1365-2141.2009.07760.x. [DOI] [PubMed] [Google Scholar]
- 10.Hanahan DJ, Ekholm JE. The preparation of red cell ghosts (membranes) Methods Enzymol. 1974;31:168–172. doi: 10.1016/0076-6879(74)31018-x. [DOI] [PubMed] [Google Scholar]
- 11.Fukuma N, Akimitsu N, Hamamoto H, Kusuhara H, Sugiyama Y, Sekimizu K. A role of the Duffy antigen for the maintenance of plasma chemokine concentrations. Biochem Biophys Res Commun. 2003;303:137–139. doi: 10.1016/s0006-291x(03)00293-6. [DOI] [PubMed] [Google Scholar]
- 12.Bäck M, Weber C, Lutgens E. Regulation of atherosclerotic plaque inflammation (published online ahead of print March 30 2015) [Accessed October 12, 2015];J Intern Med. doi: 10.1111/joim.12367. http://onlinelibrary.wiley.com/doi/10.1111/joim.12367/abstract;jsessionid=38ECD3970FD682FDD1B00D94734862E9.f04t03. [DOI] [PubMed]
- 13.Hansell CA, Hurson CE, Nibbs RJ. DARC and D6: silent partners in chemokine regulation? Immunol Cell Biol. 2011;89:197–206. doi: 10.1038/icb.2010.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Emanuele E, Falcone C, D’Angelo A, Minoretti P, Buzzi MP, Bertona M, Geroldi D. Association of plasma eotaxin levels with the presence and extent of angiographic coronary artery disease. Atherosclerosis. 2006;186:140–145. doi: 10.1016/j.atherosclerosis.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 15.Rousselle A, Qadri F, Leukel L, Yilmaz R, Fontaine JF, Sihn G, Bader M, Ahluwalia A, Duchene J. CXCL5 limits macrophage foam cell formation in atherosclerosis. J Clin Invest. 2013;123:1343–1347. doi: 10.1172/JCI66580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Döring Y, Pawig L, Weber C, Noels H. The CXCL12/CXCR4 chemokine ligand/receptor axis in cardiovascular disease. Front Physiol. 2014;5:212. doi: 10.3389/fphys.2014.00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dasgupta SK, Abdel-Monem H, Guchhait P, Nagata S, Thiagarajan P. Role of lactadherin in the clearance of phosphatidylserine-expressing red blood cells. Transfusion. 2008;48:2370–2376. doi: 10.1111/j.1537-2995.2008.01841.x. [DOI] [PubMed] [Google Scholar]
- 18.Hale JP, Winlove CP, Petrov PG. Effect of hydroperoxides on red blood cell membrane mechanical properties. Biophys J. 2011;101:1921–1929. doi: 10.1016/j.bpj.2011.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hardeman MR, Ince C. Clinical potential of in vitro measured red cell deformability, a myth? Clin Hemorheol Microcirc. 1999;21:277–284. [PubMed] [Google Scholar]
- 20.Grau M, Pauly S, Ali J, Walpurgis K, Thevis M, Bloch W, Suhr F. RBC-NOS-dependent S-nitrosylation of cytoskeletal proteins improves RBC deformability. PLoS One. 2013;8:e56759. doi: 10.1371/journal.pone.0056759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Price JE, Barth RF, Johnson CW, Staubus AE. Injection of cells and monoclonal antibodies into mice: comparison of tail vein and retroorbital routes. Proc Soc Exp Biol Med. 1984;177:347–353. doi: 10.3181/00379727-177-41955. [DOI] [PubMed] [Google Scholar]
- 23.Dijkstra CD, Van Vliet E, Döpp EA, van der Lelij AA, Kraal G. Marginal zone macrophages identified by a monoclonal antibody: characterization of immuno- and enzyme-histochemical properties and functional capacities. Immunology. 1985;55:23–30. [PMC free article] [PubMed] [Google Scholar]
- 24.Safeukui I, Buffet PA, Deplaine G, Perrot S, Brousse V, Ndour A, Nguyen M, Mercereau-Puijalon O, David PH, Milon G, Mohandas N. Quantitative assessment of sensing and sequestration of spherocytic erythrocytes by the human spleen. Blood. 2012;120:424–430. doi: 10.1182/blood-2012-01-404103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wallace FA, Neely SJ, Miles EA, Calder PC. Dietary fats affect macrophage-mediated cytotoxicity towards tumour cells. Immunol Cell Biol. 2000;78:40–48. doi: 10.1046/j.1440-1711.2000.00867.x. [DOI] [PubMed] [Google Scholar]
- 26.Kleemann R, Zadelaar S, Kooistra T. Cytokines and atherosclerosis: a comprehensive review of studies in mice. Cardiovasc Res. 2008;79:360–376. doi: 10.1093/cvr/cvn120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vita JA. Endothelial function. Circulation. 2011;124:e906–e912. doi: 10.1161/CIRCULATIONAHA.111.078824. [DOI] [PubMed] [Google Scholar]
- 28.Davis N, Katz S, Wylie-Rosett J. The effect of diet on endothelial function. Cardiol Rev. 2007;15:62–66. doi: 10.1097/01.crd.0000218824.79018.cd. [DOI] [PubMed] [Google Scholar]
- 29.Hui DY, Harmony JA. Interaction of plasma lipoproteins with erythrocytes. I. Alteration of erythrocyte morphology. Biochim Biophys Acta. 1979;550:407–424. doi: 10.1016/0005-2736(79)90145-7. [DOI] [PubMed] [Google Scholar]
- 30.Cooper RA. Abnormalities of cell-membrane fluidity in the pathogenesis of disease. N Engl J Med. 1977;297:371–377. doi: 10.1056/NEJM197708182970707. [DOI] [PubMed] [Google Scholar]
- 31.Koter M, Broncel M, Chojnowska-Jezierska J, Klikczynska K, Franiak I. The effect of atorvastatin on erythrocyte membranes and serum lipids in patients with type-2 hypercholesterolemia. Eur J Clin Pharmacol. 2002;58:501–506. doi: 10.1007/s00228-002-0507-9. [DOI] [PubMed] [Google Scholar]
- 32.Jilma-Stohlawetz P, Homoncik M, Drucker C, Marsik C, Rot A, Mayr WR, Seibold B, Jilma B. Fy phenotype and gender determine plasma levels of monocyte chemotactic protein. Transfusion. 2001;41:378–381. doi: 10.1046/j.1537-2995.2001.41030378.x. [DOI] [PubMed] [Google Scholar]
- 33.Barger AC, Beeuwkes R, 3rd, Lainey LL, Silverman KJ. Hypothesis: vasa vasorum and neovascularization of human coronary arteries. A possible role in the pathophysiology of atherosclerosis. N Engl J Med. 1984;310:175–177. doi: 10.1056/NEJM198401193100307. [DOI] [PubMed] [Google Scholar]
- 34.Matsuzawa-Nagata N, Takamura T, Ando H, Nakamura S, Kurita S, Misu H, Ota T, Yokoyama M, Honda M, Miyamoto K, Kaneko S. Increased oxidative stress precedes the onset of high-fat diet-induced insulin resistance and obesity. Metabolism. 2008;57:1071–1077. doi: 10.1016/j.metabol.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 35.Connor J, Pak CC, Schroit AJ. Exposure of phosphatidylserine in the outer leaflet of human red blood cells. Relationship to cell density, cell age, and clearance by mononuclear cells. J Biol Chem. 1994;269:2399–2404. [PubMed] [Google Scholar]
- 36.Rezende AB, Neto NN, Fernandes LR, Ribeiro AC, Alvarez-Leite JI, Teixeira HC. Splenectomy increases atherosclerotic lesions in apolipoprotein E deficient mice. J Surg Res. 2011;171:e231–e236. doi: 10.1016/j.jss.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 37.Kennedy A, Gruen ML, Gutierrez DA, Surmi BK, Orr JS, Webb CD, Hasty AH. Impact of macrophage inflammatory protein-1α deficiency on atherosclerotic lesion formation, hepatic steatosis, and adipose tissue expansion. PLoS One. 2012;7:e31508. doi: 10.1371/journal.pone.0031508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galea J, Armstrong J, Gadsdon P, Holden H, Francis SE, Holt CM. Interleukin-1 beta in coronary arteries of patients with ischemic heart disease. Arterioscler Thromb Vasc Biol. 1996;16:1000–1006. doi: 10.1161/01.atv.16.8.1000. [DOI] [PubMed] [Google Scholar]
- 39.Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, Asano M, Moriwaki H, Seishima M. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2003;23:656–660. doi: 10.1161/01.ATV.0000064374.15232.C3. [DOI] [PubMed] [Google Scholar]
- 40.Ordovas-Montanes JM, Ordovas JM. Cholesterol, Inflammasomes, and Atherogenesis. Curr Cardiovasc Risk Rep. 2012;6:45–52. doi: 10.1007/s12170-011-0212-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppl. Fig 1: Weight gain (A), fasting glucose (B), and Hb1AC levels (C) in WT and DARC−/− mice maintained on chow and HFD. Animals were fed an obesogenic, 60% fat diet for 12 weeks. n=3–4; *, P<0.05.
Suppl. Fig 2: Levels of MCP-1 protein secreted by subcutaneous adipose tissues are similar in WT and DARC−/− mice maintained on chow and HFD. n=3; *, P<0.05.
Suppl. Fig 3: Levels of CCL11, CXCL5, and CXCL12 on RBC membranes of WT and DARC−/− mice maintained on chow and HFD. n=5; *, P<0.05.
Suppl. Fig 4: Levels of RBC-DARC are unaffected by HFD. A, representative flow cytometry experiment; B, calculated mean fluorescence in graph form; n=3, P<0.05.
Suppl. Fig 5: Levels of SNO-Hb as assessed by biotin switch assay are unaffected by HFD. A, representative western blot; B, calculated mean levels by analysis of relative densitometry in graph form; n=4–5, n.s. chow vs HFD.
Suppl. Fig. 6: Complete unsupervised clustering analysis, changes in gene expression (n=642) observed in macrophages isolated from WT mice maintained on CD or HFD, and exposed to either CD-RBC or HFD-RBC. Note that the bulk of the RBC response signature is recapitulated by HFD-exposed macrophages not exposed to RBC.
Suppl. Fig. 7: Pathway map depicting the major physiological and pathophysiological pathways (group color coded squares: green and brown, respectively) and target genes (red hexagons) affected by the changes in gene expression depicted in Suppl. Fig. 6. The map was compiled using ToppGene Suite (http://toppgene.cchmc.org/). In light green: pathways highly specific for the cells of myeloid lineage as per the compendium maintained by Immunological Genome Project (www.immgen.org).