

Supplemental Digital Content is available in the text.
Keywords: coronary artery disease, flow-mediated dilation, microvascular dysfunction, mitochondria, reactive oxygen species, telomerase activity, vascular biology
Abstract
Rationale:
Telomerase is a nuclear regulator of telomere elongation with recent reports suggesting a role in regulation of mitochondrial reactive oxygen species. Flow-mediated dilation in patients with cardiovascular disease is dependent on the formation of reactive oxygen species.
Objective:
We examined the hypothesis that telomerase activity modulates microvascular flow-mediated dilation, and loss of telomerase activity contributes to the change of mediator from nitric oxide to mitochondrial hydrogen peroxide in patients with coronary artery disease (CAD).
Methods and Results:
Human coronary and adipose arterioles were isolated for videomicroscopy. Flow-mediated dilation was measured in vessels pretreated with the telomerase inhibitor BIBR-1532 or vehicle. Statistical differences between groups were determined using a 2-way analysis of variance repeated measure (n≥4; P<0.05). L-NAME (Nω-nitro-L-arginine methyl ester; nitric oxide synthase inhibitor) abolished flow-mediated dilation in arterioles from subjects without CAD, whereas polyethylene glycol-catalase (PEG-catalase; hydrogen peroxide scavenger) had no effect. After exposure to BIBR-1532, arterioles from non-CAD subjects maintained the magnitude of dilation but changed the mediator from nitric oxide to mitochondrial hydrogen peroxide (% max diameter at 100 cm H2O: vehicle 74.6±4.1, L-NAME 37.0±2.0*, PEG-catalase 82.1±2.8; BIBR-1532 69.9±4.0, L-NAME 84.7±2.2, PEG-catalase 36.5±6.9*). Conversely, treatment of microvessels from CAD patients with the telomerase activator AGS 499 converted the PEG-catalase-inhibitable dilation to one mediated by nitric oxide (% max diameter at 100 cm H2O: adipose, AGS 499 78.5±3.9; L-NAME 10.9±17.5*; PEG-catalase 79.2±4.9). Endothelial-independent dilation was not altered with either treatment.
Conclusions:
We have identified a novel role for telomerase in re-establishing a physiological mechanism of vasodilation in arterioles from subjects with CAD. These findings suggest a new target for reducing the oxidative milieu in the microvasculature of patients with CAD.
Telomerase is classically known as an enzyme that maintains telomere length in nuclear DNA. Telomerase activity is inversely related to endothelial senescence,1 especially in atherosclerotic prone vessels,2 and may attenuate premature coronary artery disease (CAD) and myocardial infarction.3 Increased telomerase activity can also protect against reactive oxygen species (ROS)–induced endothelial dysfunction.4,5
In This Issue, see p 771
Editorial, see p 781
Not all of the effects of telomerase can be easily explained by its nuclear actions. Growing evidence suggests that TERT, the catalytic subunit of telomerase, can reversibly translocate from the nucleus to organelles (including the mitochondria), in a dose- and time-dependent manner, in response to stressors, such as hydrogen peroxide (H2O2) or prolonged hypoxia.6–8 Yet no physiological effects of this translocation have been described in connection with cardiovascular phenotypes.
Once translocated to the mitochondria, TERT can suppress mitochondrial ROS (mtROS) production.6,7,9 TERT-overexpressing cells demonstrate lower mitochondrial superoxide (O2–·) production,6 whereas in conditions of reduced telomerase activity, mtROS increases, contributing to cellular oxidative stress.6,10 Whether this system is active in the vascular endothelium and whether it contributes to redox regulation of vasodilation is not known.
We have shown previously that in atrial and adipose vessels from human subjects without CAD, nitric oxide (NO) is the primary mediator of endothelium-dependent dilation in response to increased shear stress (flow-mediated dilation [FMD]).11,12 In CAD, FMD persists but is mediated almost exclusively by H2O2 released from the mitochondria, independent of nitric oxide synthase (NOS).13,14 Telomerase can modulate the cellular redox state by upregulation of mitochondrial antioxidant enzymes15 and by directly reducing mtROS production,16 making it ideally suited for contributing to the switch from NO to mitochondrial-derived H2O2 as the mediator of FMD in subjects with CAD.
Eitan et al have recently described a specific activator of telomerase that reduces mtROS. The small molecule AGS 499 was confirmed to act in a telomerase-dependent manner because in telomerase-depleted cells (by shRNA or knockout), AGS 499 did not elicit protection against increased oxidative stress.17,18 These studies also showed that AGS 499 does not act nonspecifically as an antioxidant but specifically increases extranuclear levels of TERT, including mitochondrial TERT.4 AGS 499 is beneficial in a variety of disease conditions characterized by elevations in mtROS.17,18 We hypothesized that a reduction in telomerase activity, insufficient to reduce telomere length, evokes mtROS production and reduces NO synthesis, recapitulating the phenotype observed in CAD where H2O2 mediates FMD. Conversely, in vessels from subjects with CAD and elevated mtROS, we tested whether short-term telomerase activation could restore NO-mediated dilation.
Methods
Tissue Acquisition and General Protocol
All protocols were approved by the Institutional Review Board of the Medical College of Wisconsin and Froedtert Hospital. Sections of human atrial and adipose (visceral and subcutaneous) tissue were obtained in deidentified fashion as otherwise discarded tissue at the time of surgery and placed in cold 4°C HEPES (NaCl 275 mmol/L, KCL 7.99 mmol/L, MgSO4 4.9 mmol/L, CaCl2·2H2O 3.2 mmol/L, KH2PO4 2.35 mmol/L, EDTA 0.07 mmol/L, glucose 12 mmol/L, HEPES acid 20 mmol/L; adipose) or cardioplegic (atrial) buffer solution. Tissue from subjects without known cardiovascular risk factors or clinical diagnosis of CAD was used for non-CAD groups.
Cannulated Artery Preparation
Coronary and adipose arterioles (50–200 μm inner diameter) were cleaned of fat and connective tissue and prepared for continuous measurements of diameter.13,14 Briefly, in an organ chamber, both ends of the vessel were cannulated with glass micropipettes filled with physiological saline solution and pressurized (60 mm Hg) for videomicroscopy of diameter change as we have described previously.19,20
Vascular Response to Flow and Pharmacological Interventions
FMD was measured as described by Kuo et al21 using pipettes of identical impedance. Changing the height of each reservoir in equal and opposite directions generates flow without changes in vessel central pressure.21 Data are reported as diameter at a given pressure gradient. Pressure gradients of 5 to 100 cm H2O were generated, assessing steady-state diameter and flow after each change, representing estimated shear rates of 5 to 25 dynes/cm2. Two flow-response curves were generated for each vessel comparing no treatment (vehicle) to effects of pharmacological inhibitors (Nω-nitro-L-arginine methyl ester [L-NAME; 100 μmol/L]; polyethylene glycol-catalase (PEG-catalase; 500 U/mL]; rotenone [1 μmol/L]; 2-(4-carboxyphenyl)-4,5-dihydro-4,4,5,5-tetramethyl-1H-imidazol-1-yloxy-3-oxide (c-PTIO) [1 μmol/L]), actinomycin D (1.59 μmol/L), and MitoTempol (10 µmol/L). In some cases, vessels were incubated for 15 to 20 hours in endothelial cell growth medium containing 5% serum (Lonza) containing either vehicle (DMSO 2% by volume), BIBR-1532 (10 µmol/L), or AGS 499 (20 nmol/L). After incubation, media was replaced with Krebs buffer, intraluminal pressure was raised from 30 mm Hg to 60 mm Hg, and vessels were constricted with ET-1 (endothelin-1) for assessment of FMD.
All pharmacological agents were added to the external bathing solution (volume added was <1% of the circulating external bath solution). Concentrations are stated as final concentrations in organ bath.
Vessels were constricted with ET-1 (0.1–1 nmol/L) to achieve a 20% to 50% stable reduction in passive diameter. Dose response curves to the endothelial-dependent vasodilator acetylcholine (ACh; 1 nmol/L to 100 µmol/L) and the endothelial-independent vasodilator papaverine (0.1 nmol/L to 100 µmol/L) were performed to evaluate specific endothelial and smooth muscle–dependent dilation. At the end of each experiment, papaverine (100 µmol/L) was used to determine the maximal (passive) diameter at 60 mm Hg.
Quantitative Real-Time Polymerase Chain Reaction
Total mRNA was harvested from human umbilical vein endothelial cells (HUVECs) using an Ambion PureLink RNA Kit. Approximately 1500 ng of RNA was used to synthesize cDNA using the Applied Biosystems High Capacity cDNA Reverse Transcription Kit. Gene expression was quantified by real-time-quantitative polymerase chain reaction using primers (Hs_TERT_1_SG QuantiTect Primer Assay QT00073409) and SYBR green from Qiagen in a BioRad CFX96 Touch Real-Time PCR Detection System. Expression levels were normalized to 18S rRNA (Hs_RRN18S_1_SG QT00199367).
Cell Culture Experiments
HUVECs were freshly isolated from umbilical cords collected from local hospitals, processed by the hybridoma core facility of the Blood Research Institute of Wisconsin, and supplied at passage 3 to 5. Cells were exposed to AGS 499 for identical time and conditions as isolated vessels. For stressing cells in culture, heat shock treatment was used wherein culture dishes were sealed with Parafilm and immersed in a water bath at 42°C for 30 minutes. Subsequently, the cells were put into a 37°C incubator for 16 hours of recovery. Control cells were sealed for 30 minutes but remained in the incubator.
Normal human fibroblasts were cultured under standard conditions as described previously.9 In brief, cells were transiently transfected with either whole cell TERT or R3E/R6E TERT (nucTERT). Oxidative stress was generated by 30 minutes of 200 μmol/L H2O2 treatment. Purity of isolated mitochondria preparation was tested using the standard subcellular markers tubulin (cytoplasmic), mitochondrial heat shock proteins 70, and Ku80 (nuclear isoform [higher molecular weight] and mitochondrial isoform). Mitochondria were labeled with Mitotracker Red using manufacturer’s specifications.
Measurement of Mitochondrial Reactive Oxygen Species
In vessels, Mito Peroxy Yellow 1 (MitoPY1)22 was used to evaluate microvessel generation of mtH2O2. As previously described,12 after cannulation in a warmed chamber (37°C) containing HEPES buffer at pH 7.4, arterioles were perfused intraluminally with MitoPY1 (5 μmol/L, 1 hour) at low levels of flow, below the threshold for dilation, until the luminal surface was bathed in MitoPY1 containing buffer. Next the transvascular pressure gradient was changed from zero to 100 cm H2O, and fluorescence measured after 5 minutes. Experiments were performed in the presence or absence of PEG-catalase (500 U/mL). Fluorescence was evaluated with a Nikon Eclipse TE200 microscope using a krypton/argon laser at excitation wavelength of 488 nm and measured emission between 530 and 590 nm. All comparisons were made using vessels studied at the same session with constant microscope image display settings. In cultured cells, mitoSox was used to measure superoxide generation. Cells were incubated for 30 minutes with 10 μmol/L mitoSox. Relative average fluorescence intensity was normalized to background fluorescence and presented as percent change from baseline.
Telomere Length
Genomic DNA from left ventricle tissue or isolated atrial arterioles was prepared by the Medical College of Wisconsin (MCW) pathology tissue bank using Qiagen spin columns according to manufacturer’s specifications. Polymerase chain reaction reaction was performed under following conditions: 0.75 SYBR Green I (Invitrogen), 10 mmol/L Tris–HCl pH8.3, 50 mmol/L KCl, 3 mmol/L MgCl2, 0.2 mmol/L each dNTP (deoxynucleotides), 1 mmol/L DTT (dithiothreitol), and 1 mol/L betaine (US Biochemicals). For 25 mL reaction, 0.625U AmpliTaq Gold DNA polymerase (Applied Biosystems, Inc.) was used. Multiplex QPCR primer pair final concentrations were 900 nmol/L each. Telomere primers were telg, ACACTAAGGTTTGGGTTTGGGTTTGGGTTTGGGTTAGTGT; telc, TGTTAGGTATCCCTATCCCTATCCCTATCCCTATCCCT- AACA human albumin primers albu: CGGCGGCGGGCGGCGC- GGGCTGGGCGG-aaatgctgcacagaatccttg; albd: GCCCGGCCCGC-CGCGCCCGTCCCGCCG-gaaaagcatggtcgcctgtt. Polymerase chain reaction was performed as follows: step 1 (1X): 95°C for 15 minutes; Step 2 (36X): 98°C for 2 s, 48°C for 1 min, 74°C for 15 s (data acquisition), 84°C for 30 s, 85°C for 15 s (data acquisition) as previously described by Morgan et al.23
Materials
The telomerase modulators BIBR-1532 (Tocris Bioscience) and AGS 499 (Ester Priel, PhD, Israel) and the mitochondrial ROS-detecting dyes MitoPY1 (Cayman chemicals) and mitoSox (life technologies) were prepared in DMSO. ET-1 (Peninsula Laboratories) was prepared in saline with 1% bovine serum albumin. All other chemicals were obtained from Sigma-Aldrich and prepared in distilled water or physiological salt solution. All concentrations represent the final concentrations in the organ bath.
Statistical Methods
Data are presented as mean±SEM. For all flow response curves, differences between groups at each concentration were determined using a 2-way, repeated-measures analysis of variance. A post hoc Tukey’s test was used for individual comparisons. A probability value of P<0.05 was considered to be statistically significant. For fluorescence studies, telomere length, Western blot, or expression analysis, either a paired t test or 1-way analysis of variance with post hoc Tukey’s test was used to identify individual differences within groups. Values outside of 2 standard deviations of the mean were consider statistical outliers and excluded from analysis.
Results
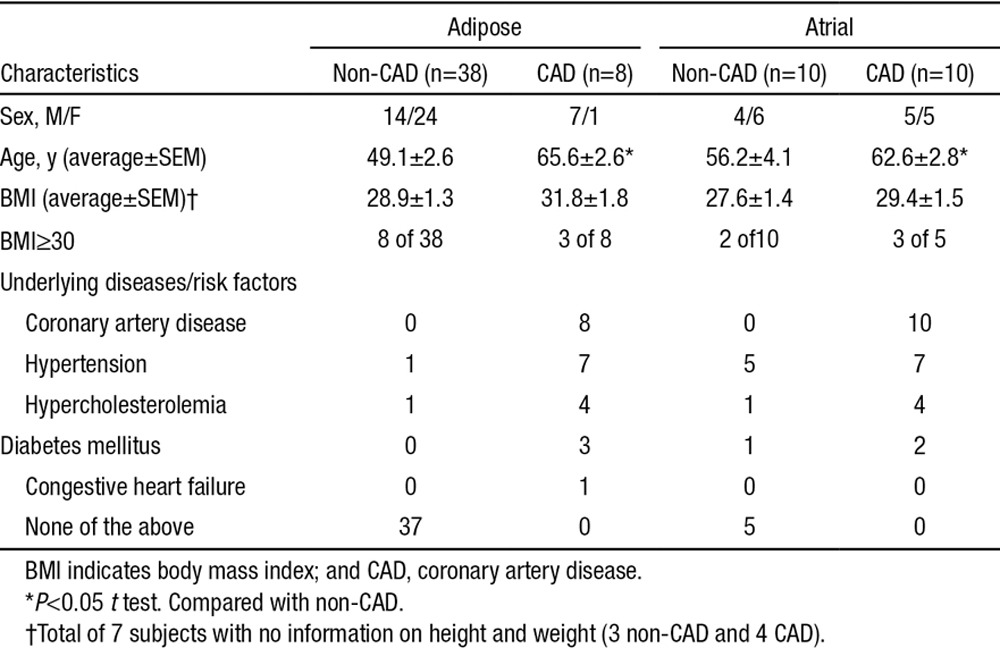
Tissue was collected from a total of 58 subjects. Results are derived from arterioles of 36 patients without CAD and 18 patients with CAD. Detailed patient demographic information is shown in Table.
Table.
Patient Characteristics for Microvessels Used in Study
Effect on FMD of Inhibiting Telomerase
After incubating atrial or adipose arterioles for 15 to 20 hours in the specific telomerase inhibitor BIBR-1532, the magnitude of FMD was not changed (Figure 1A). However, the mediator of FMD shifted from NO to H2O2 (PEG-catalase inhibitable and L-NAME-insensitive) in both vessel types after treatment with BIRB1532 for 15 to 20 hours (Figure 1B and 1C). Endothelial-dependent dilation to ACh was also reduced after telomerase inhibition (Figure 1D). Maximal endothelium-independent dilation (assessed with papaverine) was not altered by BIBR-532 (Figure 1E).
Figure 1.
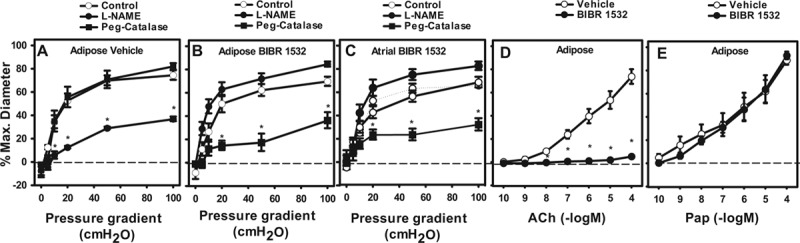
Telomerase inhibition replicates the coronary artery disease (CAD) vascular phenotype in human adipose and atrial vessels from non-CAD subjects. Flow-mediated dilation (FMD) was evaluated in isolated microvessels treated with vehicle (A; adipose), BIBR-1532 (B; adipose), and BIBR-1532 (C; atrial). In BIBR-treated vessels, the mechanism of FMD changed from nitric oxide (NO) to hydrogen peroxide (H2O2). D, Acetylcholine (ACh)-induced dilation was virtually eliminated after telomerase inhibition. E, No change in endothelium-independent dilation to papaverine was observed. N=4 to 12 adipose, N=5 to 9 atrial. *P<0.05 2-way analysis of variance (ANOVA) RM Tukey post hoc analysis. L-NAME indicates Nω-nitro-L-arginine methyl ester; and Peg-Catalase, polyethylene glycol-catalase.
Subjects With CAD Have Reduced Cardiac and Vascular TERT Levels but Normal Telomere Length
To establish whether the difference in vasodilator mechanism between CAD and non-CAD subjects correlates with telomere length, we measured telomere length in heart and microvessels from individuals with and without CAD. No difference in telomere length was observed between vessels from patients with or those without CAD. TERT protein expression was significantly higher in non-CAD subjects versus those with CAD (Figure 2), although CAD subjects tended to be older (66±4 versus 48±7 years). Immunohistochemistry of coronary vessels from subjects with and without CAD revealed expression of TERT in both smooth muscle and endothelial cells (Online Figure I).
Figure 2.
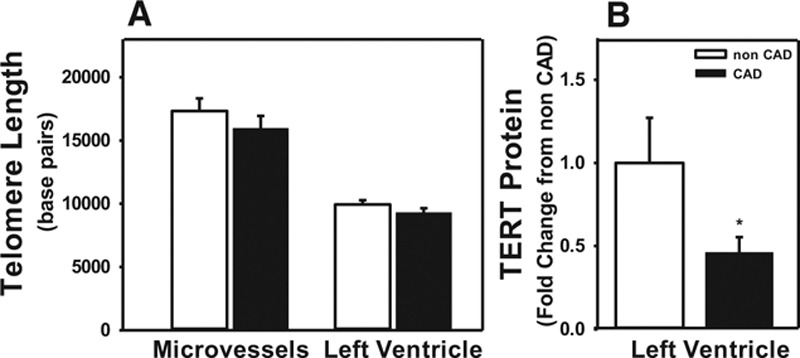
Coronary artery disease (CAD) does not cause telomere shortening in microvessels or left ventricular tissue but decreases expression of the catalytic subunit catalytic subunit of human telomerase complex (TERT). A, Total average telomere length was evaluated in genomic DNA from microvessels (adipose) and left ventricular tissue from subjects with and without CAD. B, Expression of the TERT was evaluated in left ventricular tissue from subjects with and without CAD by Western blot. Values are normalized to β-actin loading control and expressed as fold change compared with non-CAD control. N=4 to 7. *P<0.05 t test.
Role of Mitochondria As the Source of H2O2 and the Site of TERT Action in FMD
Using MitoPY1, a mitochondrial-specific fluorescent indicator for H2O2, we observed that for 15 to 20 hours, BIBR-1532 treatment in vessels from subjects without CAD caused stimulated mitochondrial release of H2O2 in response to flow (Figure 3A and 3B).
Figure 3.
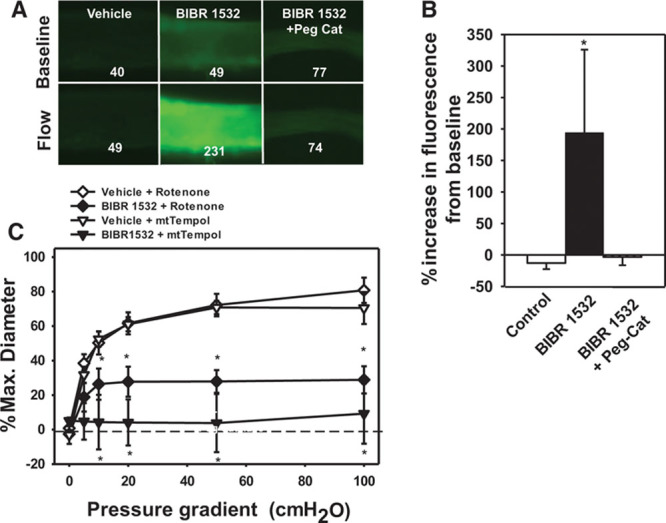
Mitochondrial hydrogen peroxide (H2O2)–mediated flow-mediated dilation (FMD) after inhibition of telomerase. H2O2 levels were analyzed using the H2O2-specific florescent probe PYI (peroxy yellow 1) targeted to mitochondria (MitoPYI) in vessels from non–coronary artery disease (CAD) subjects. A, Representative image; numbers represent florescent intensity above background. B, Summary of florescence intensity at 5 minutes after initiation of flow. Specificity of the probe for H2O2 was confirmed using polyethylene glycol-catalase (Peg-Catalase). C, An inhibitor of electron transport chain complex I (rotenone) or a mitochondrial-targeted reactive oxygen species (ROS) scavenger (MitoTempol) inhibited FMD after telomerase inhibition. N=4. *P<0.05 2-way analysis of variance (ANOVA) RM (dose response curve) or t test (fluorescence data) with Tukey post hoc.
Addition of the complex I inhibitor rotenone abolished FMD in vessels treated with BIBR-1532 (atrial and adipose) but had no effect on vehicle-treated vessels (Figure 3). Scavenging mitochondrial O2–· with the mitochondrial-specific antioxidant mitoTEMPOL (10 µmol/L) reduced FMD in vessels treated with BIBR-1532 (Figure 3C). Taken together, these data suggest that the source of ROS modulated by the telomerase inhibitor BIBR-1532 is from the mitochondrial electron transport chain.
Because of the small tissue volume of human arterioles, we were not able to quantify protein content. Therefore, we sought direct evidence for TERT translocation to the mitochondria using cultured human cells transfected with TERT-eGFP (endothelial green fluorescent protein) expression vectors as previously published.9
In cells expressing wild-type (WT) TERT, protein fused to eGFP nuclear localization of TERT was reduced following stress (application of H2O2). A corresponding increase in mitochondrial localization of TERT-GFP (green fluorescent protein) was seen by colocalization with the mitochondrial probe mitotracker red (Figure 4A).
Figure 4.

Mitochondrial translocation of catalytic subunit of human telomerase complex (TERT) after exposure to acute oxidative stress. A, Treatment of normal human fibroblasts (HF) with hydrogen peroxide (H2O2) resulted in translocation of TERT-GFP (green fluorescent protein) from the nucleus to the mitochondria (colocalized with Mitotracker Red). Cells were imaged using a 63× oil immersion objective. B, To confirm purity of isolated mitochondria of cells either transfected with whole cell (WC) TERT or R3E/R6E TERT (nucTERT) Western blots for tubulin (cytoplasmic marker), mitochondrial heat shock proteins 70 (mtHSP70), and Ku80 (nuclear isoform [higher molecular weight] and mitochondrial isoform) were performed. C, Expression of nucTERT increased mitochondrial superoxide production as measured with MitoSox. D, Expression of nucTERT decreased cellular adenosine triphosphate (ATP) production after external stress. n=3 to 5. *P<0.05 t Test.
To test whether the mitochondrial localization of TERT limits mtROS production, we used a mutant version of TERT (nucTERT) that we have previously shown to be functional (elongation of telomeres) but excluded from the mitochondria.9 Western blots confirmed the mitochondrial localization of the WT protein and the lack of mitochondrial localization of nucTERT (Figure 4B). Whole cell TERT levels were comparable among WT and nucTERT groups but only WT TERT is detected in highly purified mitochondria (Figure 4B). Generation of mtROS (mitoSOX) depended on the subcellular localization of TERT, with nucTERT cells showing consistently higher mitoSox signal (Figure 4C). To evaluate the effects of TERT localization on the response to stress, wtTERT- or nucTERT-expressing cells were plated for 16 hours then treated with 200 μmol/L H2O2 for 60 minutes. Cells expressing nucTERT (aka absence of mtTERT) show a significant decrease in the steady state amount of ATP produced compared with WT (Figure 4D), indicating that absence of mtTERT has direct effects on mitochondrial function by impairing ATP production capacity following stress.
Effects of Telomerase Inhibition Are Independent of Transcription
It is possible that the switch in mechanism of FMD based on TERT modulation is transcriptionally induced because TERT regulates gene expression (reviewed by Zhou et al24). To test this possibility, the transcription inhibitor actinomycin D (2 μg/mL)25 was used. Actinomycin D had no effect on the mediator of FMD in vessels from patients without CAD treated 15 to 20 hours with vehicle or with the telomerase inhibitor BIBR-1532 (Figure 5). Efficacy of the dose of actinomycin D was demonstrated in endothelial cells treated for 2 hours with actinomycin D or vehicle and exposed for 1 hour to heat shock (42°C). Fifteen to 20 hours later, cells in the vehicle group showed an increase in HSP 27 and 70 compared with unstressed cells. Pretreatment with actinomycin D eliminated this effect (Figure 5C and 5D). These data indicate that nuclear transcriptional properties of telomerase are not involved in the functional and redox changes observed in this study.
Figure 5.
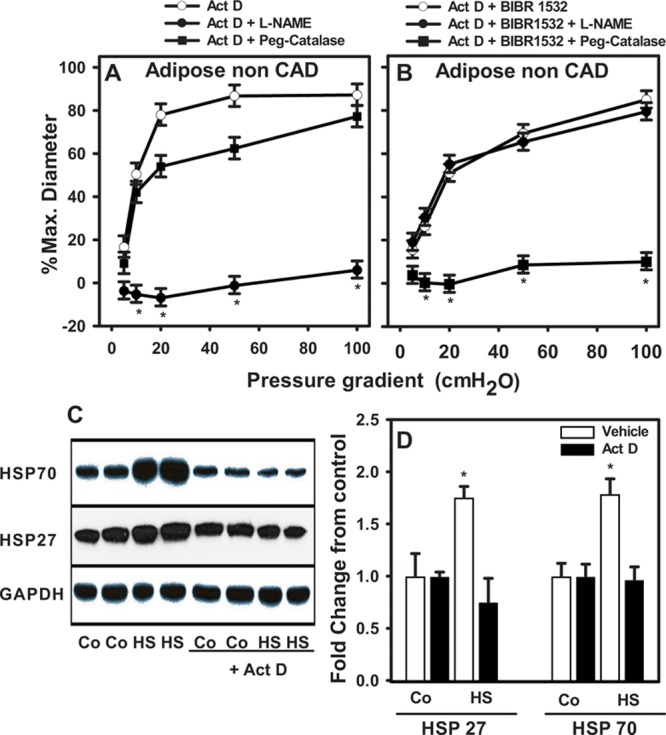
Change in vascular phenotype is independent of changes in gene expression. Flow-mediated dilation (FMD) was evaluated in isolated adipose microvessels from subjects without coronary artery disease (CAD) treated with actinomycin D, a transcriptional inhibitor. A, Act D+vehicle. B, Act D+BIBR-1532. Mechanism of FMD changed from NO (Act D)→H2O2 (Act D+BIBR-1532). C and D, Act D was sufficient to prevent transcriptional activation of heat shock proteins 27 and 70 after acute exposure to 42°C. Sample blot shows 2/5 total treatments. *P<0.05 2-way analysis of variance (ANOVA) RM Tukey post hoc analysis (functional studies). N=4 to 5. *P<0.05 vs untreated control t Test Tukey post hoc analysis (protein levels). CO indicates control; HS, heat shock; L-NAME, Nω-nitro-L-arginine methyl ester; and Peg-Catalase, polyethylene glycol-catalase.
Activation of Telomerase Restores NOS-Mediated FMD in Vessels From Subjects With CAD
Adipose and atrial vessels from subjects with CAD were exposed to AGS 499 (15–20 hours; 22 nmol/L), an activator of telomerase. AGS 499 restored the mediator of FMD from H2O2 to a NOS-derived product. Maximal dilation to flow (100 cm H2O) in vessels from subjects with CAD treated with AGS 499 was of similar magnitude to vehicle-treated vessels and was inhibited by L-NAME but not by PEG-catalase (% max diameter at 100 cm H2O in atrial vessels (n=3–4): vehicle 64.5±8.7, L-NAME 12.6±15.3*, PEG-catalase 65.2±7.8, c-PTIO 22.9±17.3*; adipose vessels (n=5–8): AGS 499+vehicle 79.5±3.9, L-NAME 10.9±17.2*, PEG-catalase 79.2±4.9; *P<0.05 2-way analysis of variance RM; Figure 6). Inhibition of telomerase activity in vessels from subjects with CAD had no effect on the magnitude or mechanism of dilation (Figure 6C). The dose of AGS 499 used was transcriptionally active because HUVECs treated with the same dose demonstrated an ≈4 fold increase of total TERT mRNA (Figure 6D).
Figure 6.

Pharmacological activation of telomerase restores nitric oxide (NO)–mediated dilation in response to flow. Flow-mediated dilation (FMD) was evaluated in isolated adipose and atrial microvessels from subjects with coronary artery disease (CAD) treated with the selective transcriptional telomerase activator AGS 499 (22 nmol/L). Vehicle-treated vessels are historic controls. A, CAD adipose+AGS 499. B, CAD atrial+AGS 499. The mechanism of FMD changed from polyethylene glycol-catalase (Peg-Catalase)–sensitive (H2O2 in CAD) to Nω-nitro-L-arginine methyl ester (L-NAME) and c-PTIO–sensitive (NO mediated in CAD+AGS 499). C, Inhibition of telomerase activity in vessels from subjects with CAD did not alter mechanism or overall dilator capacity in response to flow. D, AGS 499 resulted in transcriptional increase of catalytic subunit of human telomerase complex (TERT) mRNA. *P<0.05 vs AGS 499 control 2-way analysis of variance (ANOVA) RM Tukey post hoc analysis. N=5 to 8 adipose, N=3 to 4 atrial. *P>0.05 vs AGS 499 only–treated t Test Tukey post hoc analysis (RNA levels). #AGS 499+c-PTIO N=3 (2 adipose and 1 atrial). HUVECs indicates human umbilical vein endothelial cells.
To establish the effect of telomerase on endothelial nitric oxide synthase (eNOS), we used quantitative polymerase chain reaction and Western blotting. In HUVECs, AGS 499 increased eNOS message, whereas BIBR-1532 had no effect (Online Figure IIa). Levels of p-eNOS (ser 1117) increase after treatment with AGS 499 (Online Figure IIb). Similarly, Daf-2A (HPLC [high-performance liquid chromatography]; Methods in Online Data Supplement) significantly increased with AGS 499 treatment in HUVECs, whereas BIBR-1532 had no effect (Online Figure IIc).
Discussion
There are 3 primary novel findings of this study. First, we establish that inhibition of telomerase in vessels from subjects without CAD recapitulates the vascular phenotype seen in patients with CAD by converting the mediator of FMD from NO to H2O2. Activation of telomerase can restore an NOS-mediated FMD in vessels from patients with CAD. TERT protein levels are reduced in patients with CAD compared with those without CAD, and mtROS production is elevated when telomerase activity is reduced (mitoPY1). Second, the intracellular site for these actions of telomerase is the mitochondria and not the nucleus as demonstrated with novel mutant telomerase constructs that target either the whole cell (WT) or the nucleus alone (nucTERT). Third, the effects of telomerase inhibition are not dependent on changes in gene expression. Collectively, these data for the first time provide evidence for an extranuclear role for telomerase in modulating vascular function. Figure 7 illustrates our findings.
Figure 7.
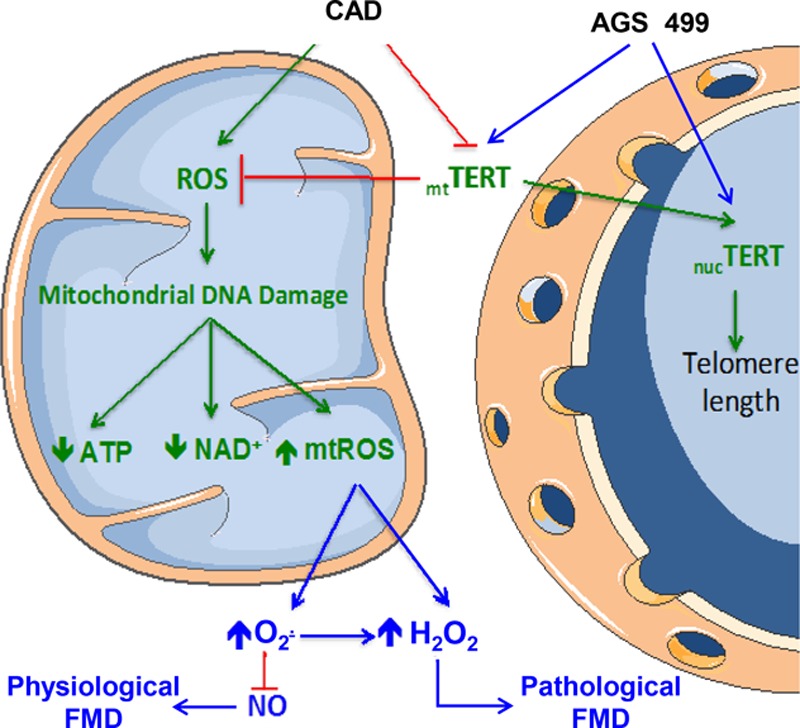
Proposed mechanism of TERT in regulating balance of endothelial mitochondrial ROS and NO to maintain FMD in human microvessels. AGS 499 indicates telomeres activator; ATP, adenosine triphosphate; CAD, coronary artery disease; FMD, flow-mediated dilation; H2O2, hydrogen peroxide; mt, mitochondrial; nuc, nuclear; NAD+, nicotinamide adenine dinucleotide; NO, nitric oxide; O2–·, superoxide; ROS, reactive oxygen species; and TERT, catalytic subunit of human telomerase complex.
An extranuclear role for telomerase suggests noncanonical sites of actions. Based on our subcellular localization experiments and prior studies in other cell types,9 we postulate an effect on mitochondria. The short timeframe of the observed changes (hours) supports such a telomere-independent mechanism. Our study provides a physiological role for the mitochondrial ROS-suppressing effects of TERT. Because TERT suppresses mtROS production26 in vascular endothelial cells, reduced TERT, as seen in CAD, would support mtROS production necessary for FMD in human arterioles.27 TERT as a regulator of mtROS is important in other diseases as well. Transcriptional activation of TERT via AGS 499 delayed the onset of amyotrophic lateral sclerosis in mice by increasing cell survival after oxidative stress.17,18 The same mechanism of action was shown in human bone marrow mesenchymal stem cells which were protected from oxidative stress by TERT.18
The role of telomerase in chronic disease is multifactorial. Traditionally, TERT protects tissues, including the vasculature, from chromosomal rearrangements by maintaining telomere length over time. For example, shortened telomeres and decreased telomerase activity are associated with atherosclerosis28–30 and activation of senescence pathways.31,32 Of recant, it has been show that telomere uncapping without change in telomere length which can also invoke cellular aging and inflammation23 or novel data show a role of TERT as a regulator of gene expression by modulating chromatin.33 In addition, some of the pathological effects of reduced telomerase related to loss of telomerase outside the nucleus, including the increase of mitochondria-free radical formation, have been established. mtROS production changes inversely with expression of TERT in the mitochondria, an action that involves extranuclear TERT.6,34
As observed previously, the magnitude of FMD in human microvessels is maintained but the mechanism changes in response to stress,35 whereas agonist induced dilation to ACh is simply lost.36 The reason for this difference is not known, but it has been speculated that ACh is but one of many agonists which can stimulate NO release, whereas shear stress-induced increases in NO involves multiple signaling pathways37 (ie, ACh binds to muscarinic receptors to induce dilation while redundant pathways may exist for FMD).
We have not determined the precise mechanism by which reduced telomerase activity modulates mtROS. However, this modulation is independent of nuclear transcriptional events and from telomere elongation, suggesting a novel nontelomeric site of action. However, we cannot exclude a role for telomere uncapping, a focus for future investigations.
In this study, we used a physiological stimulus for endothelium-dependent dilation, shear stress. The mechano-chemical signal transduction during shear that leads to dilation has been studied extensively.38 Depending on the tissue and species, the signaling pathway involves glycocalyx, TRPV4, or other mechano-sensitive ion channels, and focal adhesions tethering endothelial cells to the underlying basement membrane by cytoskeletal elements.39,40 Secondary pathways include tyrosine kinases or calcium signaling to activate eNOS and other vasoactive mediators. In human vessels from subjects with CAD, a different signaling pathway is invoked with stimulation of mitochondrial H2O2 production. The present study provides novel evidence that telomerase lies at the intersection of these pathways and regulates the switch between shear-induced mediators. We speculate that augmentation of telomerase activity may improve the coronary and peripheral vascular phenotypes in patients with CAD. However, the potential adverse effects associated with increasing telomerase activity, including accelerated tumor growth, have to be carefully considered.
Both NO and ROS are signaling pathways for mitochondrial–nuclear communication (reviewed by Kotiadis et al41and Valerio and Nisoli42), and TERT contains both mitochondrial and nuclear targeting sequences.9,43–45 Site-specific phosphorylation of TERT initiates either mitochondrial or nuclear translocation. This puts telomerase in a position to serve as a critical mediator of nuclear-mitochondrial cross-talk and implicates involvement of AKT, which phosphorylates TERT at its nuclear localization site, increasing binding affinity for the nuclear transporter importin-α, promoting translocation.43 AKT also targets mitochondrial biogenesis and NO signaling (reviewed by Miyamoto et al46and Finley and Haigis47). A critical role for non-nuclear telomerase activity in regulating this interplay represents a new field of study with implications for diseases in which mtROS play a prominent role, such as diabetes mellitus48,49 and neurodegenerative17,18 diseases and cardiovascular responses to chemotherapies, such as doxorubicin and PK inhibitors.50,51
It is well established that telomerase activity and telomere length decrease during the natural aging process. A parallel reduction in NO bioavailability is seen after the 4th decade of life, reducing vasodilator capacity.52–55 The shift toward a more oxidative environment could explain the reduction in NO bioavailability and increase in ROS production observed with CAD and represents a common feature that contributes to the development of other disease phenotypes.17,18 The telomerase activator AGS 499, which normalized FMD and ROS production in microvessels from subjects with CAD, also delays onset of ALS in rodent models.18 Thus, the ability of TERT to regulate NO production could be important beyond vasodilation.
Compared with age-matched controls, subjects with CAD show a similar degree of FMD. Importantly, the underlying mechanism of FMD changes from NO to H2O2. Inhibition of telomerase can initiate this pathological phenotype (H2O2-mediated FMD) in vessels from subjects without CAD in <24 hours. Conversely, in subjects with chronic CAD, stimulation of TERT activity is sufficient to subacutely restore NOS as critical for FMD, likely via generation of NO (L-NAME and c-PTIO inhibitable). Induced upregulation of telomerase activity may be beneficial in CAD because NO is vasoprotective, whereas H2O2 is proinflammatory in the vasculature.56,57 With growing evidence that the microcirculatory dynamics influence cardiovascular outcomes (reviewed by Lockhart et al58), understanding mechanisms of arteriolar endothelium-dependent dilation is of both physiological and clinical importance.
We have previously demonstrated that in a matter of minutes, an acute increase in intraluminal pressure can evoke the same change in mediator of FMD after telomerase inhibition or as seen in CAD subjects.35 Using ceramide as a physiological stressor, Freed et al12 showed a similar shift in vasodilator mechanism as we see with telomerase inhibition. Ceramide is known to suppress TERT expression59; thus, it is intriguing to speculate that ceramide activation of mtROS is TERT-dependent. Future studies will evaluate whether ceramide and TERT are part of the same pathway modulating mitochondrial redox function.
Study Limitations
Our functional studies point toward a critical role of NOS and NO in arteriolar FMD under normal conditions. We cannot rule out a role of prostacyclins, EETs (epoxyeicosatrienoic acid), or other EDHFs (endothelial-derived hyperpolarizing factor); however, our prior studies argue against a role of EETs because the enzyme responsible for their production, CYP450 epoxygenase, is directly inhibited by H2O2.60 Although it is not technically feasible to directly assess NO bioavailability in the small tissue samples used,61 we used DAF-FM (4-amino-5-methylamino-2',7'-difluorofluorescein diacetate) as a surrogate marker for NO production in cultured human endothelial cells. We observed an elevation in eNOS message and p-eNOS (phospho-endothelial nitric oxide synthase; ser 1117) after activation of TERT with AGS 499. NO production showed a statistically significant increase in cultured HUVECs after treatment with AGS 499. Our combined data in cultured human endothelial cells and isolated vessels strongly suggests that NOS-derived NO is responsible for FMD in vessels from patients without CAD and in vessels from patients with CAD treated with AGS 499 15 to 20 hours (dilation changes from PEG-catalase inhibitable to L-NAME and c-PTIO inhibitable). Future studies will define the underlying differences between the acute versus chronic effects of telomerase deficiency in vivo and in isolated vessel studies.
We used primary human fibroblasts to provide proof of principle data for translocation of TERT in response to oxidative stress. Because of the fact that endothelial cells are difficult to use for transient transfections, normal human fibroblasts where chosen as a surrogate. In addition, fibroblasts have higher levels of mitochondrial number, enabling us to perform ATP production assays, which are extremely difficult to perform in primary human endothelial cells.
We cannot eliminate a contribution of vascular smooth muscle cells to the observed phenotype. However, we think this is unlikely because inhibition of telomerase did not alter vascular smooth muscle cells function (Figure 1), whereas activation of telomerase restored physiological endothelial-dependent NOS-mediated (NO) dilation to flow. As telomerase activity is known to increase cellular proliferation, activation of telomerase in vascular smooth muscle cells would be counterproductive to vascular health and would be expected to impair vasoactive function. In mice with systemic deletion of telomerase, a decrease in atherosclerosis-like phenotype has been reported.62 Similarly published evidence supports that the well-established anti-inflammatory and anti-atherosclerosis properties of PPARγ are at least in part caused by suppression of telomerase activity in the vascular smooth muscle cells.63,64
Conclusions
In summary, we demonstrate direct functional evidence that decreased telomerase activity is sufficient to initiate the transition from NO to H2O2 as the primary mediator of FMD in the human resistance vasculature, recapitulating the microvascular phenotype found in patients with CAD.14,65 Furthermore, we show that increasing telomerase activity reverses the pathological phenotype seen in CAD. This pathological switch has clinical implications, especially in the coronary circulation, where beyond sharing vasodilator properties, the local influences are opposing with H2O2 exerting pro- and NO-producing anti-proliferative effects. Identifying a central and extranuclear role for telomerase in this switch suggests novel pharmacological approaches for maintaining or restoring normal endothelial function by limiting endothelial activation inflammation and vascular ROS production. This study provides support for the idea that mitochondrial telomerase activity regulates ROS production with direct physiological consequences.
Acknowledgments
We thank the Division of Cardiothoracic Surgery at the Medical College of Wisconsin, the Cardiothoracic Surgery Group of Milwaukee, the Cardiovascular Surgery Associates of Milwaukee, the Midwest Heart Surgery Institute, Cardiothoracic Surgery division at the Zablocki VA Medical Center in Milwaukee, Froedtert Memorial Lutheran Hospital, Aurora St Luke’s Medical Center, Children’s Hospital of Wisconsin, Wheaton Franciscan Healthcare’s Elmbrook Memorial Hospital, St Joseph’s Hospital, and the Wisconsin Heart Hospital, as well as the Wisconsin Donor Network, for providing tissue. We thank the Blood center of Wisconsin hybrydoma facility for isolation of umbilical vein endothelial cells and the Medical College of Wisconsin (MCW) pathology tissue bank for assistance with tissue collection and scoring of coronary artery disease (CAD) phenotypes in rejected donor hearts. We thank the Redox Biology Program at MCW for the assistance with evaluating nitric oxide (NO) via HPLC (high-performance liquid chromatography).
Sources of Funding
This work was supported by R01 HL113612 (D.D. Gutterman) and R21 OD018306 (A.M. Beyer). J.K. Freed was supported by a T-32 Physician Scientist Training Grant GM089586 (to J.R. Kersten). M.J. Durand was supported by training grants from National Institutes of Health (NIH) T32HL007792 and American Heart Association (AHA) 14POST18780022. A.J. Donato and R.G. Morgan were supported by K02 AG045339, R01 AG040297, and R21 AG0443952.
Disclosures
E. Priel filed a patent on AGS 499 and other derivatives as telomerase increasing compounds. The other authors report no conflicts.
Supplementary Material
Nonstandard Abbreviations and Acronyms
- ACh
- acetylcholine
- CAD
- coronary artery disease
- FMD
- flow-mediated dilation
- H2O2
- hydrogen peroxide
- HUVECs
- human umbilical vein endothelial cells
- mtROS
- mitochondrial reactive oxygen species
- MitoPY1
- Mito Peroxy Yellow 1
- NO
- nitric oxide
- nucTERT
- nuclear catalytic subunit of telomerase complex
- O2–·
- superoxide
- ROS
- reactive oxygen species
- TERT
- catalytic subunit of telomerase complex
- WT
- wild-type
In November 2015, the average time from submission to first decision for all original research papers submitted to Circulation Research was 15.52 days.
The online-only Data Supplement is available with this article at http://circres.ahajournals.org/lookup/suppl/doi:10.1161/CIRCRESAHA.115.307918/-/DC1.
Novelty and Significance
What Is Known?
Endothelial dysfunction predicts future cardiovascular events.
Under physiological conditions, the primary mediator of flow-mediated dilation (FMD) is nitric oxide (NO), whereas in pathology, such as coronary artery disease (CAD), the FMD is mediated by the mitochondria-derived, proinflammatory hydrogen peroxide.
The catalytic subunit of telomerase elongates telomeres in the nucleus to prevent cellular aging and promote proliferation.
A non-nuclear role for catalytic subunit of telomerase in regulating levels of mitochondrial-derived reactive oxygen species is established in other cell types but its role in vascular cells has not been examined.
What New Information Does This Article Contribute?
Telomerase activity is decreased in subjects with CAD without measurable telomere shortening.
Pharmacological inhibition of telomerase activity in arterioles from patients without CAD causes a transition in mechanism of FMD from NO to hydrogen peroxide, similar to the mechanism observed in vessels from patients with CAD.
Transcriptional activation of catalytic subunit of telomerase restores NO as the dominant mediator of FMD in subjects with CAD.
Telomerase activity regulates endothelial production of either NO (health) or hydrogen peroxide (disease) in response to endothelial shear stress in the microcirculation.
Endothelial release of NO mediates FMD under physiological conditions and serves to prevent vascular smooth muscle proliferation and inflammation. In subjects with CAD, FMD is mediated by mitochondrial-derived reactive oxygen species, specifically hydrogen peroxide, a proinflammatory and proatherosclerotic dilator. The present study identifies telomerase activity as a regulator of the underlying mechanism of FMD. Telomerase activity represents a novel target to modulate FMD and, thereby, vascular health.
References
- 1.Aviv H, Khan MY, Skurnick J, Okuda K, Kimura M, Gardner J, Priolo L, Aviv A. Age dependent aneuploidy and telomere length of the human vascular endothelium. Atherosclerosis. 2001;159:281–287. doi: 10.1016/s0021-9150(01)00506-8. [DOI] [PubMed] [Google Scholar]
- 2.Chang E, Harley CB. Telomere length and replicative aging in human vascular tissues. Proc Natl Acad Sci U S A. 1995;92:11190–11194. doi: 10.1073/pnas.92.24.11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brouilette S, Singh RK, Thompson JR, Goodall AH, Samani NJ. White cell telomere length and risk of premature myocardial infarction. Arterioscler Thromb Vasc Biol. 2003;23:842–846. doi: 10.1161/01.ATV.0000067426.96344.32. doi: 10.1161/01.ATV.0000067426.96344.32. [DOI] [PubMed] [Google Scholar]
- 4.Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation. 2002;105:1541–1544. doi: 10.1161/01.cir.0000013836.85741.17. [DOI] [PubMed] [Google Scholar]
- 5.Shaik S, Inuzuka H, Liu P, Wei W, Wang Z. Endothelium Aging and Vascular Diseases: INTECH Open Access Publisher. 2013. [Google Scholar]
- 6.Ahmed S, Passos JF, Birket MJ, Beckmann T, Brings S, Peters H, Birch-Machin MA, von Zglinicki T, Saretzki G. Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J Cell Sci. 2008;121:1046–1053. doi: 10.1242/jcs.019372. doi: 10.1242/jcs.019372. [DOI] [PubMed] [Google Scholar]
- 7.Santos JH, Meyer JN, Van Houten B. Mitochondrial localization of telomerase as a determinant for hydrogen peroxide-induced mitochondrial DNA damage and apoptosis. Hum Mol Genet. 2006;15:1757–1768. doi: 10.1093/hmg/ddl098. doi: 10.1093/hmg/ddl098. [DOI] [PubMed] [Google Scholar]
- 8.Minamino T, Mitsialis SA, Kourembanas S. Hypoxia extends the life span of vascular smooth muscle cells through telomerase activation. Mol Cell Biol. 2001;21:3336–3342. doi: 10.1128/MCB.21.10.3336-3342.2001. doi: 10.1128/MCB.21.10.3336-3342.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharma NK, Reyes A, Green P, Caron MJ, Bonini MG, Gordon DM, Holt IJ, Santos JH. Human telomerase acts as a hTR-independent reverse transcriptase in mitochondria. Nucleic Acids Res. 2012;40:712–725. doi: 10.1093/nar/gkr758. doi: 10.1093/nar/gkr758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haendeler J, Hoffmann J, Brandes RP, Zeiher AM, Dimmeler S. Hydrogen peroxide triggers nuclear export of telomerase reverse transcriptase via Src kinase family-dependent phosphorylation of tyrosine 707. Mol Cell Biol. 2003;23:4598–4610. doi: 10.1128/MCB.23.13.4598-4610.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miura H, Wachtel RE, Liu Y, Loberiza FR, Jr, Saito T, Miura M, Gutterman DD. Flow-induced dilation of human coronary arterioles: important role of Ca(2+)-activated K(+) channels. Circulation. 2001;103:1992–1998. doi: 10.1161/01.cir.103.15.1992. [DOI] [PubMed] [Google Scholar]
- 12.Freed JK, Beyer AM, LoGiudice JA, Hockenberry JC, Gutterman DD. Ceramide changes the mediator of flow-induced vasodilation from nitric oxide to hydrogen peroxide in the human microcirculation. Circ Res. 2014;115:525–532. doi: 10.1161/CIRCRESAHA.115.303881. doi: 10.1161/CIRCRESAHA.115.303881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC, Gutterman DD. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ Res. 2003;93:573–580. doi: 10.1161/01.RES.0000091261.19387.AE. doi: 10.1161/01.RES.0000091261.19387.AE. [DOI] [PubMed] [Google Scholar]
- 14.Miura H, Bosnjak JJ, Ning G, Saito T, Miura M, Gutterman DD. Role for hydrogen peroxide in flow-induced dilation of human coronary arterioles. Circ Res. 2003;92:e31–e40. doi: 10.1161/01.res.0000054200.44505.ab. [DOI] [PubMed] [Google Scholar]
- 15.Sahin E, Colla S, Liesa M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–365. doi: 10.1038/nature09787. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vasa M, Breitschopf K, Zeiher AM, Dimmeler S. Nitric oxide activates telomerase and delays endothelial cell senescence. Circ Res. 2000;87:540–542. doi: 10.1161/01.res.87.7.540. [DOI] [PubMed] [Google Scholar]
- 17.Tichon A, Eitan E, Kurkalli BG, Braiman A, Gazit A, Slavin S, Beith-Yannai E, Priel E. Oxidative stress protection by novel telomerase activators in mesenchymal stem cells derived from healthy and diseased individuals. Curr Mol Med. 2013;13:1010–1022. doi: 10.2174/1566524011313060013. [DOI] [PubMed] [Google Scholar]
- 18.Eitan E, Tichon A, Gazit A, Gitler D, Slavin S, Priel E. Novel telomerase-increasing compound in mouse brain delays the onset of amyotrophic lateral sclerosis. EMBO Mol Med. 2012;4:313–329. doi: 10.1002/emmm.201200212. doi: 10.1002/emmm.201200212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larsen BT, Bubolz AH, Mendoza SA, Pritchard KA, Jr, Gutterman DD. Bradykinin-induced dilation of human coronary arterioles requires NADPH oxidase-derived reactive oxygen species. Arterioscler Thromb Vasc Biol. 2009;29:739–745. doi: 10.1161/ATVBAHA.108.169367. doi: 10.1161/ATVBAHA.108.169367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phillips SA, Bian JT, Church EC, Das EK, Vidovich M, Gutterman DD. Hydrogen peroxide prevents impaired endothelium-dependent dilation following acute exertion in chronic exercising but not in sedentary subjects. Circulation. 2009;120:S1013. [Google Scholar]
- 21.Kuo L, Chilian WM, Davis MJ. Interaction of pressure- and flow-induced responses in porcine coronary resistance vessels. Am J Physiol. 1991;261:H1706–H1715. doi: 10.1152/ajpheart.1991.261.6.H1706. [DOI] [PubMed] [Google Scholar]
- 22.Dickinson BC, Chang CJ. A targetable fluorescent probe for imaging hydrogen peroxide in the mitochondria of living cells. J Am Chem Soc. 2008;130:9638–9639. doi: 10.1021/ja802355u. doi: 10.1021/ja802355u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morgan RG, Ives SJ, Lesniewski LA, Cawthon RM, Andtbacka RH, Noyes RD, Richardson RS, Donato AJ. Age-related telomere uncapping is associated with cellular senescence and inflammation independent of telomere shortening in human arteries. Am J Physiol Heart Circ Physiol. 2013;305:H251–H258. doi: 10.1152/ajpheart.00197.2013. doi: 10.1152/ajpheart.00197.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou J, Ding D, Wang M, Cong YS. Telomerase reverse transcriptase in the regulation of gene expression. BMB Rep. 2014;47:8–14. doi: 10.5483/BMBRep.2014.47.1.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rill RL, Hecker KH. Sequence-specific actinomycin D binding to single-stranded DNA inhibits HIV reverse transcriptase and other polymerases. Biochemistry. 1996;35:3525–3533. doi: 10.1021/bi9530797. doi: 10.1021/bi9530797. [DOI] [PubMed] [Google Scholar]
- 26.Haendeler J, Dröse S, Büchner N, Jakob S, Altschmied J, Goy C, Spyridopoulos I, Zeiher AM, Brandt U, Dimmeler S. Mitochondrial telomerase reverse transcriptase binds to and protects mitochondrial DNA and function from damage. Arterioscler Thromb Vasc Biol. 2009;29:929–935. doi: 10.1161/ATVBAHA.109.185546. doi: 10.1161/ATVBAHA.109.185546. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Bubolz AH, Mendoza S, Zhang DX, Gutterman DD. H2O2 is the transferrable factor mediating flow-induced dilation in human coronary arterioles. Circ Res. 2011;108:566–573. doi: 10.1161/CIRCRESAHA.110.237636. doi: 10.1161/CIRCRESAHA.110.237636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogami M, Ikura Y, Ohsawa M, Matsuo T, Kayo S, Yoshimi N, Hai E, Shirai N, Ehara S, Komatsu R, Naruko T, Ueda M. Telomere shortening in human coronary artery diseases. Arterioscler Thromb Vasc Biol. 2004;24:546–550. doi: 10.1161/01.ATV.0000117200.46938.e7. doi: 10.1161/01.ATV.0000117200.46938.e7. [DOI] [PubMed] [Google Scholar]
- 29.Samani NJ, Boultby R, Butler R, Thompson JR, Goodall AH. Telomere shortening in atherosclerosis. Lancet. 2001;358:472–473. doi: 10.1016/S0140-6736(01)05633-1. doi: 10.1016/S0140-6736(01)05633-1. [DOI] [PubMed] [Google Scholar]
- 30.Benetos A, Gardner JP, Zureik M, Labat C, Xiaobin L, Adamopoulos C, Temmar M, Bean KE, Thomas F, Aviv A. Short telomeres are associated with increased carotid atherosclerosis in hypertensive subjects. Hypertension. 2004;43:182–185. doi: 10.1161/01.HYP.0000113081.42868.f4. doi: 10.1161/01.HYP.0000113081.42868.f4. [DOI] [PubMed] [Google Scholar]
- 31.Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol Cell. 2004;14:501–513. doi: 10.1016/s1097-2765(04)00256-4. [DOI] [PubMed] [Google Scholar]
- 32.Gao B, Li K, Wei YY, Zhang J, Li J, Zhang L, Gao JP, Li YY, Huang LG, Lin P, Wei YQ. Zinc finger protein 637 protects cells against oxidative stress-induced premature senescence by mTERT-mediated telomerase activity and telomere maintenance. Cell Death Dis. 2014;5:e1334. doi: 10.1038/cddis.2014.298. doi: 10.1038/cddis.2014.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qing H, Aono J, Findeisen HM, Jones KL, Heywood EB, Bruemmer D. Differential regulation of telomerase reverse transcriptase promoter activation and protein degradation by histone deacetylase inhibition [published online ahead of print October 27, 2015]. J Cell Physiol. doi: 10.1002/jcp.25226. doi: 10.1002/jcp.25226. http://onlinelibrary.wiley.com/doi/10.1002/jcp.25226/abstract;jsessionid=3E2BCE708E47739D04E75790D1E59474.f04t03. [DOI] [PubMed] [Google Scholar]
- 34.Indran IR, Hande MP, Pervaiz S. hTERT overexpression alleviates intracellular ROS production, improves mitochondrial function, and inhibits ROS-mediated apoptosis in cancer cells. Cancer Res. 2011;71:266–276. doi: 10.1158/0008-5472.CAN-10-1588. doi: 10.1158/0008-5472.CAN-10-1588. [DOI] [PubMed] [Google Scholar]
- 35.Beyer AM, Durand MJ, Hockenberry J, Gamblin TC, Phillips SA, Gutterman DD. An acute rise in intraluminal pressure shifts the mediator of flow-mediated dilation from nitric oxide to hydrogen peroxide in human arterioles. Am J Physiol-Heart Circ Physiol. 2014;307:H1587–H1593. doi: 10.1152/ajpheart.00557.2014. doi: 10.1152/ajpheart.00557.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Durand MJ, Phillips SA, Widlansky ME, Otterson MF, Gutterman DD. The vascular renin-angiotensin system contributes to blunted vasodilation induced by transient high pressure in human adipose microvessels. Am J Physiol Heart Circ Physiol. 2014;307:H25–H32. doi: 10.1152/ajpheart.00055.2014. doi: 10.1152/ajpheart.00055.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 38.Davies PF, Zilberberg J, Helmke BP. Spatial microstimuli in endothelial mechanosignaling. Circ Res. 2003;92:359–370. doi: 10.1161/01.RES.0000060201.41923.88. doi: 10.1161/01.RES.0000060201.41923.88. [DOI] [PubMed] [Google Scholar]
- 39.Banes AJ, Tsuzaki M, Yamamoto J, Fischer T, Brigman B, Brown T, Miller L. Mechanoreception at the cellular level: the detection, interpretation, and diversity of responses to mechanical signals. Biochem Cell Biol. 1995;73:349–365. doi: 10.1139/o95-043. [DOI] [PubMed] [Google Scholar]
- 40.Sun D, Huang A, Sharma S, Koller A, Kaley G. Endothelial microtubule disruption blocks flow-dependent dilation of arterioles. Am J Physiol Heart Circ Physiol. 2001;280:H2087–H2093. doi: 10.1152/ajpheart.2001.280.5.H2087. [DOI] [PubMed] [Google Scholar]
- 41.Kotiadis VN, Duchen MR, Osellame LD. Mitochondrial quality control and communications with the nucleus are important in maintaining mitochondrial function and cell health. Biochim Biophys Acta. 2014;1840:1254–1265. doi: 10.1016/j.bbagen.2013.10.041. doi: 10.1016/j.bbagen.2013.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valerio A, Nisoli E. Nitric oxide, interorganelle communication, and energy flow: a novel route to slow aging. Front Cell Dev Biol. 2015;3:6. doi: 10.3389/fcell.2015.00006. doi: 10.3389/fcell.2015.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jeong SA, Kim K, Lee JH, Cha JS, Khadka P, Cho H-S, Chung IK. Akt-mediated phosphorylation increases the binding affinity of htert for importin α to promote nuclear translocation. J Cell Sci. 2015;128:2287–2301. doi: 10.1242/jcs.166132. doi: 10.1242/jcs.166132. [DOI] [PubMed] [Google Scholar]
- 44.Kovalenko OA, Caron MJ, Ulema P, Medrano C, Thomas AP, Kimura M, Bonini MG, Herbig U, Santos JH. A mutant telomerase defective in nuclear-cytoplasmic shuttling fails to immortalize cells and is associated with mitochondrial dysfunction. Aging Cell. 2010;9:203–219. doi: 10.1111/j.1474-9726.2010.00551.x. doi: 10.1111/j.1474-9726.2010.00551.x. [DOI] [PubMed] [Google Scholar]
- 45.Chung J, Khadka P, Chung IK. Nuclear import of hTERT requires a bipartite nuclear localization signal and Akt-mediated phosphorylation. J Cell Sci. 2012;125:2684–2697. doi: 10.1242/jcs.099267. doi: 10.1242/jcs.099267. [DOI] [PubMed] [Google Scholar]
- 46.Miyamoto S, Rubio M, Sussman MA. Nuclear and mitochondrial signalling akts in cardiomyocytes. Cardiovasc Res. 2009;82:272–285. doi: 10.1093/cvr/cvp087. doi: 10.1093/cvr/cvp087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Finley LW, Haigis MC. The coordination of nuclear and mitochondrial communication during aging and calorie restriction. Ageing Res Rev. 2009;8:173–188. doi: 10.1016/j.arr.2009.03.003. doi: 10.1016/j.arr.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qi Nan W, Ling Z, Bing C. The influence of the telomere-telomerase system on diabetes mellitus and its vascular complications. Expert Opin Ther Targets. 2015;19:849–864. doi: 10.1517/14728222.2015.1016500. doi: 10.1517/14728222.2015.1016500. [DOI] [PubMed] [Google Scholar]
- 49.Mendelsohn AR, Larrick JW. Telomerase reverse transcriptase and peroxisome proliferator-activated receptor γ co-activator-1α cooperate to protect cells from DNA damage and mitochondrial dysfunction in vascular senescence. Rejuvenation Res. 2015;18:479–483. doi: 10.1089/rej.2015.1780. doi: 10.1089/rej.2015.1780. [DOI] [PubMed] [Google Scholar]
- 50.Yan J, Zhou Y, Chen D, Li L, Yang X, You Y, Ling X. Effects of mitochondrial translocation of telomerase on drug resistance in hepatocellular carcinoma cells. J Cancer. 2015;6:151–159. doi: 10.7150/jca.10419. doi: 10.7150/jca.10419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yurtcu E, Darcansoy Iseri O, Iffet Sahin F. Effects of silymarin and silymarin-doxorubicin applications on telomerase activity of human hepatocellular carcinoma cell line HepG2. J BUON. 2015;20:555–561. [PubMed] [Google Scholar]
- 52.Schrage WG, Eisenach JH, Joyner MJ. Ageing reduces nitric-oxide- and prostaglandin-mediated vasodilatation in exercising humans. J Physiol. 2007;579:227–236. doi: 10.1113/jphysiol.2006.124313. doi: 10.1113/jphysiol.2006.124313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cox DA, Vita JA, Treasure CB, Fish RD, Alexander RW, Ganz P, Selwyn AP. Atherosclerosis impairs flow-mediated dilation of coronary arteries in humans. Circulation. 1989;80:458–465. doi: 10.1161/01.cir.80.3.458. [DOI] [PubMed] [Google Scholar]
- 54.Nakajima M, Hashimoto M, Wang F, Yamanaga K, Nakamura N, Uchida T, Yamanouchi K. Aging decreases the production of PGI2 in rat aortic endothelial cells. Exp Gerontol. 1997;32:685–693. doi: 10.1016/s0531-5565(97)00089-2. [DOI] [PubMed] [Google Scholar]
- 55.Sato I, Morita I, Kaji K, Ikeda M, Nagao M, Murota S. Reduction of nitric oxide producing activity associated with in vitro aging in cultured human umbilical vein endothelial cell. Biochem Biophys Res Commun. 1993;195:1070–1076. doi: 10.1006/bbrc.1993.2153. doi: 10.1006/bbrc.1993.2153. [DOI] [PubMed] [Google Scholar]
- 56.Durand MJ, Gutterman DD. Diversity in mechanisms of endothelium-dependent vasodilation in health and disease. Microcirculation. 2013;20:239–247. doi: 10.1111/micc.12040. doi: 10.1111/micc.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Han J, Shuvaev VV, Muzykantov VR. Targeted interception of signaling reactive oxygen species in the vascular endothelium. Ther Deliv. 2012;3:263–276. doi: 10.4155/tde.11.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lockhart CJ, Hamilton PK, Quinn CE, McVeigh GE. End-organ dysfunction and cardiovascular outcomes: the role of the microcirculation. Clin Sci (Lond) 2009;116:175–190. doi: 10.1042/CS20080069. doi: 10.1042/CS20080069. [DOI] [PubMed] [Google Scholar]
- 59.Ogretmen B, Kraveka JM, Schady D, Usta J, Hannun YA, Obeid LM. Molecular mechanisms of ceramide-mediated telomerase inhibition in the A549 human lung adenocarcinoma cell line. J Biol Chem. 2001;276:32506–32514. doi: 10.1074/jbc.M101350200. doi: 10.1074/jbc.M101350200. [DOI] [PubMed] [Google Scholar]
- 60.Larsen BT, Gutterman DD, Sato A, Toyama K, Campbell WB, Zeldin DC, Manthati VL, Falck JR, Miura H. Hydrogen peroxide inhibits cytochrome p450 epoxygenases: interaction between two endothelium-derived hyperpolarizing factors. Circ Res. 2008;102:59–67. doi: 10.1161/CIRCRESAHA.107.159129. doi: 10.1161/CIRCRESAHA.107.159129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Uhlenhut K, Högger P. Pitfalls and limitations in using 4,5-diaminofluorescein for evaluating the influence of polyphenols on nitric oxide release from endothelial cells. Free Radic Biol Med. 2012;52:2266–2275. doi: 10.1016/j.freeradbiomed.2012.03.006. doi: 10.1016/j.freeradbiomed.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 62.Poch E, Carbonell P, Franco S, Díez-Juan A, Blasco MA, Andrés V. Short telomeres protect from diet-induced atherosclerosis in apolipoprotein E-null mice. FASEB J. 2004;18:418–420. doi: 10.1096/fj.03-0710fje. doi: 10.1096/fj.03-0710fje. [DOI] [PubMed] [Google Scholar]
- 63.Ota H, Eto M, Kano MR, Ogawa S, Iijima K, Akishita M, Ouchi Y. Cilostazol inhibits oxidative stress-induced premature senescence via upregulation of Sirt1 in human endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28:1634–1639. doi: 10.1161/ATVBAHA.108.164368. doi: 10.1161/ATVBAHA.108.164368. [DOI] [PubMed] [Google Scholar]
- 64.Ogawa D, Nomiyama T, Nakamachi T, Heywood EB, Stone JF, Berger JP, Law RE, Bruemmer D. Activation of peroxisome proliferator-activated receptor gamma suppresses telomerase activity in vascular smooth muscle cells. Circ Res. 2006;98:e50–e59. doi: 10.1161/01.RES.0000218271.93076.c3. doi: 10.1161/01.RES.0000218271.93076.c3. [DOI] [PubMed] [Google Scholar]
- 65.Sato A, Sakuma I, Gutterman DD. Mechanism of dilation to reactive oxygen species in human coronary arterioles. Am J Physiol Heart Circ Physiol. 2003;285:H2345–H2354. doi: 10.1152/ajpheart.00458.2003. doi: 10.1152/ajpheart.00458.2003. [DOI] [PubMed] [Google Scholar]