

Abstract
Treatment with BRAF kinase inhibitors leads to rapid resistance and tumor regression in BRAF V600E mutant melanoma patients. However, the underlying mechanism of the developed tumor resistance is not fully clear. In this issue of The EMBO Journal, Kim and colleagues show that melanoma cells acquire resistance to BRAF inhibitors by changing cell shape, modifying their cytoskeleton and, in turn, activating the YAP/TAZ mechanotransduction pathway (Kim et al, 2016).
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Molecular Biology of Disease
Our way to understand tumor initiation and progression is centered around the notion that cancer is a genetic disease, whereby tumor cells become addicted to specific mutations in “driving” oncogenes that empower novel, often highly aberrant phenotypes. This notion has led to the expectation that blocking the activity of these mutant genes with targeted therapies should ultimately defeat tumors. This ideal scenario, however, clashes with the much grimmer reality of the clinical evidence, as such targeted therapies generally produce mere transient responses that are ultimately overthrown by the emergence of resistant cells dominating the tumor and its metastases (Flaherty et al, 2012). The typical clinical history of melanoma patients is a point in case. A large fraction of melanomas carry activating BRAF mutations, with the majority of these (about 80%) being V600E mutations (Fedorenko et al, 2015). Treatment with BRAF kinase inhibitors, such as PLX4720 (vemurafenib), causes rapid regression of many tumors that invariably recur after few months as refractory to BRAF inhibitors. Such resistance is, in most cases (> 70%), associated to reactivation of MAPK signaling; and yet, resistance still occurs in patients treated with the combination of BRAF and MEK inhibitors (Fedorenko et al, 2015).
So, what is the nature of tumor resistance? Current models typically associate resistance to targeted therapies to darwinian selection of mutant clones pre‐existing within the tumor. In contrast, recent evidence indicates that melanoma cells escape MAPK inhibition by PLX4720 in few days, that is, too quickly to be genetic (Hirata et al, 2015). In fact, BRAF inhibitors do not only act in tumor cells but also in the surrounding tumor fibroblasts, paradoxically activating them to produce a stiff, collagen‐rich extracellular matrix (ECM). Melanoma cells rapidly respond to this new microenvironment by increased integrin‐mediated ECM attachment, in turn reactivating MAPK signaling in a BRAF‐independent manner (Hirata et al, 2015). In other words, changing ECM composition and, more specifically, increasing ECM rigidity, serve as “safe haven” for melanoma cells, allowing them to readily escape the effects of PLX4720; as such, these drug‐induced biomechanical niches foster tumor growth and residual disease and thus epigenetically prelude to the installment of any type of genetic resistance (Hirata et al, 2015). Clearly, capturing the molecular nature of such adaptive responses is essential to target tumor resistance at its incipit.
Cells respond to external biomechanical inputs by rewiring their cytoskeleton, increasing contractility, and changing cell shape. In this issue of The EMBO Journal, Joon Kim and colleagues break new ground in this area by connecting these cytoskeletal changes to activation of YAP/TAZ in the emergence of melanoma resistance to BRAF inhibitors (Kim et al, 2016). Indeed, YAP/TAZ are closely related transcriptional activators that serve as central mediators of multiple cellular mechanotransduction pathways (Dupont et al, 2011). In line, F‐actin integrity and organization are leading regulators of YAP/TAZ nuclear localization in most, if not all, mammalian cells (Dupont et al, 2011; Aragona et al, 2013). Importantly, expression of YAP/TAZ is associated to tumor progression in several human solid tumors, and their activation endows tumor cells with cancer stem cell properties that include chemoresistance (Cordenonsi et al, 2011). In the present paper, the authors find that prolonged exposure to PLX4032 in vitro leads to the emergence of insensitive cell clones that survive and proliferate in spite of continuous drug administration. The uprising of drug resistance is accompanied by an increased polymerization of actin stress fibers, which becomes evident after 1 week of drug exposure. At the same time, the authors observed a shift toward a preferential nuclear localization of YAP/TAZ, together with the activation of their transcriptional target genes. YAP overexpression is actually able to confer vemurafenib resistance to sensitive cells, whereas YAP/TAZ depletion sensitizes cells to vemurafenib. Small molecules that inhibit actin polymerization (cytochalasin D) or actomyosin contractility (blebbistatin) induce YAP/TAZ redistribution to the cytoplasm and inhibit YAP/TAZ transcriptional effects. The same F‐actin inhibitors reduce the viability of vemurafenib‐resistant cells, either on their own, or in combination with vemurafenib. Based on the observation that suppression of actin remodeling can overcome survival to BRAF inhibitors, the authors performed a screening to identify kinases whose inhibition can synergize with BRAF inhibition to achieve a successful therapy. The screening revealed that TESK1, a kinase that inactivates Cofilin [an actin‐severing protein previously reported to inhibit YAP/TAZ activity (Aragona et al, 2013)], is relevant to keep YAP/TAZ in the nucleus of vemurafenib‐resistant cells and that its depletion sensitizes cells to BRAF inhibitors. Interestingly, with the onset of drug resistance, melanoma cells gain a higher dependency on YAP/TAZ for their survival and proliferation, to the extent that YAP/TAZ inhibition (by siRNAs, or treatment with cytochalasin D or blebbistatin) is sufficient to block cell growth, even in the absence of the BRAF inhibitor. TESK1 itself is not activated by prolonged exposure to vemurafenib, but its function becomes essential as cells become independent of oncogenic BRAF, and dependent on YAP/TAZ.
The work of Kim et al (2016) is consistent with prior reports on the role of YAP/TAZ in chemoresistance to RAF and MEK inhibitors, and to other chemotherapy agents (Cordenonsi et al, 2011; Lin et al, 2015a,b). For example, multiple tumor types, irrespectively of their genetic background, are vulnerable to combined inhibition of YAP and the MAPK pathway (Lin et al, 2015b). However, YAP/TAZ are never activated by mutations and only rarely amplified. Kim et al (2016) have the merit to add a new layer to this picture, indicating that increased cellular mechano‐responsiveness may be the culprit of YAP/TAZ‐induced drug resistance.
It is thus tempting to combine different studies and propose the following model as mainframe of drug resistance (Fig 1): inactivation of MAPK signaling with BRAF/MEK inhibitors bears the side effect of increasing ECM deposition by the tumor stroma, in turn enhancing mechanotransduction and YAP/TAZ activation in cancer cells. YAP/TAZ are potent triggers of cellular fitness, likely as part of their CSC‐endowing repertoire (Cordenonsi et al, 2011).
Figure 1. Biomechanical pathways to vemurafenib resistance.
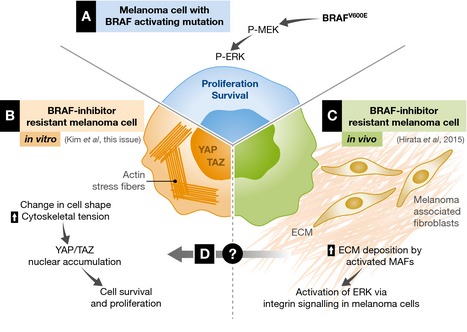
(A) In a melanoma cell harboring BRAF oncogenic mutation, the MAPK/ERK signaling pathway is constitutively activated and fosters uncontrolled cell survival and proliferation. Pharmacological BRAF inhibition silences this pathway, but the effect is transient and it is quickly subverted by the emergence of alternative survival mechanisms. (B) Kim and colleagues show that melanoma cells that acquire resistance to the BRAF inhibitor PLX4032 in vitro activate YAP/TAZ in response to increased cytoskeletal tension. (C) In vivo, therapy with PLX4032 leads to hyperactivation of the tumor stroma, with increased ECM deposition by melanoma‐associated fibroblasts. (D) An intriguing hypothesis is that the intrinsic increase in mechano‐sensitivity of melanoma cells and the alterations in ECM properties due to stroma activation may cooperate to boost YAP/TAZ activation, providing an alternative survival pathway when oncogenic BRAF is inhibited.
This scenario is at least consistent with a series of preliminary observations in the clinical context given that YAP levels anticorrelate with the clinical efficacy of BRAF and MEK inhibitors; also, YAP levels raise after a tumor develops drug resistance (Lin et al, 2015b). Furthermore, histological re‐examination of residual disease specimens confirmed that BRAF inhibition modulates the fibrous ECM and induces an elongated melanoma cell morphology (Hirata et al, 2015).
We also note that a piece of evidence is however still missing to make all this a full circle, as there is not yet formal proof that the non‐cell‐autonomous “safe haven” provided by the fibroblast and their ECM nest is actually inducing the cytoskeleton‐driven, cell autonomous activation of YAP/TAZ studied by Kim et al (2016).
Melanoma immune therapy has made headlines because of its remarkable efficacy in a substantial fraction of patients (Flaherty et al, 2012). However, for those who do not respond, melanoma remains a devastating disease raising the need of better biomarkers and combinatorial therapies. It is tempting to speculate that attenuation of YAP/TAZ signaling—either by targeting matrix stiffness, melanoma cytoskeletal organization, or YAP/TAZ directly—may slow down proliferation and help the immune system to overcome the tumor.
Irrespectively of these remaining gaps, the paper of Kim et al (2016), and the other set of related evidence, greatly emphasize the role of YAP/TAZ and mechanotransduction as prime targets of cancer therapy.
See also: MH Kim et al (March 2016)
Contributor Information
Francesca Zanconato, Email: francesca.zanconato@unipd.it.
Stefano Piccolo, Email: piccolo@bio.unipd.it.
References
- Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, Piccolo S (2013) A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin‐processing factors. Cell 154: 1047–1059 [DOI] [PubMed] [Google Scholar]
- Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti AR, Poletti A, Daidone MG, Dupont S, Basso G, Bicciato S, Piccolo S (2011) The Hippo transducer TAZ confers cancer stem cell‐related traits on breast cancer cells. Cell 147: 759–772 [DOI] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S (2011) Role of YAP/TAZ in mechanotransduction. Nature 474: 179–183 [DOI] [PubMed] [Google Scholar]
- Fedorenko IV, Gibney GT, Sondak VK, Smalley KS (2015) Beyond BRAF: where next for melanoma therapy? Br J Cancer 112: 217–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Hodi FS, Fisher DE (2012) From genes to drugs: targeted strategies for melanoma. Nat Rev Cancer 12: 349–361 [DOI] [PubMed] [Google Scholar]
- Hirata E, Girotti MR, Viros A, Hooper S, Spencer‐Dene B, Matsuda M, Larkin J, Marais R, Sahai E (2015) Intravital imaging reveals how BRAF inhibition generates drug‐tolerant microenvironments with high integrin beta1/FAK signaling. Cancer Cell 27: 574–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MH, Kim J, Hong H, Lee S‐H, Lee J‐K, Jung E, Kim J (2016) Actin remodeling confers BRAF inhibitor resistance to melanoma cells through YAP/TAZ activation. EMBO J 35: 462–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Pelissier FA, Zhang H, Lakins J, Weaver VM, Park C, LaBarge MA (2015a) Microenvironment rigidity modulates responses to the HER2 receptor tyrosine kinase inhibitor lapatinib via YAP and TAZ transcription factors. Mol Biol Cell 26: 3946–3953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Sabnis AJ, Chan E, Olivas V, Cade L, Pazarentzos E, Asthana S, Neel D, Yan JJ, Lu X, Pham L, Wang MM, Karachaliou N, Cao MG, Manzano JL, Ramirez JL, Torres JM, Buttitta F, Rudin CM, Collisson EA et al (2015b) The Hippo effector YAP promotes resistance to RAF‐ and MEK‐targeted cancer therapies. Nat Genet 47: 250–256 [DOI] [PMC free article] [PubMed] [Google Scholar]