

Abstract
The activation of transcriptional coactivators YAP and its paralog TAZ has been shown to promote resistance to anti‐cancer therapies. YAP/TAZ activity is tightly coupled to actin cytoskeleton architecture. However, the influence of actin remodeling on cancer drug resistance remains largely unexplored. Here, we report a pivotal role of actin remodeling in YAP/TAZ‐dependent BRAF inhibitor resistance in BRAF V600E mutant melanoma cells. Melanoma cells resistant to the BRAF inhibitor PLX4032 exhibit an increase in actin stress fiber formation, which appears to promote the nuclear accumulation of YAP/TAZ. Knockdown of YAP/TAZ reduces the viability of resistant melanoma cells, whereas overexpression of constitutively active YAP induces resistance. Moreover, inhibition of actin polymerization and actomyosin tension in melanoma cells suppresses both YAP/TAZ activation and PLX4032 resistance. Our siRNA library screening identifies actin dynamics regulator TESK1 as a novel vulnerable point of the YAP/TAZ‐dependent resistance pathway. These results suggest that inhibition of actin remodeling is a potential strategy to suppress resistance in BRAF inhibitor therapies.
Keywords: actin cytoskeleton, BRAF inhibitor resistance, melanoma, TAZ, YAP
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Molecular Biology of Disease
Introduction
Systematic studies on genetic alterations in human malignancies have enabled the development of genotype‐driven targeted therapy for several types of cancers. However, the emergence of acquired resistance to targeted anti‐cancer agents poses a critical hurdle for improving cancer patient prognosis. BRAF or BRAF/MEK combined inhibition initially decreases tumor burden of BRAF V600E mutant melanoma patients, but the vast majority of them encounter emerging resistance within 1 or 2 years (Chapman et al, 2011; Robert et al, 2015). Several studies have identified potential mechanisms of BRAF inhibitor resistance: EGFR upregulation driven by SOX10 loss (Girotti et al, 2013; Sun et al, 2014), acquired NRAS and MEK1 mutations (Van Allen et al, 2014), WNT5A upregulation (Anastas et al, 2014), and BRAF alternative splicing (Poulikakos et al, 2011). However, clinical genetic screening studies on BRAF inhibitor resistance have repeatedly reported that a considerable proportion of patients have unrevealed resistance mechanisms (Rizos et al, 2014; Van Allen et al, 2014).
The Hippo/YAP (Yes‐associated protein) signaling pathway is a key determinant of organ size control, stem cell homeostasis, and cellular differentiation (Zhao et al, 2011b). Two Hippo pathway transducers, YAP and its paralog TAZ (transcriptional coactivator with PDZ‐binding motif), are coactivators of transcription factors, such as TEADs, SMADs, and RUNX. Previous studies have highlighted tumorigenic potential of YAP and TAZ in mouse model (Zhou et al, 2009) and their substantial contribution to the acquisition of both cancer stem cell‐related traits in breast cancer and pro‐invasive properties in melanoma (Cordenonsi et al, 2011; Nallet‐Staub et al, 2014). Furthermore, YAP rescues cancer cells from oncogenic KRAS deprivation in transgenic mouse model (Kapoor et al, 2014), and YAP overexpression bypasses KRAS knockdown through epithelial–mesenchymal transition in a c‐FOS‐dependent manner (Shao et al, 2014). Remarkably, Lin et al (2015) recently showed that YAP is implicated in RAF and MEK inhibitor resistance, and suppression of YAP activity improves drug sensitivities in BRAF and KRAS mutant cancer cells. This growing evidence indicates that YAP/TAZ activation in cancer cells is oncogenic and also promotes drug resistance. Revealing the upstream and downstream molecular mechanisms of YAP/TAZ will facilitate the development of therapeutic targeting of the YAP/TAZ pathway.
YAP/TAZ activity in mammalian cells is influenced by various signaling inputs including GPCR, WNT signaling, EGF, mechanical stress, and cell–cell/cell–matrix contact (Halder et al, 2012; Moroishi et al, 2015). These inputs converge on two mutually interacting YAP/TAZ regulators, the Hippo signaling pathway and actin cytoskeleton architecture. Hippo signaling kinases LATS1/2 negatively regulate YAP/TAZ activity by phosphorylating serine residues which mediates YAP/TAZ cytoplasmic retention and degradation. The actin cytoskeleton is another important axis of YAP/TAZ activity regulation. Increased actin cytoskeletal tension promotes YAP/TAZ nuclear translocation and prevents degradation in both Hippo‐dependent (Zhao et al, 2012) and Hippo‐independent ways (Dupont et al, 2011; Calvo et al, 2013). In addition, recent studies have suggested that an increase in actin filaments releases YAP from Angiomotin to enable YAP nuclear translocation (Zhao et al, 2011a; Mana‐Capelli et al, 2014). The relationship between the actin cytoskeleton and YAP/TAZ localization/activity allows us to understand how cells translate mechanical stimuli and cytoskeletal tension into their transcriptional programs. Importantly, cancer cells are subjected to continuous actin cytoskeletal remodeling that adapts themselves to environmental mechanical stimuli and matrix stiffness, promoting their survival, migration, and metastasis (Paszek et al, 2005; Butcher et al, 2009). However, the potential linkage between actin cytoskeleton remodeling and acquisition of drug resistance in cancer has not been uncovered, although actin dynamics has been suggested to interact with receptor tyrosine kinase pathways, and regulate gene transcriptions via the MRTF‐SRF circuit (Olson & Nordheim, 2010).
Here, we show remodeling of the actin cytoskeleton in two BRAF V600E mutant melanoma cell lines during the acquisition of resistance to the BRAF inhibitor PLX4032 (vemurafenib). We demonstrate that nuclear accumulation of YAP/TAZ is accompanied by the increase in actin stress fiber formation. Actin‐dependent YAP/TAZ activation appears to play a critical role in PLX4032 resistance establishment, promoting E2F‐related cell cycle progression and upregulation of EGFR, AKT, and c‐MYC. YAP/TAZ depletion suppresses PLX4032‐resistant cell growth, whereas constitutively active YAP expression raises PLX4032 resistance in melanoma cells. Moreover, inhibition of actin filament assembly and tension suppresses both YAP/TAZ activation and PLX4032 resistance. Consistent with the importance of the actin cytoskeleton, our kinome‐wide RNAi screening identifies TESK1, a kinase regulating actin dynamics, as a potential synthetic lethal target of PLX4032‐resistant melanoma cells. TESK1 depletion affects YAP/TAZ localization and decreases the survival of resistant cells. These data collectively point to an important role of actin remodeling and YAP/TAZ activation in the acquisition of resistance to BRAF inhibition.
Results
PLX4032 treatment induces actin remodeling in BRAF mutant melanoma cells
We established PLX4032‐resistant melanoma cell lines which harbor BRAF V600E mutation. SKMEL28 and WM3248 cells were treated with 2 μM of PLX4032 for 2 months, and PLX4032 resistance of surviving clones was confirmed by dose–response analyses (Fig 1A). We confirmed that resistant cells maintained BRAF V600E mutation (Fig 1B). As expected, PLX4032 treatment efficiently blocked the proliferation of parental cells, whereas resistant SKMEL28 and WM3248 cells continued to proliferate in the presence of PLX4032 (Fig 1C and D). It has been shown recently that adaptive resistance to PLX4032 can be induced by SOX10 suppression, which leads to increased EGFR and decreased MITF expression (Sun et al, 2014). Similarly, we observed suppression of both SOX10 and MITF expression (Fig 1E), and elevation of EGFR levels in resistant cells (Fig 1F). In addition, resistant SKMEL28 and WM3248 cells showed higher levels of phospho‐ERK than parental cells, and increased ERK activity in resistant WM3248 cells was maintained even after 24‐h PLX4032 treatment (Fig 1F). As shown in Fig 1F, PLX4032 treatment suppressed c‐MYC in parental cells, whereas PLX4032 did not change c‐MYC levels in resistant cells.
Figure 1. Establishment and characterization of PLX4032‐resistant melanoma cells.
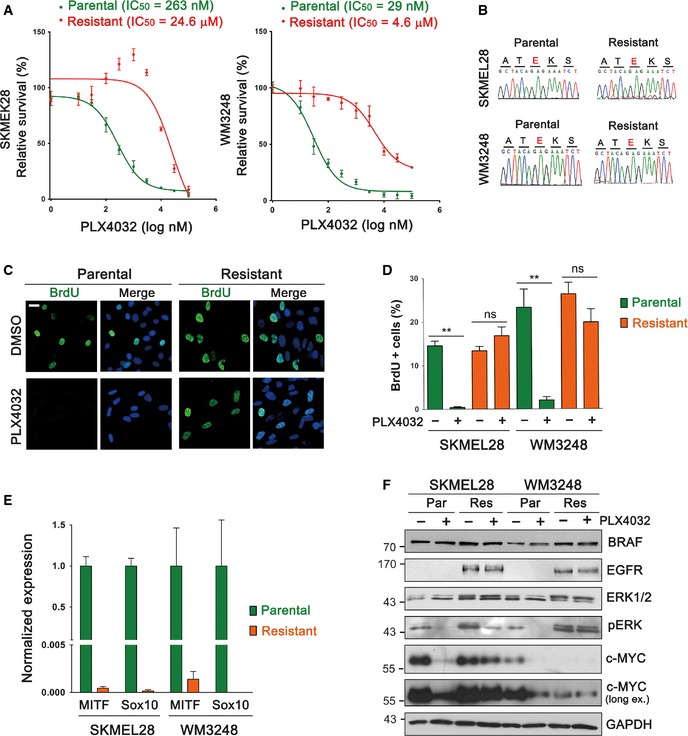
- PLX4032 dose–response curves of parental and resistant melanoma cells. Relative cell viability, compared to DMSO control, was measured by CCK8 assay after PLX4032 treatment for 72 h.
- Sequencing results confirming BRAF V600E mutation (c.1799T>A) in resistant cells.
- Immunofluorescence images showing BrdU incorporation in parental and resistant WM3248 cells after PLX4032 (2 μM) or DMSO treatment for 24 h.
- Quantification of the experiment presented in (C). Both SKMEL28 and WM3248 cells were analyzed.
- qRT–PCR analysis comparing SOX10 and MITF expression levels in parental and resistant cells.
- Immunoblotting for the indicated proteins. Cells were plated and incubated without drugs for 24 h, and then treated with PLX4032 (2 μM) or DMSO for 24 h.
We noticed a gradual change in the shape of melanoma cells during the establishment of PLX4032 resistance. Both SKMEL28 and WM3248 cells spread more extensively in adherent monolayer culture after the acquisition of resistance (Fig 2A). The significance of the morphological change was confirmed by visualizing cell boundaries and measuring the area (Fig 2B and C). The actin cytoskeleton is a major determinant of cell morphology, and melanoma cells have been shown to dynamically change their shapes along with the actin architecture upon Rho/Rac activity switch (Sanz‐Moreno et al, 2008). Therefore, we reasoned that PLX4032‐resistant cells might display altered actin structures and dynamics. Interestingly, we observed substantial increases in basal actin stress fiber formation in both SKMEL28 and WM3248 cells resistant to PLX4032 (Fig 2D). Resistant SKMEL28 cells showed an increase in stress fiber formation throughout the cell body, whereas thickening of transverse arc and dorsal stress fibers was dominant in resistant WM3248 cells (Fig 2D). As shown in Fig 2E, resistant cells exhibited much decreased apical actin pool and remarkably flattened cell shape, which may contribute to the extensive spreading of resistant cells. Next, we examined the time‐course of actin cytoskeleton remodeling upon PLX4032 treatment in parental SKMEL28 and WM3248 cells. PLX4032 treatment for 12 h or 24 h did not cause noticeable changes in the actin cytoskeleton, indicating that actin remodeling is not an acute cellular response to PLX4032. Interestingly, an increase in actin stress fibers was apparent after 7 days, and prolonged PLX4032 treatment provoked robust stress fiber formation (Fig 2F).
Figure 2. PLX4032‐resistant melanoma cells exhibit actin cytoskeleton remodeling.
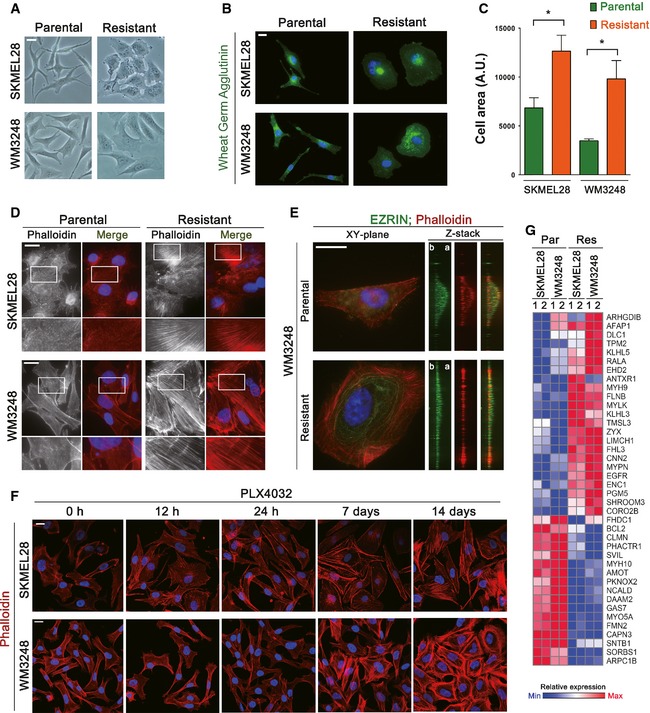
- Phase‐contrast images showing morphological changes in resistant cell lines.
- Immunofluorescence staining for visualizing cell boundaries. Cells were plated at low densities to prevent cell–cell contacts and labeled with fluorescent wheat germ agglutinin conjugates.
- Quantification of the area of cells prepared as in (B).
- Fluorescence micrographs visualizing actin filaments stained with phalloidin‐Alexa Fluor 594. Magnified views of boxed areas are shown below.
- Z‐plane views of phalloidin‐labeled actin filaments in WM3248 cells (b, basal; a, apical). The cell apex was marked by anti‐Ezrin staining. Z‐stack images were generated by 3D volume rendering of deconvoluted XY‐plane images.
- Confocal fluorescence micrographs showing actin filaments in parental SKMEL28 and WM3248 cells treated with PLX4032 (2 μM) for the indicated times.
- A heatmap showing expression levels of significantly altered actin cytoskeleton‐related genes identified by a microarray analysis comparing parental and resistant cells. These genes were selected by gene ontology analysis described in Fig EV1B.
To test whether actin remodeling in resistant melanoma cells involves changes in gene expression patterns, we performed an expression microarray analysis comparing parental and resistant cells of both SKMEL28 and WM3248. We found 829 significantly altered genes common to both resistant cell lines [fold change > 2.0 and P‐value < 0.05 by local‐pooled‐error (LPE) test; Fig EV1A and Table EV1], and gene ontology analyses for the altered genes revealed multiple actin‐related functional categories as significantly altered in resistant cells (Fig EV1B and Table EV2). Actin cytoskeleton regulator genes upregulated in resistant cells include Coronin (CORO2B), Tropomyosin (TPM2) and Zyxin (ZYX), and downregulated genes include ARP2/3 complex subunit (ARPC1B), Formin (FMN2) and Supervilin (SVIL) (Fig 2G). The expression levels of non‐muscle myosins MYO5A, MYH9, and MYH10 were also significantly changed. These results suggest that remodeling of actin cytoskeletal architecture in resistant cell lines is driven by a global change in the expression levels of genes associated with actin organization. Because actin cytoskeleton architecture and resulting mechanical properties impact on diverse aspects of cellular behavior (Vogel & Sheetz, 2006; Jaalouk & Lammerding, 2009), we hypothesized that actin remodeling in melanoma cells may contribute to the induction of adaptive resistance to BRAF inhibitors.
Figure EV1. Comparison of expression profiles between PLX4032‐resistant and parental cells reveals significant alterations of multiple genes encoding actin cytoskeleton regulators.

- Venn diagram showing the number of significantly altered genes in a microarray analysis comparing parental and resistant cells of SKMEL28 and WM3248.
- A list showing significantly enriched gene ontology terms (Benjamini–Hochberg FDR < 0.05) related to the actin cytoskeleton on functional annotation charts which analyze 829 significantly altered genes.
YAP/TAZ nuclear localization and their transcriptional activity are promoted in PLX4032‐resistant cells
Recent studies have identified actin cytoskeletal structure and tension as dominant upstream regulators of YAP/TAZ activity in mammalian cells (Dupont et al, 2011; Zhao et al, 2012). To test whether YAP/TAZ provide a link between actin remodeling and PLX4032 resistance, we first compared the localization of YAP/TAZ in parental and resistant cells. We used an antibody detecting both YAP and TAZ. Importantly, resistant WM3248 cells exhibited an increase in nuclear YAP/TAZ localization when compared with parental WM3248 cells (Fig 3A and B). An increase in nuclear YAP/TAZ localization was also observed in resistant SKMEL28 cells cultured in mid to high confluency, which normally induces cytoplasmic retention of YAP/TAZ (Fig EV2A and B). Moreover, we observed a gradual increase in nuclear YAP/TAZ enrichment in parental SKMEL28 and WM3248 cells after PLX4032 treatment (Figs 3C and D, and EV2C and D). Nuclear YAP/TAZ localization peaks after 7 days of PLX4032 treatment in SKMEL28 cells and after 14 days in WM3248 cells, which correspond to the progression of actin remodeling shown in Fig 2F. To further clarify YAP/TAZ localization status in resistant cells, we fractionated nuclear and cytoplasmic proteins and found an increase in nuclear YAP/TAZ in resistant SKMEL28 cells, and nuclear YAP in resistant WM3248 cells (Fig 3E and F).
Figure 3. PLX4032‐resistant melanoma cells exhibit both nuclear localization and elevated transcriptional activity of YAP/TAZ .
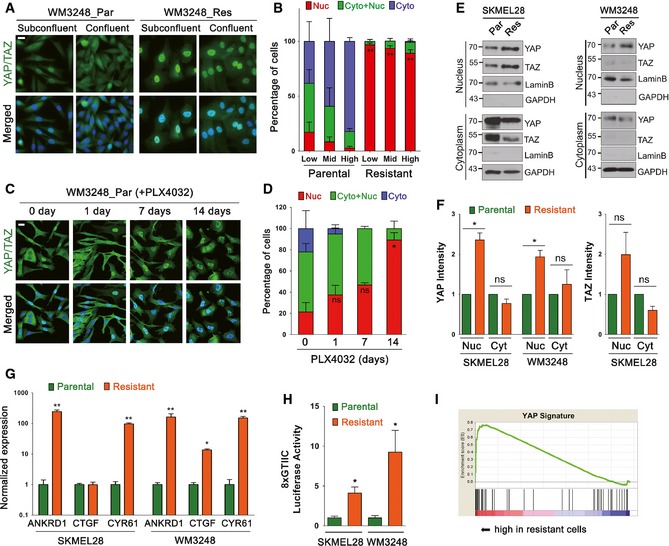
- Immunofluorescence micrographs showing YAP/TAZ localization in parental and resistant WM3248 cells in subconfluent and confluent cultures. Cells were labeled with anti‐YAP/TAZ antibody.
- Quantification of the experiment presented in (A). Cells were classified as nuclear (Nuc), nucleocytoplasmic (Cyto+Nuc), and cytoplasmic (Cyto) according to the subcellular localization of YAP/TAZ (150–250 cells were examined for each cell line). Cells were seeded at low, middle, and high confluency. Proportion of nuclear YAP/TAZ in resistant cells was compared with that in parental cells of the same confluency.
- Immunofluorescence micrographs showing YAP/TAZ localization. Parental WM3248 cells were treated with PLX4032 (2 μM) for the indicated times before labeling with anti‐YAP/TAZ antibody.
- Quantification of the experiment presented in (C). Proportion of nuclear YAP/TAZ was compared to that of control (0 day) by t‐test.
- Immunoblotting for the indicated proteins. Cell lysates were fractionated into nuclear and cytoplasmic fractions. Lamin B and GAPDH were shown as loading controls for each fractionation.
- Quantification of band intensities of the immunoblots presented in (E).
- qRT–PCR analyses of relative normalized expression of YAP/TAZ target genes.
- Luciferase reporter assay of YAP/TAZ transcriptional activity. Cells were transfected with 8xGTIIC reporter vector for 24 h before the analysis.
- GSEA enrichment plots demonstrating upregulation of YAP signature genes in resistant cells compared with parental cells (normalized enrichment score = 1.42, nominal P‐value < 0.001, and FDR q‐value = 0.174).
Figure EV2. Resistant melanoma cells exhibit both YAP/TAZ nuclear localization and YAP signature gene upregulation.
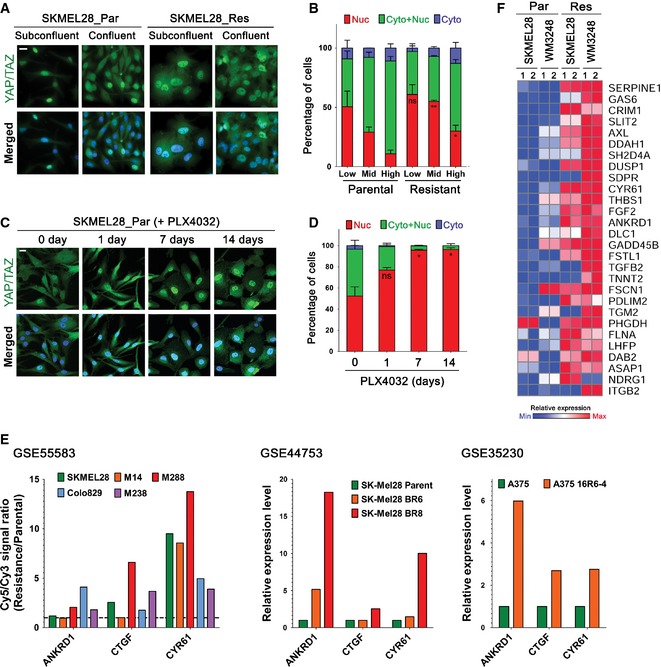
- Immunofluorescence micrographs showing YAP/TAZ localization in parental and resistant SKMEL28 cells in subconfluent and confluent cultures. Cells were labeled with anti‐YAP/TAZ antibody.
- Quantification of the experiment presented in (A). Cells were seeded at low, middle, and high confluency. Proportion of nuclear YAP/TAZ in resistant cells was compared with that in parental cells of the same confluency.
- Immunofluorescence micrographs showing YAP/TAZ localization. Parental SKMEL28 cells were treated with PLX4032 (2 μM) for the indicated times before labeling with anti‐YAP/TAZ antibody.
- Quantification of the experiment presented in (C).
- Comparison of YAP/TAZ target gene expressions (ANKRD1, CTGF, and CYR61) between parental and BRAF inhibitor‐resistant melanoma cells in published gene expression datasets (GSE55583, GSE44753, and GSE35230 from GEO database).
- A heatmap showing upregulated YAP signature genes in resistant cells identified by GSEA analysis (the leading edge subset) comparing expression profiles of resistant and parental cells.
Data information: All data are mean and SEM [two biologic replicates (D) or three biologic replicates (B)], and P‐values were determined by t‐test (*P < 0.05 and **P < 0.01). The nuclei were stained with DAPI (blue), and scale bars represent 20 μm.
Next, we examined transcriptional activation of YAP/TAZ target genes ANKRD1, CTGF, and CYR61 in resistant cells. As shown in Fig 3G, expression levels of the three target genes were significantly increased in resistant WM3248 cells, and ANKRD1 and CYR61 were increased in resistant SKMEL28 cells. We also performed a luciferase assay using a YAP/TAZ‐responsive TEAD reporter, 8XGTIIC‐luciferase (Dupont et al, 2011), which contains eight TEAD binding sites. The reporter activity was significantly higher in both SKMEL28 and WM3248 cells resistant to PLX4032, confirming increased transcriptional activity of YAP/TAZ (Fig 3H). We next explored published expression array studies, analyzing BRAF inhibitor‐resistant melanoma cell lines, in Gene Expression Omnibus (GEO) database (Edgar et al, 2002). Three independent GEO datasets (GSE55583, GSE44753, and GSE35230) show increased levels of ANKRD1, CTGF, and CYR61 expression in resistant cell lines, further confirming our results (Fig EV2E). We next performed a gene set enrichment analysis (GSEA) (Subramanian et al, 2005) on our expression microarray data comparing resistant SKMEL28 and WM3248 versus parental SKMEL28 and WM3248 cells. The GSEA using MSigDB (C6 oncogenic signatures) on the microarray data revealed significant enrichment of YAP signature genes (nominal P‐value < 0.05 and FDR < 0.25) in resistant cells (Figs 3I and EV2F, and Appendix Table S1). Together, these results suggest that enhanced YAP/TAZ nuclear localization in resistant cells induces a YAP/TAZ‐dependent transcriptional activity which might confer drug resistance.
YAP/TAZ knockdown reduces resistant cell viability, whereas constitutively active YAP induces PLX4032 resistance
To address the role of YAP/TAZ activation in BRAF inhibitor resistance, we next asked whether YAP/TAZ knockdown can suppress the survival of PLX4032‐resistant melanoma cells. Dose–response analyses revealed that double knockdown of YAP and TAZ, using two distinct siRNA sets, causes shifts in PLX4032 sensitivity in resistant cells (Figs 4A and EV3A). A significant restoration of PLX4032 sensitivity after YAP/TAZ knockdown was noted in resistant SKMEL28 cells and resistant WM3248 cells showed lower levels of restoration of PLX4032 sensitivity. Growth arrest of both resistant SKMEL28 and resistant WM3248 cells was maintained until 7 days after siRNA transfection (Figs 4B and EV3B). Parental cell lines also showed a slight increase in PLX4032 sensitivity upon YAP/TAZ knockdown although only WM3248 cells transfected with YAP/TAZ siRNA set 1 (siYT #1) showed statistical significance (Fig 4A). Next, we confirmed that YAP/TAZ knockdown efficiently suppresses the proliferation of PLX4032‐resistant cells, using BrdU incorporation assay (Figs 4C and D, and EV3C). YAP/TAZ knockdown alone in parental cells did not cause a significant reduction in the number of BrdU positive cells, whereas YAP/TAZ knockdown was sufficient to inhibit cell proliferation even in the absence of PLX4032 treatment in both resistant cell lines. These results suggest that PLX4032 resistance is associated with higher YAP/TAZ dependency.
Figure 4. YAP/TAZ knockdown causes growth arrest in melanoma cells resistant to PLX4032.
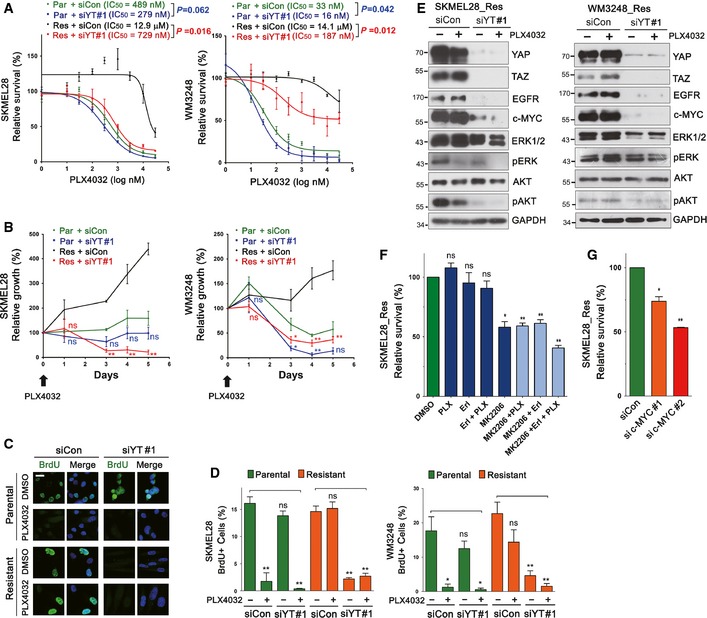
- PLX4032 dose–response curves of parental and resistant cells after YAP/TAZ knockdown. Cells were transfected with either control (siCon) or YAP plus TAZ (siYT#1) siRNAs for 72 h, and then treated with PLX4032 at the indicated concentrations for additional 72 h. Relative cell viability, compared with DMSO control, was measured by CCK8 assay. Sigmoidal dose–response curves were fitted to data, and significance of the difference in IC50 values was determined by extra sum‐of‐squares F test.
- Time‐course analyses of cell viability after YAP/TAZ siRNA #1 knockdown. Cells were transfected with the indicated siRNAs for 48 h, and then treated with PLX4032 (2 μM) for the indicated times. Relative cell viability, compared with initial cell viability, was measured by CCK8 assay. Significance of the difference between control and YAP/TAZ depletion for parental or resistant cells was determined by t‐test.
- Immunofluorescence micrographs identifying BrdU‐incorporated cells. WM3248 cells were transfected with the indicated siRNAs for 48 h, and then treated with PLX4032 (2 μM) or DMSO for additional 24 h before 45‐min BrdU labeling.
- Quantification of the experiment presented in (C). Both SKMEL28 and WM3248 cells were analyzed.
- Immunoblotting for the indicated proteins after transfection of siRNAs. Resistant SKMEL28 and WM3248 cells were transfected with the indicated siRNAs for 48 h, and then treated with PLX4032 (2 μM) or DMSO for additional 24 h.
- Cell viability assay of resistant SKMEL28 cells treated with PLX4032 (PLX, 2 μM), Erlotinib (Erl, 2.5 μM), MK‐2206 (2.5 μM), or their combinations. Relative cell viability, compared with DMSO control, was analyzed by CCK8 assay after drug treatment for 72 h.
- Cell viability analyses of resistant SKMEL28 cells. Cells were transfected with the indicated siRNAs for 5 days. Relative cell viability, compared with that of control siRNA‐transfected cells, was measured by CCK8 assay.
Figure EV3. Validation of the specificity of YAP/TAZ knockdown, and comparison of YAP/TAZ knockdown sensitivity between parental and resistant melanoma cells.
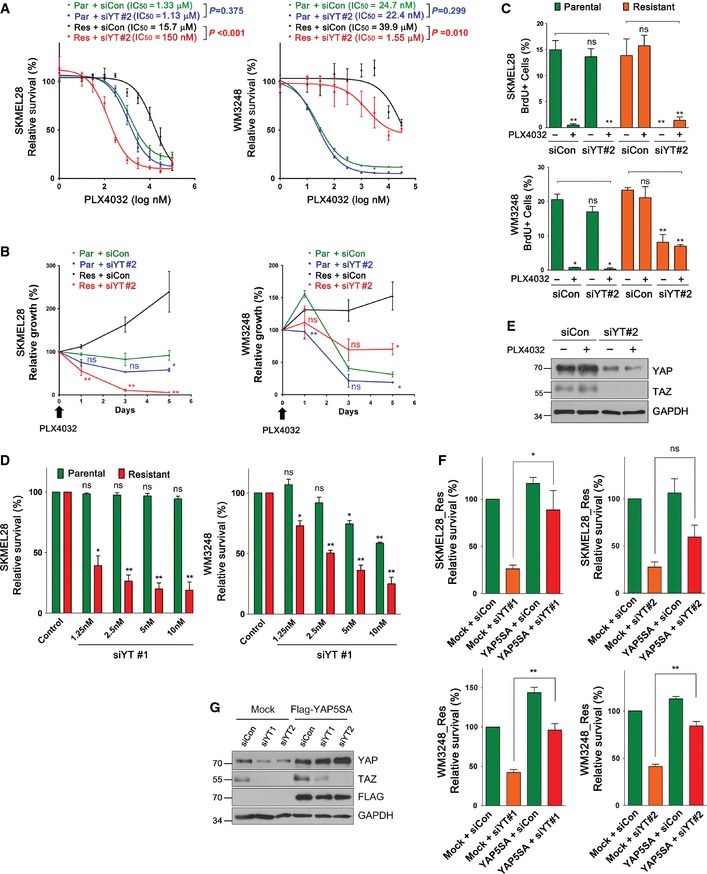
- PLX4032 dose–response curves of parental and resistant cells after YAP/TAZ knockdown. Cells were transfected with either control or YAP/TAZ siRNA set 2 (siYT#2) for 72 h, and then treated with PLX4032 at the indicated concentrations for additional 72 h. Relative cell viability, compared to DMSO control, was measured by CCK8 assay. Sigmoidal dose–response curves were fitted to data, and significance of the difference in IC 50 values was determined by extra sum‐of‐squares F test.
- Time‐course analyses of cell viability after YAP/TAZ knockdown. Cells were transfected with the indicated siRNAs for 48 h, and then treated with PLX4032 (2 μM) for the indicated times. Relative cell viability, compared to initial cell viability, was measured by CCK8 assay. Significance of the difference between control and YAP/TAZ depletion for parental or resistant cells were determined by t‐test.
- Quantification of BrdU assay of SKMEL28 and WM3248 cells transfected with either control siRNA or siYT#2. Cells were treated as in Fig 4C.
- Cell viability analyses of SKMEL28 and WM3248 cells transfected with variable doses of YAP/TAZ siRNAs and control siRNA. Cells were transfected with siRNAs for 144 h and cell viability, compared with that of control siRNA‐transfected cells, was measured by CCK8 assay.
- Immunoblotting for the indicated proteins 48 h after transfection of siRNAs followed by 24‐h PLX4032 or DMSO treatment.
- Cell viability analyses of resistant SKMEL28 and WM3248 cells transfected with siRNAs and siRNA‐resistant flag‐YAP‐5SA retrovirus. Cells were transfected with retrovirus for 24 h, and then transfected with the indicated siRNAs (forward transfection) for 72 h (SKMEL28) or 144 h (WM3248). Relative cell viability, compared with that of mock and control siRNA‐transfected cells, was measured by CCK8 assay.
- Immunoblotting of the indicated proteins in transfected HEK293T. HEK293T cells were transfected with siRNA‐resistant Flag‐YAP5SA retrovirus for 24 h, and then transfected with the indicated siRNAs for 72 h (reverse transfection).
Data information: All data are mean and SEM (three biologic replicates). In all graphs except (A), P‐values were determined by t‐test (*P < 0.05 and **P < 0.01). Source data are available online for this figure.
To further demonstrate that resistant cells gained higher dependency on YAP/TAZ for their survival, we compared cell viability of parental and resistant cells after transfection with variable doses of siRNAs. As shown in Fig EV3D, resistant cells showed significant loss of cell viability even at low siRNA doses (1.25 and 2.5 nM), which did not affect the viability of parental cells. Therefore, we conclude that PLX4032‐resistant cells are highly dependent on YAP/TAZ activity for both survival and proliferation. RNAi‐mediated YAP/TAZ knockdown was confirmed by Western blot analyses (Figs 4E and EV3E). In addition, we performed a rescue experiment of YAP/TAZ knockdown, using a vector expressing YAP‐5SA, a constitutively active YAP containing five serine‐to‐alanine substitutions that prevent its phosphorylation and cytoplasmic retention. To disrupt the siRNA recognition site, we introduced silent mutations in YAP‐5SA cDNA. Expression of siRNA‐resistant YAP‐5SA rescued the suppression of cell viability caused by YAP/TAZ siRNAs in both SKMEL28 and WM3248 cells, confirming the specificity of YAP/TAZ knockdown (Fig EV3F and G).
We subsequently examined changes of molecular pathways in resistant cells upon YAP/TAZ depletion. Remarkably, YAP/TAZ knockdown caused a reduction in EGFR, c‐MYC, and phospho‐AKT (pAKT) levels in both SKMEL28 and WM3248 cells (Fig 4E). Reduction in c‐MYC protein and mRNA was further confirmed by immunofluorescence staining and quantitative RT–PCR (qRT–PCR), respectively (Fig EV4A and B). To test the significance of these changes on BRAF inhibitor resistance, we treated resistant cells with EGFR inhibitor Erlotinib, and AKT inhibitor MK‐2206 (Figs 4F and EV4C). MK‐2206 partially suppressed resistant cell survival, and combined treatment with Erlotinib and PLX4032 resulted in slightly higher suppression of cell viability. We also depleted c‐MYC expression by two independent c‐MYC siRNAs (Fig EV4D), and observed a reduction in cell viability in resistant SKMEL28 cells (Fig 4G). These results suggest that AKT, EGFR, and c‐MYC contribute to the survival of resistant cells, although inhibition of the pathways does not seem to be sufficient to overcome BRAF inhibitor resistance. Consistent with low c‐MYC levels in resistant WM3248 cells (Fig 1F), the survival of resistant WM3248 cells was not affected by c‐MYC knockdown (Fig EV4E). In addition, we found that overexpression of c‐MYC is not sufficient for rescuing cell viability loss caused by YAP/TAZ knockdown (Fig EV4F and G).
Figure EV4. YAP/TAZ knockdown downregulates c‐MYC expression in resistant cells, and AKT inhibition suppresses the viability of resistant WM3248 cells.
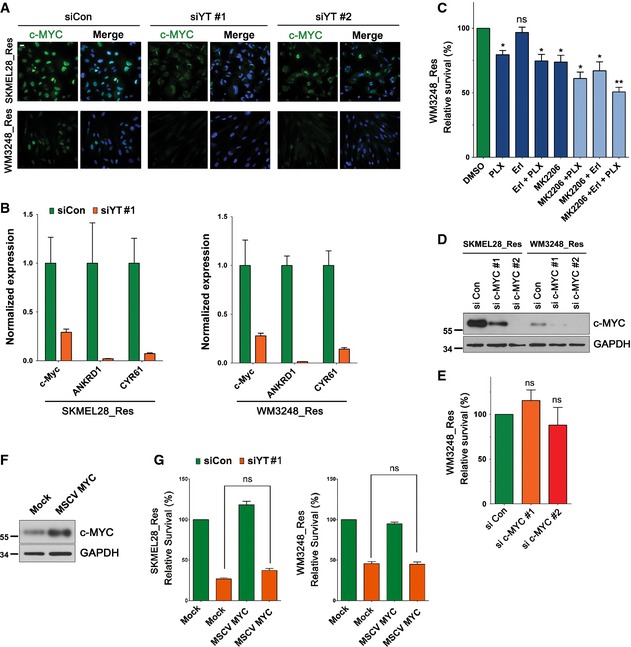
- Immunofluorescence micrographs showing c‐MYC in resistant SKMEL28 and WM3248 cells 72 h after transfection of control siRNA or YAP/TAZ siRNAs. Cells were labeled with anti‐c‐MYC antibody.
- qRT–PCR analyses of the expression of c‐MYC and YAP/TAZ target genes. Resistant SKMEL28 and WM3248 cells were transfected with the indicated siRNAs for 72 h.
- Cell viability analyses of resistant WM3248 cells treated with PLX4032 (PLX, 2 μM), Erlotinib (Erl, 2.5 μM), MK‐2206 (2.5 μM) or their combinations. Relative cell viability, compared with DMSO control, was analyzed by CCK8 assay after drug treatment for 72 h.
- Immunoblotting for c‐MYC proteins 72 h after transfection of the indicated siRNAs.
- Cell viability analyses of resistant WM3248 cells. Cells were transfected with the indicated siRNAs for 5 days. Relative cell viability, compared with that of control siRNA‐transfected cells, was measured by CCK8 assay.
- Immunoblotting for c‐MYC proteins 48 h after retrovirus transfection of MSCV mock or MSCV‐c‐MYC.
- Cell viability analyses of resistant SKMEL28 and WM3248 cells. Cells were transfected with the indicated retrovirus for 24 h, and then transfected with the indicated siRNAs for additional 72 h (forward transfection). Relative cell viability, compared with that of mock‐ and control siRNA‐transfected cells, was measured by CCK8 assay.
Data information: All data are mean and SEM [two biological replicates (G) or three biological replicates (B, C, and E)], and P‐values were determined by t‐test (*P < 0.05 and **P < 0.01). The nuclei were stained with DAPI (blue), and scale bars represent 20 μm. Source data are available online for this figure.
To further test whether YAP/TAZ hyperactivation could confer BRAF inhibitor resistance, we performed gain‐of‐function studies, using YAP expression vectors. As shown in Fig 5A, parental cells expressing YAP‐5SA, but not wild‐type YAP, acquired resistance to PLX4032 treatment. Moreover, YAP‐5SA‐expressing parental cells showed higher levels of EGFR compared with mock or wild‐type YAP transfected cells. The cells also retained phospho‐ERK and c‐MYC levels upon PLX4032 treatment (Fig 5B). Our qRT–PCR analyses demonstrated that the expression of YAP‐5SA, but not wild‐type YAP, upregulates the levels of YAP target gene mRNAs in parental cell lines (Fig 5C). In addition, the expression of MITF and SOX10 was downregulated by YAP‐5SA (Fig 5C). The properties of parental cells transfected with YAP‐5SA recapitulate those of PLX4032‐resistant cells independently generated by chronic drug treatment. Altogether, these data suggest that YAP/TAZ play a key role in the establishment of resistance to PLX4032.
Figure 5. Overexpression of YAP‐5SA induces PLX4032 resistance in parental melanoma cell lines.
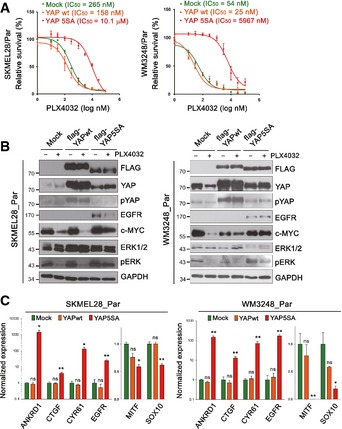
- PLX4032 dose–response curves of parental cells expressing wild‐type YAP or YAP‐5SA. Mock‐transfected cells were used as a control.
- Immunoblotting for the indicated proteins. Lysates of mock, wild‐type YAP‐ and YAP‐5SA‐expressing cells were analyzed. Cells were treated either with PLX4032 (2 μM) or DMSO for 24 h.
- qRT–PCR analyses of YAP target genes (ANKRD1, CTGF, and CYR61), MITF, and SOX10 in wild‐type YAP‐ or YAP‐5SA‐expressing melanoma cells compared with mock‐transfected cells.
Expression microarray analysis identifies cell cycle‐related genes and EGFR, E2F1, and MYC pathway signature genes as potential YAP/TAZ effectors
To explore transcriptome‐wide signatures supported by YAP/TAZ activation in resistant cells, we performed an expression microarray analysis, comparing resistant cells transfected with either YAP/TAZ siRNAs or non‐targeting siRNA. In total, 598 and 545 significantly downregulated genes (311 overlapping genes) upon YAP/TAZ knockdown were identified from the expression profiles of SKMEL28 and WM3248, respectively (fold change > 2.0 and P‐value < 0.05 by LPE test) (Fig 6A and Table EV3). A gene ontology analysis using Biologic Process Gene Ontology Database (GOTERM_BP_FAT) revealed that the cell cycle and mitosis were most enriched functional categories of downregulated genes after YAP/TAZ knockdown (Fig 6B and Table EV4). In addition, we searched for enriched transcriptional motifs from the promoters of downregulated genes, using TransFind algorithm (Kielbasa et al, 2010). Interestingly, E2F binding motif was the most significantly enriched transcriptional motif in both cell lines (Fig 6C). These results suggest that E2F‐related cell cycle progression is a central mechanism of YAP/TAZ‐dependent PLX4032 resistance. In line with our results, a recently published study also demonstrated that E2F1 and YAP cooperate to enable bypass of oncogenic KRAS addiction (Kapoor et al, 2014).
Figure 6. YAP/TAZ knockdown decreases the expression of target genes of EGFR, E2F1, and c‐MYC pathways in resistant cell lines.
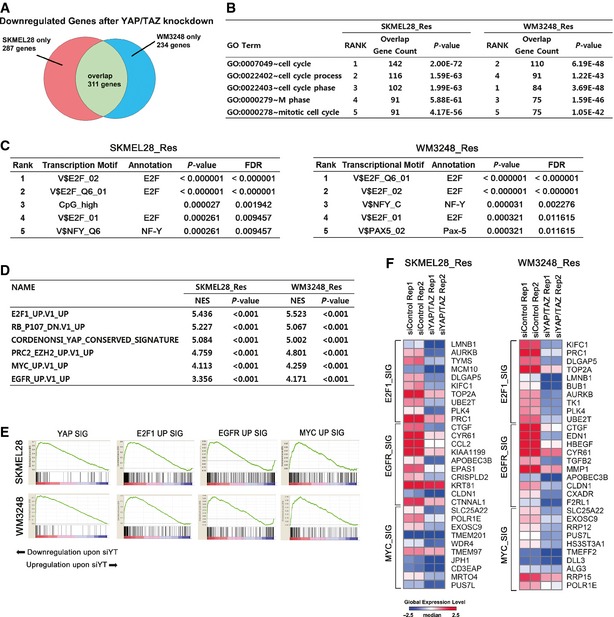
- Venn diagram showing the number of significantly downregulated genes upon YAP/TAZ knockdown in resistant SKMEL28 and WM3248 cells.
- Gene ontology analysis of significantly downregulated genes upon YAP/TAZ knockdown in resistant SKMEL28 and WM3248 cells. The list of significantly downregulated genes (fold change > 2.0 and P‐value < 0.05 by LPE test) was submitted to functional annotation chart analysis of DAVID, using Biological Process Gene Ontology database. Top 5 enriched gene ontology terms are shown.
- Significantly enriched transcription motifs in promoters (from − 800 to + 200 of transcription start sites) of downregulated genes upon YAP/TAZ knockdown, predicted by TransFind software.
- GSEA analysis of enriched gene sets downregulated in response to YAP/TAZ knockdown.
- GSEA enrichment plots demonstrating downregulation of YAP, E2F1, EGFR and c‐MYC pathway signatures after YAP/TAZ knockdown in resistant SKMEL28 and WM3248 cells.
- A heatmap of top 10 E2F1, EGFR, and c‐MYC pathway signature genes on GSEA. Expression levels are shown in log2 scale. Red color indicates higher expression levels and blue color indicates lower expression levels than median.
To find enriched oncogenic gene signatures affected by YAP/TAZ knockdown, we performed GSEA using MSigDB (C6 oncogenic signatures) on our expression array data. As expected, YAP signature gene sets were significantly downregulated upon YAP/TAZ knockdown (Fig 6D and E). The analysis also revealed that E2F1, PRC2/EZH2, MYC, and EGFR signature genes were significantly downregulated in response to YAP/TAZ knockdown (Fig 6D–F and Appendix Table S2). This finding corresponds with the immunoblotting results shown in Fig 4E and identifies E2F1, EGFR, and c‐MYC as possible downstream effectors of YAP/TAZ activation which contribute to BRAF inhibitor resistance.
Increased actin stress fibers and cytoskeletal tension are necessary for YAP/TAZ activity and PLX4032 resistance
Substantial influence of YAP/TAZ activity on PLX4032 resistance prompted us to investigate upstream regulators of YAP/TAZ, which might provide molecular targets for preventing adaptive resistance to PLX4032. Previously, it has been shown that changes in mechanical stimuli and cytoskeletal tension directly affect YAP/TAZ localization in mammalian cells (Dupont et al, 2011; Aragona et al, 2013). Therefore, we speculated that increased actin stress fiber assembly and contractility are the key regulators of YAP/TAZ activation in PLX4032‐resistant melanoma cells, promoting YAP/TAZ nuclear translocation. First, we asked whether extensive spreading of resistant cells was causally related to YAP/TAZ nuclear localization. We restricted the spreading area of resistant cells, using adhesive micropatterns (Thery, 2010). As expected, restriction of cell spreading suppressed YAP/TAZ nuclear enrichment in both resistant cell lines (Fig 7A and B). In addition, YAP/TAZ nuclear localization was sensitive to pharmacologic inhibition of actin polymerization mediated by cytochalasin D in both resistant cell lines (Fig 7C and D). Blocking actomyosin contraction with blebbistatin also provoked YAP/TAZ cytoplasmic retention. These results suggest that an increase in both actin stress fibers and cytoskeletal tension is the key inducer of YAP/TAZ activation in PLX4032‐resistant melanoma cells.
Figure 7. YAP activation in PLX4032‐resistant cells depends on increased actin stress fiber formation and actomyosin tension.
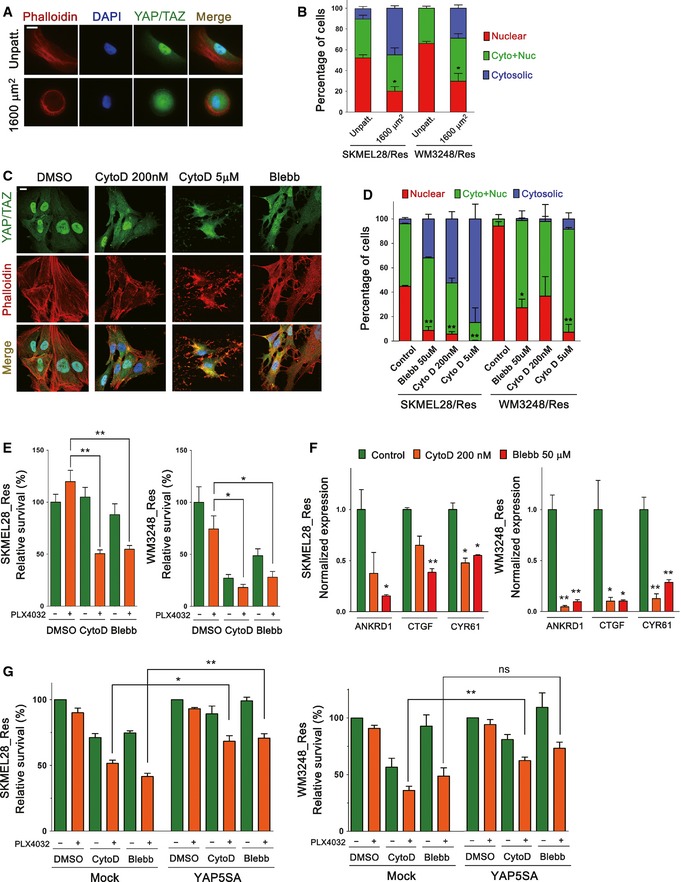
- Fluorescence micrographs of resistant WM3248 cells plated on unpatterned slides (control) and fibronectin‐coated micropattern (1,600 μm2) slides. Cells were double‐stained with phalloidin‐Alexa Fluor 594 and anti‐YAP/TAZ antibody.
- Quantification of the experiment presented in (A). Both SKMEL28 and WM3248 cells were analyzed.
- Fluorescence micrographs showing YAP/TAZ and actin filaments in resistant WM3248 cells treated with the indicated drugs for 3 h. Blebbistatin (Blebb) was added at 50 μM.
- Quantification of YAP/TAZ localization in the experiment presented in (C). Both SKMEL28 and WM3248 cells were analyzed.
- Cell viability assay of resistant melanoma cells treated with 2 μM PLX4032 alone or in combination with either cytochalasin D (200 nM) or blebbistatin (50 μM). Relative cell viability was analyzed by CCK8 assay after drug treatment for 72 h.
- qRT–PCR analyses of YAP target genes in resistant SKMEL28 and WM3248 cells after the treatment of cytochalasin D (200 nM) or blebbistatin (50 μM) for 12 h.
- Cell viability assay of mock‐ or YAP5SA retrovirus‐transfected resistant melanoma cells. Cells were transfected with the indicated retrovirus for 24 h, and then treated with 2 μM of PLX4032 alone or in combination with either cytochalasin D (200 nM) or blebbistatin (50 μM). Relative cell viability was analyzed by CCK8 assay after drug treatment for 72 h.
Because PLX4032 resistance was dependent on YAP/TAZ activity, we next tested whether cytochalasin D or blebbistatin treatment could be effective in overcoming PLX4032 resistance. As shown in Fig 7E, cytochalasin D or blebbistatin alone did not significantly affect the viability of resistant SKMEL28 cells, while resistant WM3248 cells exhibited inherent sensitivity to the drugs. Remarkably, PLX4032 treatment either with cytochalasin D or blebbistatin induced significantly stronger growth inhibition than PLX4032 alone in both resistant cell lines (Fig 7E). Moreover, the expression of YAP/TAZ target genes was suppressed by cytochalasin D and blebbistatin (Fig 7F). Lastly, the retroviral YAP5SA expression rescued cell viability loss caused by cytochalasin D or blebbistatin, suggesting that the effect of cytochalasin D and blebbistatin on cell survival is related to YAP (Fig 7G). These results suggest that actin modulation can suppress PLX4032 resistant cell viability, although resistant WM3248 cells did not show a clear synergistic effect due to their inherent sensitivity to cytochalasin D and blebbistatin. With these results, we conclude that increased actin stress fibers and actomyosin tension mediate YAP/TAZ‐dependent PLX4032 resistance, and suppression of actin remodeling is a potential strategy to overcome BRAF inhibitor resistance.
A kinome siRNA library screening identified TESK1 as a synthetic lethal target of melanoma cells exhibiting YAP/TAZ‐dependent PLX4032 resistance
Next, we employed an RNAi‐based synthetic lethal approach to further elucidate the mechanisms of PLX4032 resistance and to identify targets for preventing the resistance. Kinome siRNAs (targeting 607 human kinases; four different siRNAs per gene) were transfected to resistant WM3248 cells for 48 h, and cell viability was measured after additional 72‐h incubation with 2 μM of PLX4032 (Fig 8A and Table EV5). siRNA targets with Z scores < −2 in both replicates were considered as synthetic lethal hits, and siRNA targets with Z scores > 2 were considered as growth promoting hits (Fig 8B). The screening identified actin regulators (TESK1 and MYLK) as well as cell cycle regulators (BUB1, PLK1, and CDK9) and cell metabolism regulators (SAST and IHPK3) as genes supporting the survival of resistant WM3248 cells.
Figure 8. A kinome siRNA library screening identifies TESK1 as a synthetic lethal target in melanoma cells resistant to PLX4032.
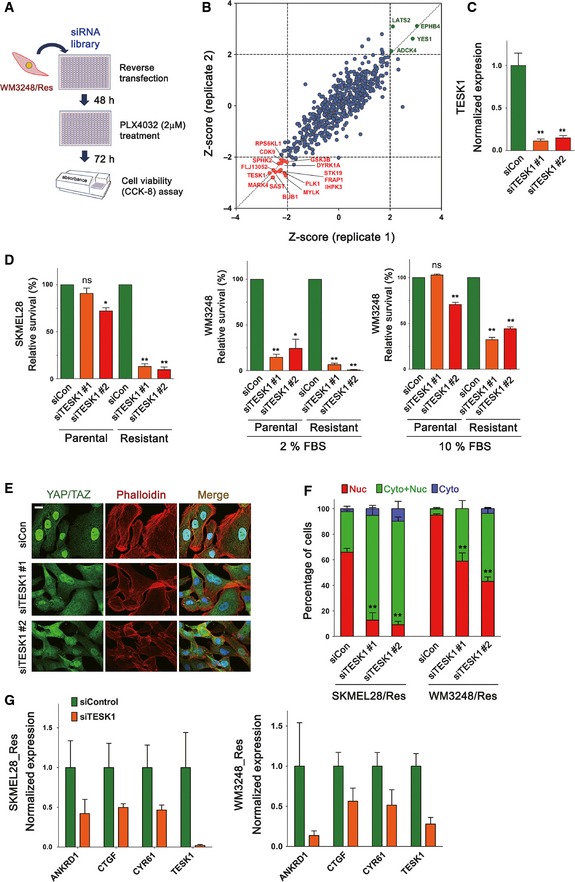
- Schematic illustration of kinome siRNA library screening for identifying synthetic lethal targets in resistant WM3248 cells.
- A graph plotting the Z scores of normalized cell viability after siRNA transfection (two replicates). siRNA targets with Z scores < −2 in both replicates were considered as synthetic lethal hits.
- qRT–PCR analyses confirming knockdown of TESK1 mRNAs in resistant WM3248 cells after transfection of two distinct TESK1 siRNAs for 48 h.
- Cell viability analyses of parental and resistant cells. Cells were transfected with the indicated siRNAs for 6 days. WM3248 cells were cultured in media containing either 2 or 10% FBS before cell viability assay. Relative cell viability was measured by CCK8 assay.
- Fluorescence micrographs showing YAP/TAZ and actin filaments in resistant WM3248 cells. Cells were transfected with the indicated siRNAs for 72 h before staining.
- Quantification of YAP/TAZ localization in the experiment presented in (E). Both SKMEL28 and WM3248 cells were analyzed.
- qRT–PCR analyses of the expression of YAP/TAZ target genes (ANKRD1, CTGF, and CYR61) and TESK1. Resistant SKMEL28 and WM3248 cells were transfected with either control siRNA or TESK1 siRNA pool (1 + 2) for 48 h.
We were particularly interested in TESK1, because previous studies have shown that TESK1 knockdown leads to actin cytoskeletal changes resulting in the reduction in YAP/TAZ activity (Mohseni et al, 2014; Kim et al, 2015). TESK1 is a serine/threonine kinase which is known to inactivate the actin‐severing protein Cofilin via phosphorylation, and also has been suggested to play a role in stem cell fate determination and ciliogenesis (Sakurai et al, 2014; Kim et al, 2015). Therefore, TESK1 knockdown was a good candidate to prevent actin remodeling which is linked to YAP/TAZ activation and resulting drug resistance. First, we confirmed the reduction in TESK1 expression, as well as Cofilin phosphorylation after transfection of two independent TESK1 siRNAs (Figs 8C and EV5A and B). We next examined the effect of TESK1 knockdown on the survival of melanoma cells. As shown in Fig 8D, TESK1 depletion caused a significant reduction in cell viability in resistant SKMEL28, whereas cell viability loss was much milder in parental SKMEL28 cells. TESK1 knockdown caused considerable cell viability loss in parental WM3248 cells as well as in resistant WM3248 cells (Fig 8D). We reasoned that the inherent sensitivity of WM3248 cells to TESK1 knockdown might be alleviated by increasing the concentration of serum, which is known to promote YAP/TAZ activity (Yu et al, 2012). When we increased serum content of the media from 2 to 10%, cell survival improved and higher sensitivity of resistant WM3248 cells in comparison to parental WM3248 cells became apparent.
Figure EV5. TESK1 knockdown decreased phospho‐Cofilin levels, and TESK1 knockdown‐mediated cell viability loss is rescued by YAP‐5SA .
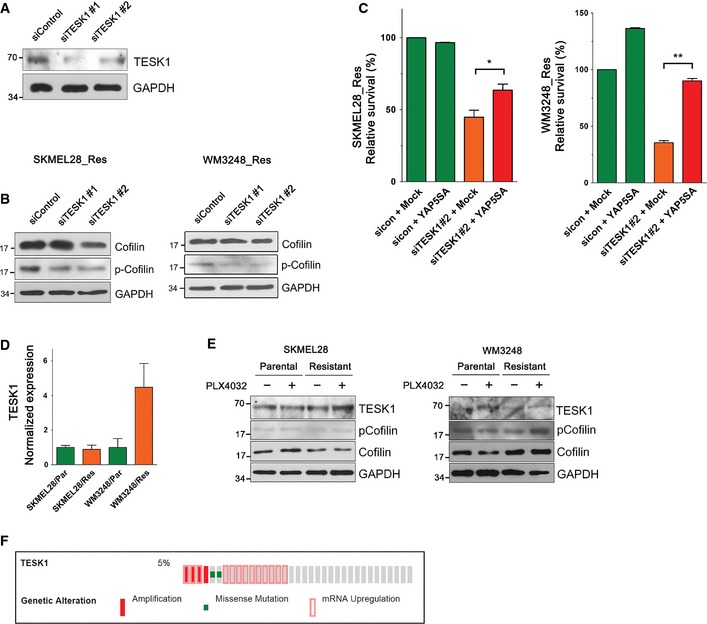
- Immunoblotting of the indicated proteins in SKMEL28 cells transfected with either control siRNA or TESK1 siRNAs for 60 h.
- Immunoblotting with anti‐Cofilin and anti‐phospho‐Cofilin antibodies. Lysates of resistant SKMEL28 and WM3248 cells were collected after transfection with the indicated siRNAs for 72 h (SKMEL28) or 48 h (WM3248).
- Cell viability analyses of resistant SKMEL28 and WM3248 cells. Cells were transfected with mock or YAP‐5SA retrovirus for 24 h, and then transfected with the indicated siRNAs for additional 144 h (forward transfection). Relative cell viability, compared with that of mock‐ and control siRNA‐transfected cells, was measured by CCK8 assay.
- TESK1 expression levels in parental and resistant cells measured by qRT–PCR.
- Immunoblotting of the indicated proteins in parental and resistant cells. Cells were plated and incubated without drugs for 24 h, and then treated with PLX4032 (2 μM) or DMSO for 24 h.
- A cBioPortal oncomap for genetic alterations of TESK1 in human melanoma tumor samples in TCGA database.
Data information: All data are mean and SEM (three biologic replicates), and P‐values were determined by t‐test (*P < 0.05 and **P < 0.01). Source data are available online for this figure.
To understand the underlying mechanism of high TESK1 dependency of resistant cells, we evaluated changes in the actin cytoskeleton and YAP/TAZ activity in resistant cells upon TESK1 knockdown. TESK1 knockdown decreased both actin stress fiber formation and nuclear enrichment of YAP/TAZ (Fig 8E and F). Moreover, the expression of YAP/TAZ target genes was decreased in cells depleted of TESK1 (Fig 8G). These results suggest that TESK1 knockdown decreases resistant cell viability through inhibition of YAP/TAZ activity. Supporting the involvement of YAP in TESK1 knockdown effect, YAP‐5SA expression rescued cell viability loss caused by TESK1 knockdown (Fig EV5C). To examine possible correlation between TESK1 expression levels and PLX4032 resistance, we next measured TESK1 mRNA and protein levels as well as phospho‐Cofilin levels. As shown in Fig EV5D, resistant WM3248 cells showed an increase in TESK1 mRNA levels. However, TESK1 mRNA levels were unaltered in resistant SKEML28 cells. In addition, we observed clear differences in neither TESK1 nor phospho‐Cofilin protein levels between parental and resistant cells of both SKMEL28 and WM3248 (Fig EV5E). This result suggests that changes in TESK1 expression levels are not causally linked to the establishment of PLX4032 resistance, although TESK1 serves as a significant vulnerable point for resistant cells which depend on actin remodeling and YAP/TAZ activation. Collectively, these data indicate that viability of melanoma cells resistant to PLX4032 is highly sensitive to TESK1 knockdown, and TESK1 inhibition might be a potential strategy to overcome PLX4032 resistance.
Discussion
Cytoskeletal tension and stiffness have been shown to affect chemotherapeutic drug sensitivity of cancer cells (Nguyen et al, 2014; Sharma et al, 2014). Moreover, a recent study found that remodeling of the extracellular matrix, which alters matrix elasticity and stiffness, confers BRAF inhibitor tolerance to melanoma cells (Hirata et al, 2015). Therefore, molecular mechanisms that link cytoskeletal architecture and physical properties of cells to cancer drug resistance are a critical issue of cancer biology. Our study demonstrates both actin cytoskeletal remodeling upon PLX4032 treatment in BRAF V600E mutant melanoma cells and its pivotal contribution to adaptive resistance to PLX4032.
Recent studies have consistently reported that BRAF inhibitor‐resistant melanoma cells have higher migratory and metastatic potential both in vitro and in vivo models (Girotti et al, 2013; Sanchez‐Laorden et al, 2014). Moreover, Girotti et al (2013) presented significant differences in phosphorylation levels of diverse cytoskeletal proteins between BRAF inhibitor‐resistant cells and parental melanoma cells. Interestingly, it has also been suggested that appropriate RAS/RAF/ERK activity levels are important for maintaining actin stress fiber integrity (Sahai et al, 2001; Pritchard et al, 2004). However, it is not clear whether PLX4032 treatment directly modulates the activities of actin dynamics regulators in melanoma cells. Changes in the actin cytoskeleton were apparent 7 to 14 days after PLX4032 treatment, whereas short‐term exposure to the drug did not affect basal actin filament patterns. It is noteworthy that the expression levels of multiple actin regulator genes are altered in resistant cells when compared with parental cells. We speculate that complex cellular responses to BRAF inhibition gradually induce epigenetic changes, redirecting global expression patterns of actin cytoskeleton‐related genes. Another important issue is whether the Hippo pathway is implicated in BRAF inhibitor resistance. Both the interaction between the Hippo pathway and the cytomechanics/F‐actin pathway and their relative contribution to YAP control are still being debated. We suggest that the actin cytoskeleton and the Hippo pathway may cooperatively support YAP/TAZ activation in PLX4032‐resistant cells, because recent findings suggest the Hippo pathway and the actin cytoskeleton mutually regulate each other (Matsui & Lai, 2013). Our study does not address Hippo pathway alterations in resistant cells. Further studies are needed to determine the relationship between the Hippo pathway and the actin cytoskeleton in YAP/TAZ‐dependent BRAF inhibitor resistance.
YAP/TAZ have been frequently implicated in the pathogenesis of various human cancers, driving chemoresistance and stemness‐related traits (Cordenonsi et al, 2011; Kapoor et al, 2014; Shao et al, 2014; Lin et al, 2015). However, only a few somatic or germline mutations in the Hippo pathway components have been identified in human malignancies, and YAP/TAZ‐regulating mechanisms in the majority of cancer species remain unclear. Interestingly, overexpression of wild‐type YAP did not change BRAF inhibitor sensitivity of melanoma cells, whereas constitutively active YAP (YAP‐5SA) efficiently provoked BRAF inhibitor resistance (Fig 5A). This result implies that an increase in YAP/TAZ expression levels is not sufficient to drive cancer cell drug resistance, probably because of strong negative regulators suppressing YAP/TAZ activity. Importantly, a recent study demonstrated that activating mutation in GNAQ gene stimulates YAP through the promotion of actin polymerization, independent of the Hippo pathway, in uveal melanoma (Feng et al, 2014) This study suggests the possibility that actin‐dependent YAP/TAZ regulation might be dominant in certain types of cancer. Our data further support the importance of actin‐dependent YAP/TAZ regulation in cancer cells. Our study could not formally exclude genetic alterations during the acquisition of BRAF inhibitor resistance in SKMEL28 and WM3248 cells. However, induction of actin cytoskeletal remodeling and YAP/TAZ nuclear enrichment within 1 or 2 weeks suggests an epigenetic mechanism of drug resistance.
Our expression microarray analysis revealed that cell cycle‐ and mitosis‐related genes are significantly enriched among downregulated genes after YAP/TAZ knockdown in PLX4032‐resistant cells. This result corresponds with a recent study that highlighted YAP‐TEAD‐E2F1 interaction in rescuing pancreatic cancer cells from oncogenic KRAS deprivation (Kapoor et al, 2014). In addition, we observed strong influence of YAP/TAZ knockdown on EGFR and pAKT levels. A number of studies have reported upregulation of EGFR and AKT as a key resistance mechanism of BRAF inhibitor (Prahallad et al, 2012; Su et al, 2012; Girotti et al, 2013; Sun et al, 2014), and a recent study showed that SOX10 loss in melanoma cells mediates EGFR upregulation (Sun et al, 2014). Interestingly, YAP‐5SA overexpression suppressed the expression of SOX10 and MITF, while upregulating EGFR (Fig 5B and C). Our finding suggests that YAP may serve as an important regulator of EGFR and AKT pathway activity in BRAF inhibitor resistance models, although additional pathways are likely to be involved in mediating resistance to BRAF inhibition. Our data also showed both c‐MYC protein and mRNA levels are dependent on YAP/TAZ. However, because c‐MYC depletion caused cell viability suppression only in SKMEL28 cells, we suggest that the role of c‐MYC in BRAF inhibitor response depends on cellular context.
We propose TESK1 as a potential synthetic lethal target for melanoma cells resistant to BRAF inhibitors. TESK1 has come into the spotlight after the discovery that TESK1 knockdown can block YAP/TAZ activity (Mohseni et al, 2014) and TESK1 knockdown promotes pluripotency acquisition in somatic cell reprogramming (Sakurai et al, 2014). TESK1 regulates actin depolymerization by phosphorylating Cofilin, which disassembles actin filaments. Although actin cytoskeletal regulators have been the subject of much interest due to their importance in metastasis, their role is widely unrecognized in oncologic clinical practice. In our study, TESK1 expression levels are not consistently dysregulated in resistant cells compared with parental cells. However, TESK1 knockdown suppressed both cell survival and YAP/TAZ activation in resistant melanoma cells. We suggest that TESK1 may not be a driver of actin remodeling, but serves as an important weak point of YAP/TAZ‐dependent BRAF inhibitor resistance in melanoma cells. However, we also found an inconsistency in the sensitivity to TESK1 inhibition between the two cell lines used, and a change in serum concentration influenced overall effect of TESK1 inhibition. These results raise the possibility that melanoma cells could evolve adaptive resistant mechanisms to TESK1 inhibition. Lastly, we surveyed TCGA melanoma database using cBioportal for Cancer Genomics (Cerami et al, 2012; Gao et al, 2013) and found TESK1 alterations (mutation, amplification, or mRNA upregulation) in 5% of evaluated tumor samples (Fig EV5F). Further studies are needed to characterize TESK1 alterations in terms of BRAF inhibitor response and YAP/TAZ activation in cancer.
In summary, we propose a new BRAF inhibitor resistance mechanism of melanoma cells, in which actin cytoskeletal remodeling upon BRAF inhibitor treatment confers YAP/TAZ activation, allowing cancer cell survival and proliferation. Our findings prompt a need for developing new therapeutic strategy to inhibit actin cytoskeletal remodeling and YAP/TAZ activation to overcome BRAF inhibitor resistance.
Materials and Methods
See Appendix Supplementary Methods for details.
Reagents, cell culture and PLX4032‐resistance establishment
PLX4032 (Vemurafenib) was purchased from Selleckchem. Cytochalasin D (Sigma‐Aldrich) was used either at 200 nM or 5 μM, and blebbistatin (Enzo Life Science) was used at 50 μM. SKMEL28 cells were purchased from ATCC, and WM3248 cells were purchased from Coriell Institute. To establish PLX4032‐resistant cell lines, SKMEL28 and WM3248 cells were continuously exposed to 2 μM of PLX4032 for 2 months. PLX4032‐containing media were replaced every 3–4 days. Survived resistant cells were maintained in the presence of 2 μM PLX4032.
Plasmid, transfection, and retroviral infection
8xGTIIC‐luciferase cDNA was provided by Dr. Stefano Piccolo (University of Padua; Addgene plasmid # 34615), and MSCV‐c‐MYC‐IRES‐GFP was provided by Dr. John Cleveland (Moffitt Cancer Center; Addgene plasmid # 18119). Flag‐YAP wild‐type and flag‐YAP‐5SA cDNAs cloned into retroviral pMSCV‐puro vector were provided by Dr. Dae‐Sik Lim (KAIST). We generated siRNA‐resistant pMSCV‐puro flag‐YAP‐5SA vector by performing site directed mutagenesis that incorporates silent mutations on YAP siRNA target site. The sequence of siRNAs used in this study is described in Appendix Table S3. For retroviral particle assembly, retroviral constructs and two packaging plasmids (pCMV‐VSV‐G and pCMV‐Gag‐Pol) were co‐transfected into HEK293T cells, and retroviral supernatant was collected 24 h after transfection. The supernatants were filtered through a 0.45‐μm filter, and infected into melanoma cells with 4 μg/ml Polybrene.
Immunofluorescence, microscopy and image analysis
For indirect immunofluorescence, cells were plated either in 8‐well Lab‐Tek II chamber slides (Nunc) or 96‐well clear bottom plates (BD Falcon), and fixed with 4% paraformaldehyde for 8 min at room temperature. After fixation, 0.1% Triton X‐100 (Sigma‐Aldrich) was applied for permeabilization. Cells were incubated with primary antibodies for 1 h at room temperature. Bound primary antibodies were detected by incubating with Alexa Fluor 488‐ or 594‐conjugated secondary antibodies (Life Technologies) for 1 h at room temperature. For staining actin filaments, Alexa Fluor 594‐conjugated Phalloidin (Life Technologies) was used according to the manufacturer's protocol. Primary antibodies and dilutions used in this study are described in Appendix Table S4. Fluorescence images were acquired using a DeltaVision Spectris Imaging System (Applied Precision). Vertical images of Fig 2E were generated by stacking deconvoluted images of melanoma cells with 0.3 μm z‐step size, and processing by SoftWorx volume rendering tool.
Micropattern experiment
All micropatterns used in this study were produced by CYTOO (http://www.cytoo.com/). Cells were seeded on coverslip chips containing fibronectin‐coated disk‐shaped 1,600 μm2 micropatterns (CYTOOchips™ DC‐L‐FN) or on unpatterned Lab‐Tek chamber slides. Cells were incubated for 30 min, and unattached cells were flushed by changing medium. After flushing, cells were further incubated for 3 h for allowing spreading, and fixed with 4% paraformaldehyde.
BrdU incorporation assay
Cells were seeded at 5,000 cells/well in 8‐well Lab‐Tek II chamber slides and incubated for 24 h with FBS‐supplemented media. Cells were treated with DMSO or PLX4032 (2 μM) for 24 h, and 3 μg/ml of BrdU (Sigma‐Aldrich) was applied for 45 min before fixation. After fixation, slides were treated with 2 N HCl for 30 min to denature DNA. After denaturation, cells were stained with anti‐BrdU antibody.
Cell viability assay
For PLX4032 dose–response analyses, cells were incubated for 24 h after plating, and then treated with variable doses of PLX4032 for 72 h. Viable cells were quantified by 450 nm absorbance measurement 2 h after treatment of Cell Counting Kit‐8 reagent (CCK8; Dojindo). For PLX4032 dose–response analyses after siRNA transfection, cells were seeded with siRNAs (reverse transfection). After transfection for 72 h, cells were treated with PLX4032 for 72 h before CCK8 assay.
Immunoblotting
For immunoblotting, cells were lysed with RIPA lysis buffer supplemented with protease and phosphatase inhibitors (Merck Millipore) on ice. Cell lysates were sonicated and centrifuged for 10 min at 4°C, 13,000 rpm. Aliquots of each protein lysate (10–20 μg) were applied to SDS–polyacrylamide gel electrophoresis. After electrophoresis, proteins were transferred to nitrocellulose membranes and incubated for 30 min with blocking solution (TBST buffer with 5% skim milk). For phosphorylated protein detection, membranes were blocked with 2% BSA in PBS. Blocked membranes were incubated with primary antibodies overnight at 4°C, and then incubated with peroxidase‐coupled secondary antibody (Santa Cruz Biotechnology) for 1 h at 4°C. Target proteins were detected using enhanced chemiluminescence Western blot detection solution (Thermo Scientific).
Subcellular fractionation
Nuclear and non‐nuclear fractional lysates were prepared using NE‐PER nuclear and cytoplasmic extraction reagent (Thermo Scientific) according to the manufacturer's instructions. Cells were harvested for lysis by cell scrapers, instead of trypsin–EDTA, because changes in cell morphologies and mechanical tension during trypsinization process can affect the localization of YAP/TAZ.
Quantitative RT–PCR
Total RNA was extracted from cells using RNeasy kit (Qiagen) according to the manufacturer's instructions. A total of 1 μg of extracted RNA was transcribed into cDNA using M‐MLV reverse transcriptase (Promega) in the presence of RNasin Plus RNase Inhibitor (Promega). cDNA was mixed with primers and iQ SYBR Green Supermix (Bio‐Rad), and mRNA expression levels were measured by real‐time qRT–PCR on CFX96 system (Bio‐Rad). The primers were designed using Primer‐BLAST or adopted from previously published studies. The list of qRT–PCR primers used in this study is described in Appendix Table S5. Primer reaction specificity was confirmed by both agarose gel electrophoresis and melting curve analysis. Relative gene expression was analyzed by ΔΔC t method using the CFX Manager software (Bio‐Rad).
Luciferase assay
SKMEL28 and WM3248 cells were plated on 96‐well plates at 5,000 cells/well, and co‐transfected with 8XGTIIC‐luciferase vector (1 μg/ml) and pcDNA3.1‐His‐lacZ (1 μg/ml) for 24 h using Lipofectamine LTX and PLUS reagent. Cells were harvested with reporter lysis buffer, and luciferase activity was measured by luciferase assay kit (Promega). β‐Galactosidase activity was also measured using a β‐Galactosidase enzyme assay system (Promega) for transfection efficiency normalization.
Expression microarray analysis
Total RNAs were extracted from melanoma cells using the RNeasy kit (Qiagen). Total RNA was amplified and purified using TargetAmp‐Nano Labeling Kit for Illumina Expression BeadChip (EPICENTRE) to yield biotinylated cRNA. 750 ng of labeled cRNA samples were hybridized to Human HT‐12 v4.0 Expression Beadchip for 17 h at 58°C. Detection of array signal was carried out using Amersham fluorolink streptavidin‐Cy3 (GE Healthcare Bio‐Sciences) following the bead array manual. Arrays were scanned with an Illumina bead array Reader confocal scanner according to the manufacturer's instructions. DAVID gene‐enrichment and functional annotation analysis was performed for characterizing (i) significantly altered genes common in resistant SKMEL28 and WM3248 cells compared to each parental cell, and (ii) significantly downregulated genes upon YAP/TAZ siRNA knockdown. The list of significantly downregulated genes upon YAP/TAZ siRNA knockdown was submitted to TransFind algorithm (Kielbasa et al, 2010) to query enriched transcription factor motifs. GSEA (Subramanian et al, 2005) was performed using C6 MSigDB gene set database (i) to test whether YAP signature is enriched on resistant cells compared with parental cells, and (ii) to identify enriched oncogenic signature gene sets suppressed by YAP/TAZ knockdown.
Kinome siRNA library screening
A kinome‐wide siRNA library targeting 607 human kinases was purchased in 384‐well plate format from Dharmacon. Four different siRNAs targeting each kinase were pooled. Polystyrene flat bottom 384‐well plates (Greiner) were spotted with 3 μl of 0.25 μM siRNA library pools (62.5 nM for each siRNA) using the Biomek FX Laboratory Automation Workstation (Beckman Coulter), and 0.1 μl of Lipofectamine RNAiMAX dissolved in 7 μl of Opti‐MEM (Gibco) was mixed in assay plates to perform reverse transfection (total siRNA concentration was 15 nM). Resistant WM3248 cells were seeded onto assay plates at 1,000 cells/well with a final volume of 50 μl. PLX4032 was added 48 h after transfection, and cells were further incubated for 72 h. CCK8 reagent was applied for 2 h to measure cell viability. Z scores of normalized target siRNA viability were calculated. siRNA targets with Z scores < −2 in both replicates were considered as significant synthetic lethal hits, and siRNA targets with Z scores > 2 were taken as growth promoting hits.
Quantification and statistical analysis
The quantification of YAP/TAZ localization was performed by inspecting at least 150–200 cells stained with anti‐YAP/TAZ immunofluorescence, using ImageJ software. Images were processed and placed in figures using Adobe Photoshop CS6. The data of qRT–PCR and luciferase assay were normalized by dividing all values of control and treatment groups by mean of the control. Data analysis was performed using GraphPad Prism version 6 (GraphPad Software), and statistical significance was considered when P‐value was < 0.05 in two‐sided unpaired Student's t‐test (*P < 0.05; **P < 0.01). Paired t‐test was used for evaluating relative survival after drug treatment or siRNA transfection (survival values were normalized by control cell survival value before the test).
Data accession
Raw expression array data have been deposited at NCBI Gene Expression Omnibus (GEO) database (Accession Number GSE68599).
Author contributions
MHK and JooK conceived the project. MHK performed the majority of the experiments and analyzed the data. JonK, HH, SHL, JKL, and EJ performed some experiments and contributed to data analyses. MHK and JooK interpreted the data and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank Dr. Dae‐Sik Lim (KAIST) for providing reagents and comments, and Dr. Mi Young Kim (KAIST) for providing devices for kinome siRNA library screening. This study was supported by Outstanding Young Investigator Grant from the National Research Foundation of Korea (20120004114), and by a grant from the National R&D Program for Cancer Control, Korean Ministry of Health & Welfare (1120040). M.H.K was supported by the KAIST Venture Research Program for Graduate and PhD Students (VRPGP).
The EMBO Journal (2016) 35: 462–478
See also: F Zanconato & S Piccolo (March 2016)
References
- Anastas JN, Kulikauskas RM, Tamir T, Rizos H, Long GV, von Euw EM, Yang PT, Chen HW, Haydu L, Toroni RA, Lucero OM, Chien AJ, Moon RT (2014) WNT5A enhances resistance of melanoma cells to targeted BRAF inhibitors. J Clin Investig 124: 2877–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, Piccolo S (2013) A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin‐processing factors. Cell 154: 1047–1059 [DOI] [PubMed] [Google Scholar]
- Butcher DT, Alliston T, Weaver VM (2009) A tense situation: forcing tumour progression. Nat Rev Cancer 9: 108–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo F, Ege N, Grande‐Garcia A, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary E, Charras G, Sahai E (2013) Mechanotransduction and YAP‐dependent matrix remodelling is required for the generation and maintenance of cancer‐associated fibroblasts. Nat Cell Biol 15: 637–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2: 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O'Day SJ, Sosman JA, Kirkwood JM, Eggermont AM et al (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364: 2507–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti AR, Poletti A, Daidone MG, Dupont S, Basso G, Bicciato S, Piccolo S (2011) The Hippo transducer TAZ confers cancer stem cell‐related traits on breast cancer cells. Cell 147: 759–772 [DOI] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S (2011) Role of YAP/TAZ in mechanotransduction. Nature 474: 179–183 [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE (2002) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30: 207–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, Degese MS, Iglesias‐Bartolome R, Vaque JP, Molinolo AA, Rodrigues M, Zaidi MR, Ksander BR, Merlino G, Sodhi A, Chen Q, Gutkind JS (2014) Hippo‐independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio‐regulated rho GTPase signaling circuitry. Cancer Cell 25: 831–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6: pl1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girotti MR, Pedersen M, Sanchez‐Laorden B, Viros A, Turajlic S, Niculescu‐Duvaz D, Zambon A, Sinclair J, Hayes A, Gore M, Lorigan P, Springer C, Larkin J, Jorgensen C, Marais R (2013) Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov 3: 158–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder G, Dupont S, Piccolo S (2012) Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat Rev Mol Cell Biol 13: 591–600 [DOI] [PubMed] [Google Scholar]
- Hirata E, Girotti MR, Viros A, Hooper S, Spencer‐Dene B, Matsuda M, Larkin J, Marais R, Sahai E (2015) Intravital Imaging Reveals How BRAF Inhibition Generates Drug‐Tolerant Microenvironments with High Integrin beta1/FAK Signaling. Cancer Cell 27: 574–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaalouk DE, Lammerding J (2009) Mechanotransduction gone awry. Nat Rev Mol Cell Biol 10: 63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, Zhong Y, Wu CJ, Sadanandam A, Hu B, Chang Q, Chu GC, Al‐Khalil R, Jiang S, Xia H, Fletcher‐Sananikone E, Lim C, Horwitz GI, Viale A, Pettazzoni P et al (2014) Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 158: 185–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielbasa SM, Klein H, Roider HG, Vingron M, Bluthgen N (2010) TransFind–predicting transcriptional regulators for gene sets. Nucleic Acids Res 38: W275–W280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Jo H, Hong H, Kim MH, Kim JM, Lee JK, Heo WD, Kim J (2015) Actin remodelling factors control ciliogenesis by regulating YAP/TAZ activity and vesicle trafficking. Nat Commun 6: 6781 [DOI] [PubMed] [Google Scholar]
- Lin L, Sabnis AJ, Chan E, Olivas V, Cade L, Pazarentzos E, Asthana S, Neel D, Yan JJ, Lu X, Pham L, Wang MM, Karachaliou N, Cao MG, Manzano JL, Ramirez JL, Torres JM, Buttitta F, Rudin CM, Collisson EA et al (2015) The Hippo effector YAP promotes resistance to RAF‐ and MEK‐targeted cancer therapies. Nat Genet 47: 250–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mana‐Capelli S, Paramasivam M, Dutta S, McCollum D (2014) Angiomotins link F‐actin architecture to Hippo pathway signaling. Mol Biol Cell 25: 1676–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui Y, Lai ZC (2013) Mutual regulation between Hippo signaling and actin cytoskeleton. Protein Cell 4: 904–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohseni M, Sun J, Lau A, Curtis S, Goldsmith J, Fox VL, Wei C, Frazier M, Samson O, Wong KK, Kim C, Camargo FD (2014) A genetic screen identifies an LKB1‐MARK signalling axis controlling the Hippo‐YAP pathway. Nat Cell Biol 16: 108–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroishi T, Hansen CG, Guan KL (2015) The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer 15: 73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nallet‐Staub F, Marsaud V, Li L, Gilbert C, Dodier S, Bataille V, Sudol M, Herlyn M, Mauviel A (2014) Pro‐invasive activity of the Hippo pathway effectors YAP and TAZ in cutaneous melanoma. J Invest Dermatol 134: 123–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TV, Sleiman M, Moriarty T, Herrick WG, Peyton SR (2014) Sorafenib resistance and JNK signaling in carcinoma during extracellular matrix stiffening. Biomaterials 35: 5749–5759 [DOI] [PubMed] [Google Scholar]
- Olson EN, Nordheim A (2010) Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol 11: 353–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart‐King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM (2005) Tensional homeostasis and the malignant phenotype. Cancer Cell 8: 241–254 [DOI] [PubMed] [Google Scholar]
- Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, Salton M, Dahlman KB, Tadi M, Wargo JA, Flaherty KT, Kelley MC, Misteli T, Chapman PB, Sosman JA, Graeber TG et al (2011) RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 480: 387–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R (2012) Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 483: 100–103 [DOI] [PubMed] [Google Scholar]
- Pritchard CA, Hayes L, Wojnowski L, Zimmer A, Marais RM, Norman JC (2004) B‐Raf acts via the ROCKII/LIMK/cofilin pathway to maintain actin stress fibers in fibroblasts. Mol Cell Biol 24: 5937–5952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, Haydu LE, Mijatov B, Becker TM, Boyd SC, Howle J, Saw R, Thompson JF, Kefford RF, Scolyer RA, Long GV (2014) BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res 20: 1965–1977 [DOI] [PubMed] [Google Scholar]
- Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, Chiarion‐Sileni V, Drucis K, Krajsova I, Hauschild A, Lorigan P, Wolter P, Long GV, Flaherty K, Nathan P, Ribas A et al (2015) Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 372: 30–39 [DOI] [PubMed] [Google Scholar]
- Sahai E, Olson MF, Marshall CJ (2001) Cross‐talk between Ras and Rho signalling pathways in transformation favours proliferation and increased motility. EMBO J 20: 755–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai K, Talukdar I, Patil VS, Dang J, Li Z, Chang KY, Lu CC, Delorme‐Walker V, Dermardirossian C, Anderson K, Hanein D, Yang CS, Wu D, Liu Y, Rana TM (2014) Kinome‐wide functional analysis highlights the role of cytoskeletal remodeling in somatic cell reprogramming. Cell Stem Cell 14: 523–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Laorden B, Viros A, Girotti MR, Pedersen M, Saturno G, Zambon A, Niculescu‐Duvaz D, Turajlic S, Hayes A, Gore M, Larkin J, Lorigan P, Cook M, Springer C, Marais R (2014) BRAF inhibitors induce metastasis in RAS mutant or inhibitor‐resistant melanoma cells by reactivating MEK and ERK signaling. Sci Signal 7: ra30 [DOI] [PubMed] [Google Scholar]
- Sanz‐Moreno V, Gadea G, Ahn J, Paterson H, Marra P, Pinner S, Sahai E, Marshall CJ (2008) Rac activation and inactivation control plasticity of tumor cell movement. Cell 135: 510–523 [DOI] [PubMed] [Google Scholar]
- Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, Schinzel AC, Sood S, Rosenbluh J, Kim JW, Zwang Y, Roberts TM, Root DE, Jacks T, Hahn WC (2014) KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 158: 171–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Santiskulvong C, Rao J, Gimzewski JK, Dorigo O (2014) The role of Rho GTPase in cell stiffness and cisplatin resistance in ovarian cancer cells. Integr Biol 6: 611–617 [DOI] [PubMed] [Google Scholar]
- Su F, Bradley WD, Wang Q, Yang H, Xu L, Higgins B, Kolinsky K, Packman K, Kim MJ, Trunzer K, Lee RJ, Schostack K, Carter J, Albert T, Germer S, Rosinski J, Martin M, Simcox ME, Lestini B, Heimbrook D et al (2012) Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation. Cancer Res 72: 969–978 [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Wang L, Huang S, Heynen GJ, Prahallad A, Robert C, Haanen J, Blank C, Wesseling J, Willems SM, Zecchin D, Hobor S, Bajpe PK, Lieftink C, Mateus C, Vagner S, Grernrum W, Hofland I, Schlicker A, Wessels LF et al (2014) Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 508: 118–122 [DOI] [PubMed] [Google Scholar]
- Thery M (2010) Micropatterning as a tool to decipher cell morphogenesis and functions. J Cell Sci 123: 4201–4213 [DOI] [PubMed] [Google Scholar]
- Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor‐Weiner A, Whittaker S, Kryukov GV, Hodis E, Rosenberg M, McKenna A, Cibulskis K, Farlow D, Zimmer L, Hillen U, Gutzmer R, Goldinger SM, Ugurel S et al (2014) The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 4: 94–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel V, Sheetz M (2006) Local force and geometry sensing regulate cell functions. Nat Rev Mol Cell Biol 7: 265–275 [DOI] [PubMed] [Google Scholar]
- Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, Zhao J, Yuan H, Tumaneng K, Li H, Fu XD, Mills GB, Guan KL (2012) Regulation of the Hippo‐YAP pathway by G‐protein‐coupled receptor signaling. Cell 150: 780–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li L, Lu Q, Wang LH, Liu CY, Lei Q, Guan KL (2011a) Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev 25: 51–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL (2012) Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev 26: 54–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Tumaneng K, Guan KL (2011b) The Hippo pathway in organ size control, tissue regeneration and stem cell self‐renewal. Nat Cell Biol 13: 877–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasler W, Lee JT, Avruch J, Bardeesy N (2009) Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 16: 425–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5