

Abstract
The Beclin1–VPS34 complex is recognized as a central node in regulating autophagy via interacting with diverse molecules such as ATG14L for autophagy initiation and UVRAG for autophagosome maturation. However, the underlying molecular mechanism that coordinates the timely activation of VPS34 complex is poorly understood. Here, we identify that PAQR3 governs the preferential formation and activation of ATG14L‐linked VPS34 complex for autophagy initiation via two levels of regulation. Firstly, PAQR3 functions as a scaffold protein that facilitates the formation of ATG14L‐ but not UVRAG‐linked VPS34 complex, leading to elevated capacity of PI(3)P generation ahead of starvation signals. Secondly, AMPK phosphorylates PAQR3 at threonine 32 and switches on PI(3)P production to initiate autophagosome formation swiftly after glucose starvation. Deletion of PAQR3 leads to reduction of exercise‐induced autophagy in mice, accompanied by a certain degree of disaggregation of ATG14L‐associated VPS34 complex. Together, this study uncovers that PAQR3 can not only enhance the capacity of pro‐autophagy class III PI3K due to its scaffold function, but also integrate AMPK signal to activation of ATG14L‐linked VPS34 complex upon glucose starvation.
Keywords: AMPK, autophagy, class III PI3K, glucose starvation, PAQR3
Subject Categories: Autophagy & Cell Death, Metabolism, Signal Transduction
Introduction
Autophagy (macroautophagy) is an evolutionarily conserved intracellular degradation process in which entire organelles, lipid vesicles, or protein aggregates are engulfed in autophagosome and eventually digested in lysosomes by acidic hydrolases (Galluzzi et al, 2014). Autophagy is not only crucial for removal of misfolded proteins and turnover of organelles for cellular homeostasis, but also important for survival of eukaryotic cells under stress conditions, development, tumorigenesis, and infection (Lamb et al, 2013). Dysregulation of autophagy has been involved in numerous diseases, such as aging, cancer, cardiovascular disorders, neurodegeneration, and metabolic disorders (Choi et al, 2013). To date, more than 30 autophagy‐related genes (ATG) have been discovered to participate in different key steps of autophagy, including initiation, vesicle nucleation, vesicle elongation, lysosome fusion, and degradation (Maiuri et al, 2007; He & Klionsky, 2009; Yang & Klionsky, 2010).
In autophagy process, the least understood step is autophagy induction and nucleation of membrane, during which Beclin1–VPS34 complex plays a central role. VPS34, the unique class III phosphatidylinositol 3‐kinase (class III PI3K) in mammals, forms a stable complex with its regulatory subunit p150 (VPS15 ortholog) and phosphorylates phosphatidylinositol to generate phosphatidylinositol 3‐phosphate (PI(3)P) (Schu et al, 1993). PI(3)P is regarded as an autophagy initiation detonator which promotes nucleation of isolation membrane (also called phagophore) and recruitment of autophagy downstream effectors, such as DFCP1 (double FYVE‐containing protein 1) and WIPI (WD‐repeat domain phosphoinositide interacting) (Simonsen & Tooze, 2009). Beclin1, a mammalian homolog of ATG6, is also a component of the core VPS34 complex and mainly responsible for recruiting multiple proteins to regulate the formation and activity of the VPS34 complex. The Beclin1–VPS34 complex can be involved in autophagy initiation and maturation by forming different complex with diverse binding partners, such as ATG14L and UVRAG (Funderburk et al, 2010). The mutually exclusive interaction between ATG14L and UVRAG with Beclin1 exhibits distinct functions of the class III PI3K (Itakura et al, 2008; Li et al, 2012). ATG14L‐linked VPS34 complex controls autophagy initiation and autophagosome formation (Matsunaga et al, 2009; Zhong et al, 2009), while UVRAG complex is mainly involved in autophagosome maturation and endocytic fusion (Liang et al, 2008).
In addition, many other Beclin–VPS34 complex binding partners have been characterized in recent years, such as Rubicon, VMP1, Ambra‐1, and Bif‐1 (Fimia et al, 2007; Ropolo et al, 2007; Takahashi et al, 2007; Matsunaga et al, 2009), suggesting that VPS34 complexes can be regulated in different manners. However, it is currently unknown what factors orchestrate the sequential activation of different VPS34‐containing pools, such as the ATG14L–Beclin1–VPS34 and UVRAG–Beclin1–VPS34 complexes.
PAQR3, as a member of the progestin and adipoQ receptors (PAQR) superfamily, is a Golgi‐resident seven‐transmembrane protein (Feng et al, 2007; Luo et al, 2008). The functional investigations of PAQR3 in the past were mainly concentrated in its negative regulation on different signaling pathways, such as Ras‐Raf‐Mek‐Erk and PI3K‐AKT cascades (Feng et al, 2007; Wang et al, 2013). However, it was poorly explored whether PAQR3 can also act as a positive mediator for certain biological pathways. In this study, we identified that PAQR3 is pivotal for autophagy initiation as it preferentially facilitates the formation of ATG14L‐associated VPS34 complex instead of UVRAG‐associated VPS34 complex. In addition, PAQR3 integrates glucose starvation signals to autophagy initiation via AMPK‐mediated phosphorylation.
Results
PAQR3 regulates autophagosome formation without altering the activities of AMPK and mTOR
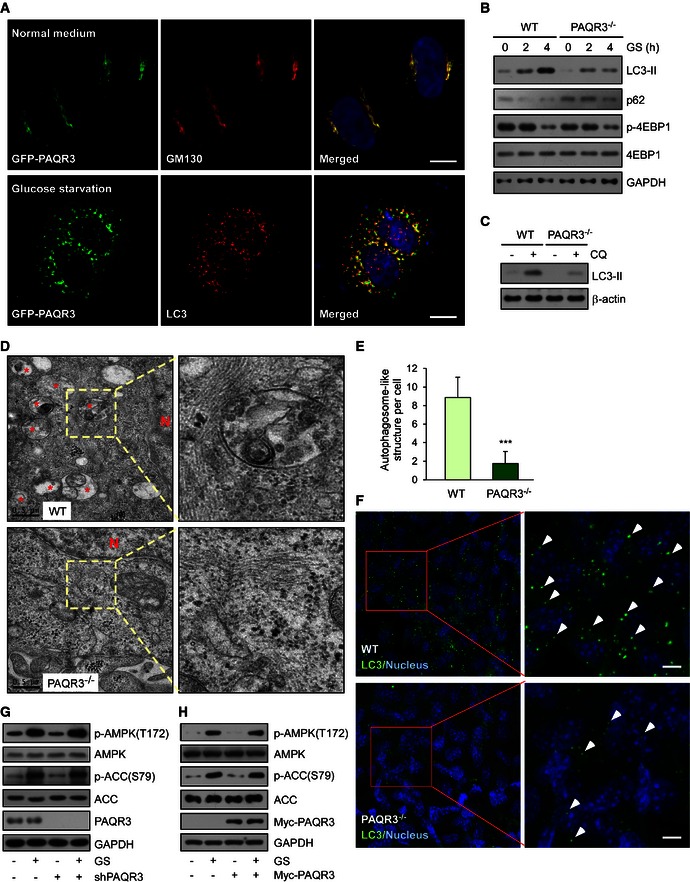
This study was kindled by an accidental discovery that under starvation conditions, PAQR3 was mostly redistributed into punctiform structures, different from our previous report that PAQR3 is a Golgi‐resident protein under normal medium conditions (Figs 1A and EV1A) (Feng et al, 2007). As autophagy is regarded as the most important mechanism to maintain cellular homeostasis under starvation conditions and autophagosomes also exhibit as punctiform distribution in the cytoplasm, we analyzed whether PAQR3‐containing puncta were co‐localized with autophagosomes. Remarkably, upon glucose starvation, punctiform PAQR3 was no longer co‐localized with the Golgi apparatus (Fig EV1A), but basically co‐localized with the autophagosome marker, LC3 (Fig 1A), suggesting that PAQR3 is translocated from the Golgi apparatus to autophagosomes upon glucose starvation. These data led us to postulate that there might be a functional link between PAQR3 and autophagy.
Figure 1. PAQR3 regulates autophagosome formation without altering the activities of AMPK and mTOR .
-
AGFP‐PAQR3‐transfected HeLa cells were fixed for immunofluorescence staining with the indicated antibodies before or after glucose starvation (GS) for 4 h. The nuclei were stained with Hoechst 33342. Scale bar: 10 μm.
-
BImmunoblotting (IB) analysis of wild‐type (WT) and PAQR3‐deficient MEFs after GS for different times as indicated.
-
CWT and PAQR3‐deficient MEFs were treated with 80 mM chloroquine (CQ) for 2 h to induce LC3‐II accumulation. Whole‐cell lysates were harvested for IB analysis of LC3‐II.
-
DAutophagosome‐like structures in WT and PAQR3‐deleted MEFs were detected after GS for 4 h by transmission electron microscopy. * indicates autophagosome‐like structures. N, nucleus.
-
EQuantitative analysis of autophagic vacuoles in transmission electron microscopy images. Thirty cells were quantified from each independent experiment, which was repeated for three times with similar results. Values are presented as mean ± SD, ***P < 0.001.
-
FImmunofluorescence staining of LC3 in WT or PAQR3‐deficient MEFs after GS for 4 h. The arrowheads indicate representative LC3‐positive puncta. Scale bar: 10 μm.
-
G, HHeLa cells were infected by PAQR3 knockdown or overexpression of lentivirus together with their respective control virus. After GS for 1 h, the cell lysates were harvested for IB.
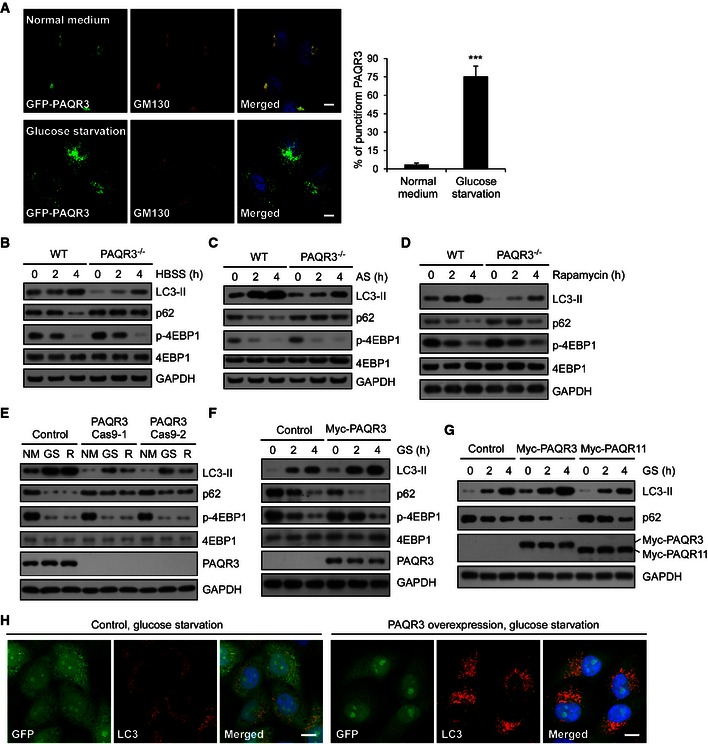
Figure EV1. PAQR3 modulates autophagy induced by multiple stress conditions.
-
AGFP‐PAQR3‐transfected HeLa cells were fixed for immunofluorescence staining with the indicated antibodies before or after glucose starvation (GS) for 4 h. At least 100 cells were counted per experiment and the data on the right represent the percentage of punctiform PAQR3 with or without GS, the results were obtained from three independent experiments. Values are presented as mean ± SD (***P < 0.001). Scale bar: 10 μm.
-
B–DImmunoblotting (IB) analysis of WT and PAQR3‐deficient MEFs after HBSS treatment, amino acid starvation (AS), or rapamycin (50 nM) treatment for different times as indicated.
-
EWT and PAQR3‐deficient HeLa (PAQR3 Cas9‐1/2) cells were incubated with normal medium (NM), GS or treated with rapamycin (50 nM) for 4 h, followed by IB.
-
FHeLa cells stably transfected with PAQR3 were incubated in GS for different times as indicated, followed by IB.
-
GHeLa cells were infected with PAQR3 or PAQR11 overexpression of lentivirus, respectively. After GS for 2 or 4 h, the whole‐cell lysates were harvested for IB.
-
HHeLa cells were infected by control or PAQR3‐expressed lentivirus. After GS for 4 h, the cells were fixed for immunofluorescence staining with LC3 antibody (red). Scale bar: 10 μm.
To analyze whether PAQR3 was able to modulate autophagy, we first examined the effect of PAQR3 deletion on autophagosome formation by investigating accumulation of LC3‐II and degradation of p62 upon glucose starvation (Klionsky et al, 2012). The accumulation of LC3‐II and degradation of p62 were drastically decreased in PAQR3‐deficient MEFs (Feng et al, 2007) (Fig 1B). We next investigated whether PAQR3 could impact autophagy induced by other autophagy‐promoting conditions. Autophagy was significantly blunted by PAQR3 deficiency in MEFs treated with Hank's balanced salt solution (HBSS) or amino acid‐free medium (Fig EV1B and C). Besides, PAQR3‐deleted MEFs also showed attenuated autophagy activity after rapamycin treatment (Fig EV1D). Then, we confirmed the regulatory function of PAQR3 on autophagy using another cell system. We utilized CRISPR/Cas9 technology to introduce mutations into PAQR3 exon with two different guide RNAs to disrupt its expression. As expected, the autophagy induced by glucose starvation or rapamycin was effectively inhibited in PAQR3‐deleted HeLa cells (Fig EV1E). In contrast, stable overexpression of PAQR3 dramatically increased the autophagic activity compared to the wild‐type cells (Fig EV1F). Furthermore, in a control experiment, glucose starvation‐induced autophagy was augmented by PAQR3, but not by PAQR11 (Jin et al, 2012), another Golgi‐localized transmembrane protein (Fig EV1G).
Accumulation of LC3‐II is mainly achieved by two mechanisms: promotion of autophagosome formation and inhibition of LC3‐II degradation. To discriminate between these two possibilities, a lysosomal inhibitor CQ (chloroquine diphosphate salt) was employed. We found that CQ‐induced LC3‐II accumulation was also reduced by PAQR3 deletion in MEFs (Fig 1C), indicating that PAQR3 might directly affect autophagosome formation. Such thought prompted us to examine whether the number of autophagosomes was altered by PAQR3. Consistent with our hypothesis, both transmission electron microscopy and LC3 immunostaining assay revealed that autophagosome formation induced by glucose starvation was strikingly blunted in PAQR3‐deficient MEFs (Fig 1D–F). These results were also confirmed in HeLa cells. The number of LC3‐containing puncta was profoundly augmented by PAQR3 overexpression in HeLa cells under the condition of glucose starvation (Fig EV1H).
The positive effect of PAQR3 on autophagy could be implemented by two possible mechanisms: 1) PAQR3 might modulate upstream autophagy‐regulatory signaling pathways, such as AMPK (Egan et al, 2011; Kim et al, 2011, 2013) or mTOR (Inoki et al, 2003; Hosokawa et al, 2009; Jung et al, 2009); 2) PAQR3 might directly affect the initial steps of vesicle nucleation during which PI(3)P was generated by activated VPS34 (Simonsen & Tooze, 2009). We found that the phosphorylation of 4‐EBP1, a downstream effector of mTORC1 (Nojima et al, 2003; Schalm et al, 2003), was not altered by PAQR3 deletion under various autophagy‐promoting conditions (Figs 1B and EV1B–F), suggesting that the regulation of PAQR3 on autophagy is not due to an alteration of mTOR activity itself. We next investigated whether PAQR3 could regulate AMPK activity by detecting AMPKα phosphorylation at Thr172 (Hawley et al, 1996) and the phosphorylation of ACC, a substrate of AMPK, at Ser79 (Davies et al, 1990). The phosphorylation levels of AMPKα and ACC were altered by neither PAQR3 knockdown (Fig 1G) nor its overexpression (Fig 1H). These data, therefore, indicated that PAQR3 regulates autophagosome formation not by directly altering the activities of AMPK and mTOR.
PAQR3 modulates the activity of ATG14L‐associated class III PI3K
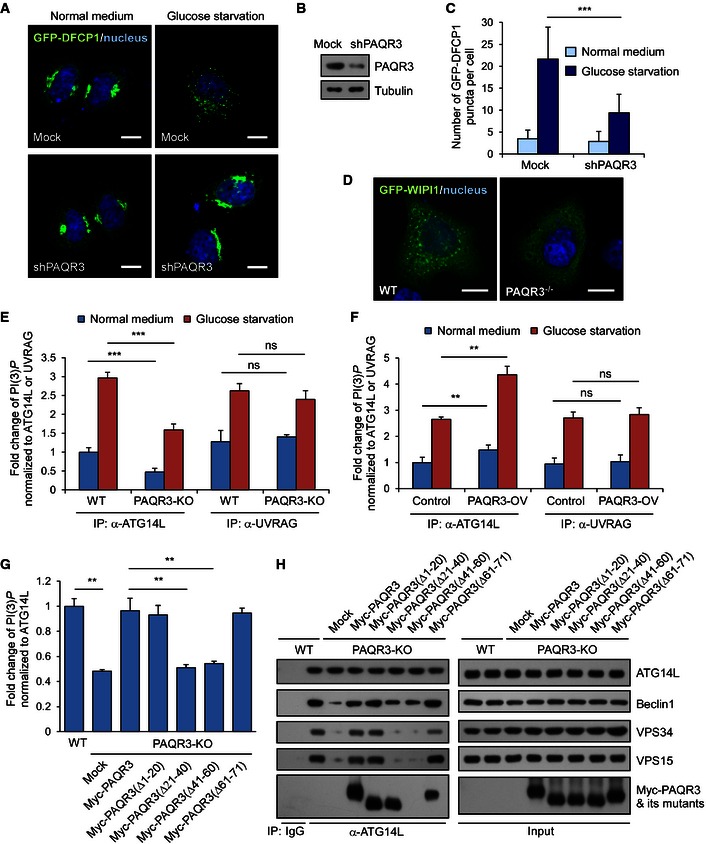
Upon triggering of autophagy by either mTOR inhibition or AMPK activation, class III PI3K is crucial for autophagy initiation by raising the concentration of PI(3)P in isolation membrane (Simonsen & Tooze, 2009). To investigate whether PAQR3 could directly modulate the activity of class III PI3K, we analyzed the effect of PAQR3 on two major downstream effectors of PI(3)P, DFCP1 and WIPI1 (Polson et al, 2010; Koyama‐Honda et al, 2013). GFP‐DFCP1 formed puncta during glucose depletion in control shRNA‐transfected cells, while PAQR3 knockdown clearly impaired the formation of GFP‐DFCP1‐positive puncta (Fig 2A–C). These data suggested that PAQR3 was essential for the activation of class III PI3K activity during the initiation phase of autophagy. Such notion was further supported by the observation that the punctiform distribution pattern of WIPI1 upon glucose starvation was also blocked by PAQR3 deletion (Fig 2D).
Figure 2. PAQR3 modulates ATG14L‐linked VPS34 lipid kinase activity.
-
A–CHeLa cells stably expressing GFP‐DFCP1 were transfected with or without PAQR3‐specific shRNA. GFP signals were observed in these cells before or after glucose starvation (GS, shown in A). The knockdown efficiency of PAQR3 is shown in (B), and the statistics of GFP‐positive puncta per cell is illustrated in (C). Scale bar: 10 μm.
-
DGFP‐WIPI1 was transfected into WT or PAQR3‐deficient MEFs. Twenty‐four hours later, the cells were incubated under GS for 4 h, followed by fluorescence observation. Scale bar: 10 μm.
-
EDifferent VPS34 complexes of WT or PAQR3 knockout HeLa cells were immunoprecipitated by ATG14L and UVRAG antibodies, respectively, in normal medium (NM) or GS. Then, PI(3)P level was determined by a quantitative ELISA. The PI(3)P level was normalized to the amount of ATG14L or UVRAG used in the assay.
-
FHeLa cells infected with control or PAQR3‐overexpressed lentivirus were treated with GS for 4 h. PI(3)P levels were determined as in Fig 2E.
-
G, HPAQR3 knockout HeLa cells were transfected with plasmids of multiple PAQR3 deletion mutants. At 24 h after transfection, the cell lysates were used to immunoprecipitate VPS34 complex by ATG14L antibody, followed by immunoblotting. The activity of ATG14L‐linked VPS34 activity was detected by a quantitative ELISA. The PI(3)P level was normalized to the amount of ATG14L.
In spite of the existence of numerous distinct VPS34 complexes according to their different modes of regulation on autophagy, only two VPS34 subpopulations have increased activity upon glucose starvation: ATG14L–Beclin1–VPS34 complex and UVRAG–Beclin1–VPS34 complex (Kim et al, 2013). To elucidate the influence of PAQR3 on VPS34 activity, the capability of PI(3)P production of these two different VPS34 complexes was determined by a quantitative ELISA analysis. In accordance with a previous report (Kim et al, 2013), glucose starvation increased the activity of both ATG14L‐ and UVRAG‐linked VPS34 complexes (Fig 2E and F). However, only ATG14L‐associated kinase activity but not UVRAG‐associated kinase activity was dramatically reduced in PAQR3‐deficient HeLa cells (Fig 2E). In contrast, PAQR3 overexpression could significantly enhance ATG14L‐associated kinase activity, without affecting UVRAG‐associated kinase activity (Fig 2F). Collectively, these data indicated that PAQR3 can specifically enhance ATG14L‐associated class III PI3K activity.
As PAQR3 was identified as a modulator of ATG14L‐associated VPS34 complex, we next investigated the essential structural motifs of PAQR3 involved in VPS34 activity regulation. Our previous studies had pinpointed that the NH2‐terminal 71 amino acid residues are critical for the functionality of PAQR3 (Feng et al, 2007; Luo et al, 2008). We therefore analyzed various PAQR3 mutants with different NH2‐terminal deletions. In PAQR3 knockout HeLa cells, the decreased ATG14L‐associated VPS34 activity could be successfully rescued by full‐length PAQR3 and its mutants Δ1–20 and Δ61–70 (Fig 2G). However, the PAQR3 mutants Δ21–40 and Δ41–60 could not rescue the decreased kinase activity (Fig 2G), suggesting that the NH2‐terminal 21–60aa of PAQR3 was indispensable for its regulation on VPS34 lipid kinase activity. In this VPS34 activity rescue assay, we also analyzed the interaction between ATG14L and other core components of the class III PI3K. Interestingly, PAQR3 deletion reduced the interaction of ATG14L with VPS34, VPS15, and Beclin1 (Fig 2H), suggesting that PAQR3 could directly modulate the formation of ATG14L‐associated VPS34 complex. In addition, the ameliorated ATG14L‐associated complex formation could be regained by wild‐type PAQR3, but not by the mutants Δ21–40 and Δ41–60 (Fig 2H). These observations, therefore, further indicated that the NH2‐terminal 21–60aa of PAQR3 is crucial for both the constitution and activity of ATG14L‐linked VPS34 activity.
PAQR3 is a constitutive component of the ATG14L–Beclin1–VPS34 complex, mutually exclusive with UVRAG
As PAQR3 could modulate the formation of ATG14L‐associated class III complex, we postulated that PAQR3 might be able to interact with the components of VPS34 complex. Consistent with our hypothesis, a co‐immunoprecipitation experiment revealed that endogenous PAQR3 could associate with VPS34, VPS15, Beclin1, and ATG14L, but not with UVRAG (Fig 3A), indicating that PAQR3 is a binding partner of the ATG14L‐linked VPS34 complex, but not the UVRAG‐associated complex. To figure out whether ATG14L‐linked VPS34 complex was the binding partner of PAQR3 cellular complex, we stably transfected Myc‐tagged PAQR3 into PAQR3‐deficient HeLa cells, in which the expression level of the exogenous PAQR3 was comparable to that in WT cells (Fig 6F). PAQR3 immunoprecipitated from these “PAQR3 rescued” HeLa cells and its associated proteins contained Beclin1, ATG14L, VPS34, and VPS15 as analyzed by mass spectrometry (Appendix Fig S1B–E), demonstrating that ATG14L‐linked VPS34 complex was the binding partner of PAQR3.
Figure 3. PAQR3 is a component of the ATG14L–Beclin1–VPS34 complex, mutually exclusive with UVRAG .
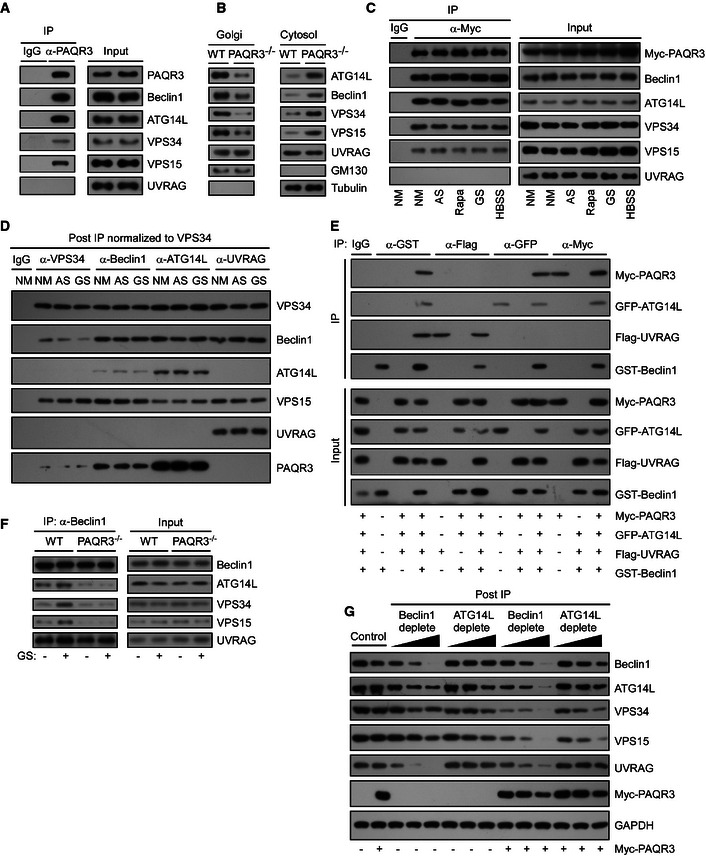
- Interaction between endogenous PAQR3 and the components of VPS34 complex in HEK293T cells. The cell lysates were used for immunoprecipitation (IP) and immunoblotting (IB) with the antibodies as indicated.
- Cytosol and Golgi fractionations were isolated from the livers of PAQR3‐deleted mice and their littermate controls. Equal protein amounts of cytosol and Golgi fractions were subjected to IB.
- HEK293T cells were transiently transfected with Myc‐tagged PAQR3. At 24 h after transfection, the cells were treated with normal medium (NM), amino acid starvation (AS) or glucose starvation (GS), HBSS solution, or rapamycin (Rapa, 50 nM) for 4 h, respectively. The cell lysates were then used in IP and IB.
- Four VPS34 complexes of MEFs were immunopurified using the indicated antibodies under NM, AS, or GS conditions. The relative abundance of VPS34‐binding partners was determined by IB. Each IP was normalized to the amount of VPS34.
- HEK293T cells were transfected with the plasmids as indicated, and the cell lysates were used in IP and IB.
- The lysates of WT or PAQR3 knockout MEFs were used in IP and IB.
- MEFs were infected with control or PAQR3‐expressing lentivirus. Then, a quantitative immunodepletion assay was performed with increasing amounts of the indicated antibodies. Supernatants of MEF lysates after immunodepletion (Post‐IP) were examined to determine the level of VPS34 complex proteins.
Figure 6. Phosphorylation of PAQR3 T32 is required for autophagy initiation upon glucose starvation.
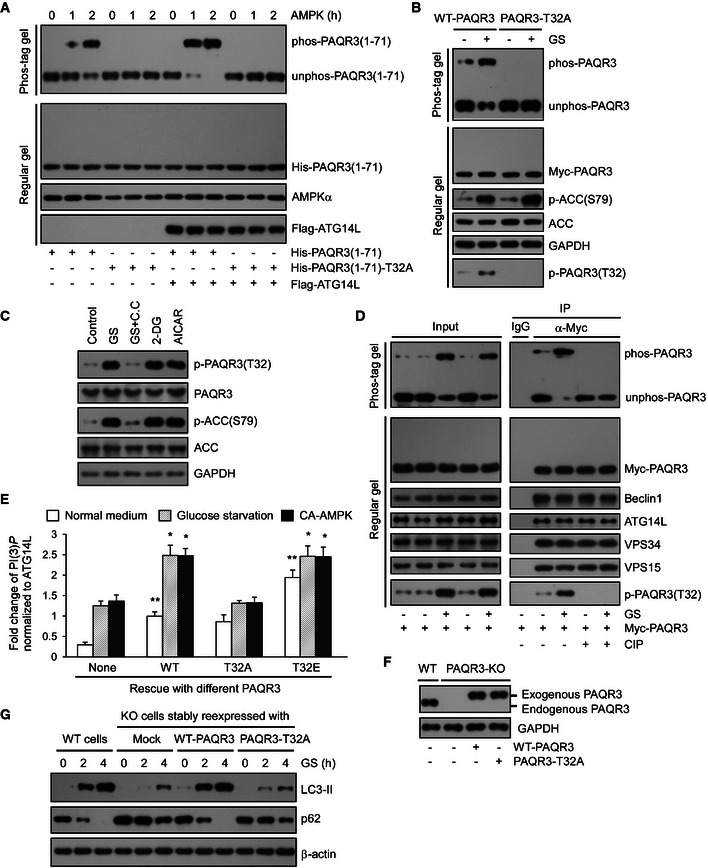
- Bacterial purified His‐tagged WT or T32A NH 2‐terminal 71aa of PAQR3 was incubated with or without Flag‐tagged ATG14L purified from HEK293T cells. Then, the complexes were incubated with AMPK for the indicated time and subjected to Phos‐tag gel or regular SDS–PAGE.
- PAQR3‐deficient HeLa cells were transfected with WT or T32A mutants of PAQR3 plasmids. Twenty‐four hours later, the cells were incubated with normal medium (NM) or glucose starvation (GS) for 4 h, followed by immunoblotting (IB) in Phos‐tag gel or regular SDS–PAGE.
- HeLa cells were incubated with NM or GS for 1 h or treated with compound C (C.C, 20 mM, 30 min) prior to GS for 1 h. In parallel, 25 mM 2‐DG or 1 mM AICAR was added in NM for 1 h. Cell lysates were analyzed by IB.
- HEK293T cells were transfected with Myc‐tagged PAQR3. At 24 h after transfection, the cells were treated with GS for 4 h. Then, the cell lysates were used in IB and immunoprecipitation (IP) with the antibodies as indicated. The immunoprecipitates were treated with CIP to remove phosphate groups and analyzed by IB in Phos‐tag gel or regular SDS–PAGE.
- WT, phosphorylation defective (T32A), or phospho‐mimetic (T32E) PAQR3 were transfected in the presence or absence of constitutively active AMPK (CA‐AMPK) into PAQR3‐deficient HeLa cells as indicated. Twenty‐four hours later, the cells without AMPK transfection were treated with GS for 4 h. About 10% of the cell lysates were subjected to IB to detect PAQR3 expression level (shown in Appendix Fig S4A), and the remaining cell lysates were used to immunoprecipitate VPS34 complexes by ATG14L antibody, followed by VPS34 activity measurement by a quantitative PI(3)P ELISA. The PI(3)P level was normalized to the amount of ATG14L. Values are presented as mean ± SD (n = 5; *P < 0.05; **P < 0.01 as compared to the first group).
- PAQR3‐deficient HeLa cells were infected with lentivirus expressing WT or T32A PAQR3. Then, PAQR3 expression levels were examined by IB.
- Both WT HeLa cells and PAQR3 knockout HeLa cells infected with lentivirus expressing WT or T32A PAQR3 were treated with GS for 2 or 4 h, respectively. Cell lysates were analyzed by IB.
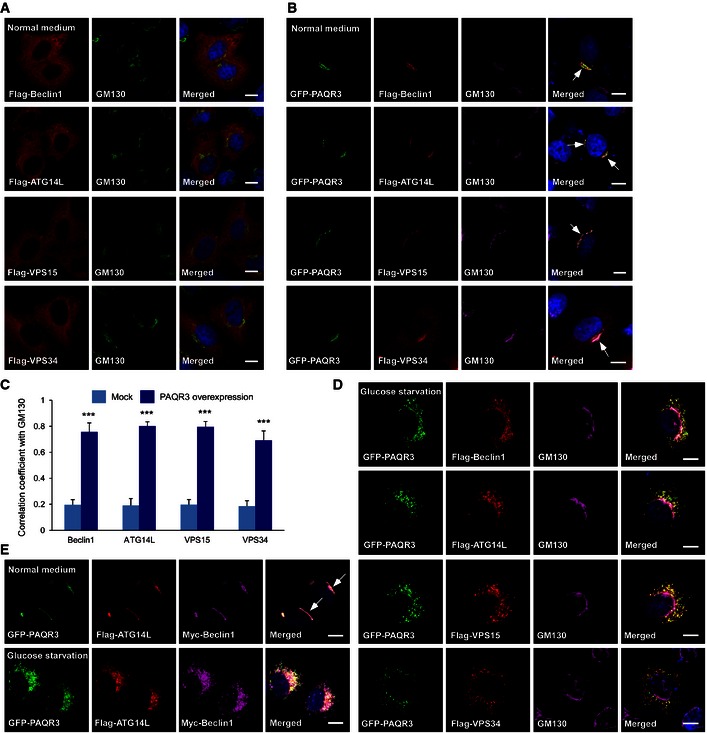
To further investigate the relationship between PAQR3 and ATG14L‐linked VPS34 complexes, we purified the Golgi complexes from mouse livers to analyze the localization of endogenous VPS34 complex using a biochemical approach as reported (Bartz et al, 2008; Wei & Seemann, 2009; Wang et al, 2013). As shown in Fig 3B, ATG14L‐linked VPS34 complex could be clearly detected in the Golgi fractions of wild‐type mice, supporting that there indeed exists Golgi localization of ATG14–Beclin1–VPS34 complex. However, PAQR3 knockout led to dramatic reduction in Golgi compartmentalization of Beclin1, ATG14L, VPS15, and VPS34, but not UVRAG (Fig 3B). These data demonstrated that PAQR3 is critical for the Golgi localization of ATG14L‐linked VPS34 complex. To further confirm this conclusion, we detected the effect of PAQR3 on cellular localization of ATG14L‐linked VPS34 complex by immunostaining assay. When transfected alone, exogenous Beclin1, ATG14L, VPS15, and VPS34 were all diffusely distributed in the cytoplasm with minimal co‐localization with the Golgi marker GM130 (Appendix Fig S3A). Remarkably, when co‐expressed with PAQR3, they were all co‐localized well with GM130 (with Pearson's correlation coefficient > 0.7, Fig EV2B and C). These data further indicated that ATG14L‐linked VPS34 complex can coexist with PAQR3 in the Golgi apparatus.
Figure EV2. ATG14L‐linked VPS34 complex coexists with PAQR3 in the Golgi apparatus.
-
A, BHeLa cells were transfected with the plasmids as indicated, followed by immunofluorescence staining with the indicated antibodies. The arrow indicates co‐localization of PAQR3 with the indicated proteins at the Golgi apparatus. Scale bar: 10 μm.
-
CCo‐localization coefficient between the indicated proteins and GM130 in the presence or absence of PAQR3 overexpression. At least fifty cells were quantified from each independent experiment, which was repeated for three times with similar results. Values are presented as mean ± SD, ***P < 0.001.
-
DHeLa cells were transfected with the plasmids as indicated. After glucose starvation for 4 h, the cells were fixed and used in immunofluorescence staining and confocal analysis. Scale bar: 10 μm.
-
EHeLa cells were co‐transfected with GFP‐fused PAQR3, Flag‐tagged ATG14L, and Myc‐tagged Beclin1 simultaneously. At 24 h after transfection, the cells were fixed for immunofluorescence staining before or after glucose starvation as indicated. The arrow indicates apparent co‐localization of ATG14L and Beclin1 with PAQR3. Scale bar: 10 μm.
Then, we asked whether the interaction between PAQR3 and ATG14L‐associated VPS34 complexes is dependent on autophagy stimulation. As shown in Fig 3C, PAQR3 could associate with the ATG14L‐linked VPS34 complex with or without autophagy‐promoting signals including amino acid depletion, glucose starvation, HBSS, and rapamycin treatment, supporting a constitutive interaction of PAQR3 with this complex. Consistently, upon glucose starvation, PAQR3 was redistributed into puncta and was no longer co‐localized with the Golgi marker (Figs EV1A and EV2D). However, the punctiform PAQR3 was still basically co‐localized with ATG14L‐associated VPS34 complex (Fig EV2D), further supporting that PAQR3 is a binding partner of ATG14L‐linked class III PI3K regardless of glucose starvation.
As the activity of distinct VPS34 complexes is differentially regulated under starvation (Kim et al, 2013), it would be necessary to determine which VPS34 pool(s) PAQR3 belongs to. We isolated various VPS34‐containing pools by immunoprecipitation with antibodies against VPS34, Beclin1, ATG14L, and UVRAG, respectively (Fig 3D). After normalization to VPS34 protein level, most PAQR3 protein was enriched in ATG14L‐associated VPS34 pool, while Beclin1‐ and VPS34‐associated pools had moderate level of PAQR3 protein. However, no detectable level of PAQR3 was found in the UVRAG pool. These results further consolidated our finding that PAQR3 could not interact with the UVRAG‐associated complex. Additionally, it was noteworthy that the distribution pattern of PAQR3 was similar to ATG14L, which acts in a mutually exclusive manner with UVRAG (Itakura et al, 2008). Therefore, we employed another co‐immunoprecipitation assay to clarify whether PAQR3 is a specific binding partner of ATG14L‐linked VPS34 complex (Fig 3E). As a positive control, Beclin1 could associate with PAQR3, ATG14L, and UVRAG, respectively. However, when the cell lysates were immunoprecipitated with GFP‐fused ATG14, only exogenous PAQR3 and Beclin1, but not UVRAG, were co‐precipitated. Similarly, PAQR3 could interact with Beclin1 and ATG14L, but not with UVRAG. Lastly, neither PAQR3 nor ATG14L could be detected in the UVRAG‐enriched precipitation. These findings indicated that PAQR3 is in the same complex with ATG14L, mutually exclusive from UVRAG.
What is the functional significance of the interaction between PAQR3 and ATG14L‐linked VPS34 complexes? We asked whether PAQR3 is required for the formation of the ATG14L‐linked VPS34 complex. Using a co‐immunoprecipitation experiment to pull down endogenous Beclin1, we found that the association of Beclin1 with ATG14L, VPS34, and VPS15 was markedly reduced by PAQR3 deletion regardless of glucose starvation. However, the association between UVRAG and Beclin1 was not affected by PAQR3 deficiency (Fig 3F). To further investigate whether the relative abundance of ATG14L‐linked VPS34 complex was modulated by PAQR3, we performed an immunodepletion assay by using increasing amounts of Beclin1 or ATG14L antibodies (Fig 3G). Overexpression of PAQR3 could profoundly increase the removal rate of ATG14L, VPS34, and VPS15 when Beclin1 was depleted. Likewise, ATG14L immunodepletion reduced VPS34 and Beclin1 more effectively in PAQR3‐overexpressed cells. However, the amount of a housekeeping protein GAPDH was not reduced after immunodepletion by Beclin1 or ATG14L antibodies. These results clearly demonstrated that PAQR3 can facilitate and stabilize the formation of ATG14L‐linked VPS34 complex.
PAQR3 is a scaffold protein of the ATG14L–Beclin1–VPS34 complex
As PAQR3 could facilitate the formation of the ATG14L‐linked VPS34 complex, we hypothesized that PAQR3 might interact with key components of this complex simultaneously. Firstly, we found that PAQR3, ATG14L, and Beclin1 could coexist in the same complex simultaneously by a two‐step co‐immunoprecipitation assay (Fig 4A). To further strengthen this conclusion, we detected the cellular co‐localization of PAQR3, Beclin1, and ATG14L. Consistent with previous reports (Matsunaga et al, 2009; Huang et al, 2012), both ATG14L and Beclin1 are mainly diffusely distributed in the cytosol when transfected alone (Fig EV2A). However, upon co‐expression of PAQR3 with ATG14L and Beclin1, these three proteins co‐localized well both under normal medium and glucose starvation (Fig EV2E), even though PAQR3 exhibited as punctiform distribution after glucose starvation, further supporting that PAQR3, Beclin1, and ATG14L could exist in the same complex regardless of glucose starvation.
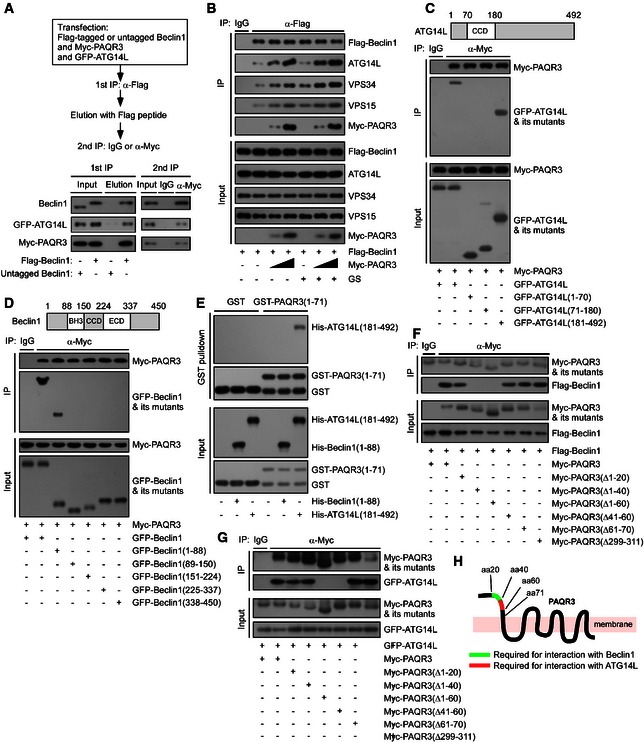
Figure 4. PAQR3 is a scaffold of the ATG14L–Beclin1–VPS34 complex.
-
AA two‐step co‐IP assay in HEK293T cells to determine the ternary complex containing PAQR3, Beclin1, and ATG14L. The procedures of the two‐step co‐IP are outlined in the top. The samples were subjected to immunoprecipitation (IP) and immunoblotting (IB) with the antibodies as indicated.
-
BHeLa cells were transiently transfected with the plasmids as indicated. At 24 h after transfection, the cells were treated with or without glucose starvation (GS) for 4 h. The cell lysates were used in IP and IB.
-
C, DDifferent truncations of ATG14L (C) or Beclin1 (D) were co‐transfected with Myc‐tagged PAQR3 into HEK293T cells as indicated, followed by IP and IB.
-
EPurified GST or GST‐fused PAQR3 (1–71) (GST‐fused NH 2‐terminal 71aa of PAQR3) on glutathione agarose beads was mixed with purified His‐tagged proteins as indicated. After incubation at 4°C for 3 h, the samples were washed extensively and subjected to IB.
-
F, GDifferent PAQR3 deletion constructs were co‐transfected with Flag‐tagged Beclin1 (F) or GFP‐fused ATG14L (G) into HEK293T cells. The cells were then subjected to IP and IB.
-
HA schematic diagram depicts critical domains of PAQR3 involved in the interaction with ATG14L and Beclin1, respectively.
As PAQR3 could stabilize the ATG14L‐linked VPS34 complex (Fig 3F and G) and coexist in the same complex with ATG14L and Beclin1 (Fig 4A), we boldly postulated that PAQR3 might function as a scaffold protein to enhance the interaction between ATG14L and Beclin1. Consistent with our hypothesis, we found that the associations of Beclin1 with ATG14L, VPS34, and VPS15 were greatly enhanced by PAQR3 in a dose‐dependent manner regardless of glucose starvation (Fig 4B).
Then, we investigated the domains of Beclin1 and ATG14L involved in the interaction with PAQR3. As shown in Fig 4C, PAQR3 could interact with the full‐length ATG14L and the region containing 181–492aa. In addition, the NH2‐terminal 1–88aa of Beclin1 was identified as the major binding region for PAQR3 (Fig 4D). We next investigated whether these interactions were direct by in vitro GST pull‐down assay. However, we could hardly purify full‐length Beclin1 and ATG14L in bacterial expression system, likely due to the instability or poor solution behavior of these proteins (Fan et al, 2011; Huang et al, 2012; Kim et al, 2013). Also, we have not obtained the purified full‐length PAQR3 in bacterial expression system until now, perhaps owing to the hydrophobic surfaces, flexibility, and instability of membrane proteins (Carpenter et al, 2008; Baker, 2010). Alternatively, based on our findings shown in Fig 4C and D, we purified GST‐tagged NH2‐terminal 71aa of PAQR3, His‐tagged 181–492aa of ATG14L, and NH2‐terminal 1–88aa of Beclin1 in vitro, respectively. As shown in Fig 4E, GST‐fused PAQR3 NH2‐terminal 71aa, but not GST alone, pulled down His‐tagged 181–492aa of ATG14L, indicating a direct interaction between PAQR3 and ATG14L. In contrast, PAQR3 could not associate with Beclin1 directly.
Similarly, we also investigated the interaction domains of VPS15 and VPS34 with PAQR3 (Appendix Fig S2A and B). Based on these mapping results, we purified the C2 and catalytic domain of VPS34, as well as the catalytic domain and WD repeats of VPS15 in vitro, respectively. As shown in the GST pull‐down assay of Appendix Fig S2C, PAQR3 directly interacted with VPS15, but not with VPS34.
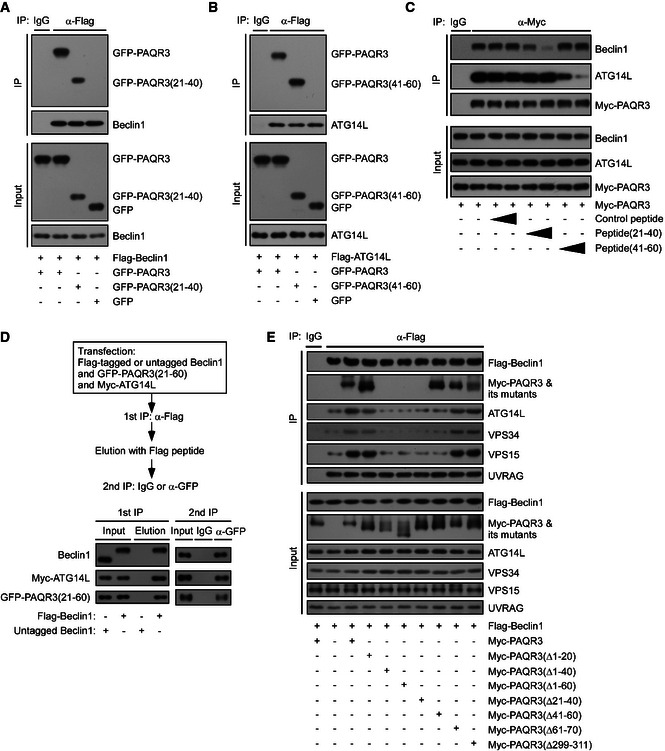
We next identified the domains of PAQR3 involved in the interaction with Beclin1 and ATG14L, respectively. We found that PAQR3 deletion mutants Δ1–40 and Δ1–60 could not interact with Beclin1, while the association of PAQR3 with Beclin1 was retained by deletion mutants Δ1–20 and Δ41–60 (Fig 4F), indicating that NH2‐terminal 21–40aa of PAQR3 was required for Beclin1 interaction. Similarly, PAQR3 deletions of amino acid residues 1–60 and 41–60, but not 1–40, lost the ability to interact with ATG14L (Fig 4G), supporting that 41–60aa of PAQR3 was essential for ATG14L binding (Fig 4H). Furthermore, 21–40aa and 41–60aa of PAQR3 were sufficient to associate with Beclin1 and ATG14L, respectively (Fig EV3A and B). To further emphasize that NH2‐terminal 21–60aa of PAQR3 is the binding motif for Beclin1 and ATG14L, we designed two kinds of synthetic peptide (P21–40 and P41–60) that covered PAQR3 NH2‐terminal 21–40 and 41–60aa, respectively. A TAT sequence (RKKRRQRRR) was added in the NH2 terminus of the peptide to facilitate membrane penetrance. As expected, P21–40 dramatically neutralized the interaction between PAQR3 and Beclin1, but not the interaction between PAQR3 and ATG14L (Fig EV3C), demonstrating the NH2‐terminal 21–40aa of PAQR3 is critical for Beclin1 interaction. In contrast, the association between PAQR3 and ATG14L was specifically ameliorated by P41–60 (Fig EV3C), supporting that the 41–60aa of PAQR3 is the binding motif for ATG14L. In addition, we performed a two‐step co‐immunoprecipitation assay and found that the 21–60aa of PAQR3 could interact with Beclin1 and ATG14L simultaneously (Fig EV3D). Furthermore, we analyzed the effects of various PAQR3 mutants on the association of Beclin1 with other components of the ATG14L‐linked VPS34 complex. As expected, wild‐type PAQR3 and its deletion mutants Δ1–20 and Δ61–70 dramatically enhanced the interaction between Beclin1 and its binding partners, such as ATG14L, while such interaction could not be promoted by PAQR3 mutants Δ21–40 and Δ41–60 (Fig EV3E). Collectively, these results illustrated that PAQR3 acts as a scaffold to promote ATG14L–Beclin1 interaction.
Figure EV3. Beclin1 and ATG14L interact with the NH 2‐terminal 21–60aa of PAQR3.
-
A, BHEK293T cells were transfected with different plasmids as indicated. At 24 h after transfection, the cells were subjected to immunoprecipitation (IP) and immunoblotting (IB) analyses.
-
CHEK293T cells were transfected with Myc‐tagged PAQR3. At 24 h after transfection, the cells were treated with P21–40 or P41–60 (4 or 20 ng/µl) for 12 h, respectively, and the cell lysates were used in IP and IB.
-
DA two‐step co‐immunoprecipitation assay to determine the complex formation among PAQR3 NH 2‐terminal 21–60aa, Beclin1, and ATG14L. The procedures of the assay are outlined in the top panel. HEK293T cells were transfected with the plasmids as indicated. At 24 h after transfection, the cells were harvested for IP and IB.
-
EHeLa cells were transfected with the plasmids as indicated. The cell lysates were used for IP and IB.
PAQR3 is phosphorylated by AMPK upon glucose starvation in an ATG14L‐dependent manner
Although we found that PAQR3 was a constitutive component of the ATG14L–Beclin1–VPS34 complex regardless of autophagy‐initiating signals (Figs 3 and 4), PAQR3 modulates autophagy process more dramatically under glucose depletion compared to energy‐rich conditions (Figs 1 and 2). We therefore postulated that glucose starvation would transduce a signal to PAQR3 to amplify its regulatory effect on autophagy. As the activity of VPS34 complex can be differentially regulated by multiple phosphorylation events of Beclin1 (Wang et al, 2012; Kim et al, 2013; Russell et al, 2013; Wei et al, 2013), we investigated whether PAQR3 could also be phosphorylated by glucose starvation. To examine PAQR3 phosphorylation, we employed a method based on Phos‐tag gels in which phosphorylated proteins are super‐shifted away from the unphosphorylated proteins (Kinoshita et al, 2006, 2009). As shown in Fig 5A, PAQR3 migrated as two major distinct bands on a Phos‐tag gel after glucose starvation, while only one band was detected on a regular SDS–PAGE gel. In addition, PAQR3 was reduced to a single band with the treatment of a phosphatase CIP (Fig 5A), suggesting that PAQR3 could be phosphorylated during starvation. Remarkably, the super‐shifted band induced by glucose starvation was dramatically attenuated in response to glucose replenishment, further demonstrating PAQR3 phosphorylation is stimulated by glucose starvation (Fig 5B).
Figure 5. PAQR3 is phosphorylated by AMPK during autophagy, dependent on ATG14L.
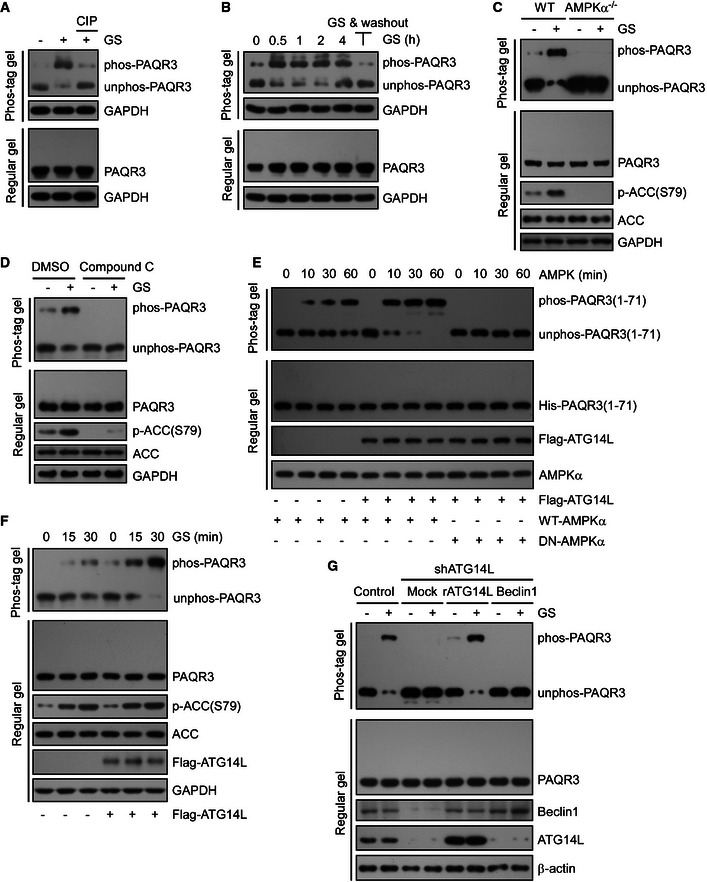
- HeLa cells were cultured in normal medium (NM) or glucose starvation (GS) for 4 h. The starved lysates were treated with calf intestinal phosphatase (CIP) to remove phosphate groups and analyzed by immunoblotting (IB) with Phos‐tag gel or regular SDS–PAGE.
- The cell lysates from HeLa cells were collected and analyzed after GS for the indicated time (lane 1–5), or GS for 2 h followed by 2‐h chase in NM (lane 6). The cell lysates were subjected to Phos‐tag gel and regular SDS–PAGE, respectively.
- WT or AMPKα1/2 double knockout MEFs were incubated under NM or GS for 4 h, followed by IB with Phos‐tag gel or regular SDS–PAGE.
- HeLa cells were incubated with compound C (20 mM, 30 min) with or without GS, followed by IB with Phos‐tag gel or regular SDS–PAGE.
- Bacterial purified His‐tagged NH 2‐terminal 71 amino acid of PAQR3 was incubated with or without purified Flag‐tagged ATG14L. Then, the complexes were treated with WT AMPK or DN‐AMPK (dominant negative AMPK) for the indicated time in vitro. PAQR3 phosphorylation was examined by Phos‐tag gel.
- HeLa cells infected with control or ATG14L‐expressing lentivirus were incubated under GS for different times as indicated. Whole‐cell lysates were analyzed by both Phos‐tag gel and regular SDS–PAGE.
- HeLa cells were infected by lentivirus expressing control shRNA or ATG14L‐specific shRNA. Then, the ATG14L knockdown cells were stably re‐expressed with shRNA‐resistant ATG14L (rATG14L) or Beclin1 and incubated under NM or GS for 4 h. Whole‐cell lysates were analyzed by both Phos‐tag gel and regular SDS–PAGE.
As glucose starvation‐induced autophagy is prevailingly mediated by the activation of AMPK signaling pathway, we next tested whether AMPK was required for PAQR3 phosphorylation. In wild‐type MEFs, glucose starvation clearly resulted in AMPK activation as verified by phosphorylation of its substrate ACC (Fig 5C). Meanwhile, PAQR3 phosphorylation was also obviously enhanced by glucose starvation in these cells (Fig 5C). However, the phosphorylation of PAQR3, together with phosphorylation of ACC, was abrogated in AMPKα‐deleted MEFs (Fig 5C). Consistently, compound C, an AMPK inhibitor, could also inhibit phosphorylation of PAQR3 and ACC induced by glucose starvation (Fig 5D). These data strongly indicated that AMPK is indispensable for glucose starvation‐induced PAQR3 phosphorylation.
Since PAQR3 modulates autophagy via association with ATG14L–Beclin1–VPS34 complex, we next examined whether or not these components impacted phosphorylation of PAQR3. Firstly, we purified his‐tagged NH2‐terminal 71aa of PAQR3 in vitro, which is regarded as the most likely phosphorylated motif for AMPK because of its cytosolic orientation in topology (Luo et al, 2008), as well as its enrichment in serine or threonine residues. Then, the purified NH2‐terminal 71aa was combined with ATG14L, Beclin1, VPS15, and VPS34, respectively, and treated with AMPK for various time (Fig 5E and Appendix Fig S3A). We found that only ATG14L but not other components of VPS34 complex enhanced the phosphorylation of PAQR3 NH2‐terminal domain by wild‐type AMPK (Fig 5E). However, PAQR3 could not be phosphorylated by the dominant negative AMPK (Fig 5E). Similarly, HeLa cells stably transfected with ATG14L showed stronger PAQR3 phosphorylation upon glucose starvation than the controls (Fig 5F). These data indicated that PAQR3 phosphorylation was specifically promoted by ATG14L. To further verify this conclusion, we examined starvation‐stimulated PAQR3 phosphorylation in wild‐type and ATG14L‐downregulated HeLa cells. As shown in Fig 5G, PAQR3 phosphorylation was almost completely diminished upon ATG14L knockdown after glucose starvation, while stable transfection of shRNA‐resistant ATG14L could dramatically restore its phosphorylation. In contrast, although Beclin1 protein level was also attenuated in ATG14L knockdown cells, PAQR3 phosphorylation failed to be rescued by Beclin1 re‐expression (Fig 5G). Furthermore, to exclude the possibility that PAQR3 phosphorylation was nonspecifically regulated by autophagy per se, other two proteins indispensable for autophagy, ATG5 and ATG7, were also knocked down. As expected, PAQR3 phosphorylation was not altered by downregulation of these two proteins (Appendix Fig S3B). Taken together, our results indicated that PAQR3 is phosphorylated by AMPK upon glucose starvation in an ATG14L‐dependent manner.
Phosphorylation of threonine 32 (T32) of PAQR3 by AMPK is required for autophagy initiation upon glucose starvation
Since the NH2‐terminal end of PAQR3 could be directly phosphorylated by AMPK, we next investigated the potential phosphorylation site within this region. There were nine serine or threonine residues (S8, S16, T32, S39, T47, S56, S62, S67, and T70) within this region and each of them was mutated into alanine, respectively. We identified that only T32A mutation could completely abolish AMPK‐mediated phosphorylation with or without overexpression of ATG14L (Figs 6A and EV4A and B). Additionally, mass spectrometry analysis provided direct evidence that T32 of PAQR3 was phosphorylated in an in vitro AMPK kinase assay (Fig EV4C). These data suggested that T32 of PAQR3 is the primary AMPK phosphorylation site. Therefore, we produced an antibody that specifically recognizes T32‐phosphorylated PAQR3. To confirm the specificity of this antibody, we reconstituted PAQR3‐deleted HeLa cells with wild‐type or T32A PAQR3 and analyzed PAQR3 phosphorylation upon glucose starvation (Fig 6B). Remarkably, starvation‐induced T32 phosphorylation was totally eliminated in cells transfected with T32A PAQR3, but not with wild‐type PAQR3. Using this phosphorylation‐specific antibody, we observed that the AMPK activators 2‐DG and AICAR increased T32 phosphorylation of PAQR3 even in glucose‐rich conditions. In contrast, an AMPK inhibitor, compound C, blocked glucose starvation‐induced PAQR3 phosphorylation (Fig 6C). Meanwhile, the constitutively activated AMPK (AMPK‐CA), rather than a dominantly negative AMPK (AMPK‐DN), enhanced PAQR3 T32 phosphorylation level (Fig EV4D). These data provided additional evidence that T32 of PAQR3 is specifically phosphorylated by AMPK upon glucose starvation. Importantly, T32 of PAQR3 was conserved throughout vertebrates (Fig EV4E). This prompted us to further dissect the potential function of this phosphorylation in the regulation of VPS34 complex. We firstly analyzed whether phosphorylation of PAQR3 could alter its association with the ATG14L‐linked VPS34 complex. As expected, glucose starvation clearly induced PAQR3 phosphorylation, which was abrogated by CIP treatment (Fig 6D). However, the changes of PAQR3 phosphorylation were not accompanied by changes of PAQR3 interaction with the components of the ATG14L‐linked VPS34 complex (Fig 6D). Notably, these results were in agreement with our previous observations that PAQR3 was a constitutive binding partner of ATG14L‐associated VPS34 complex unaffected by autophagy‐initiating signals (Fig 3C and D).
Figure EV4. PAQR3 T32 is directly phosphorylated by AMPK .
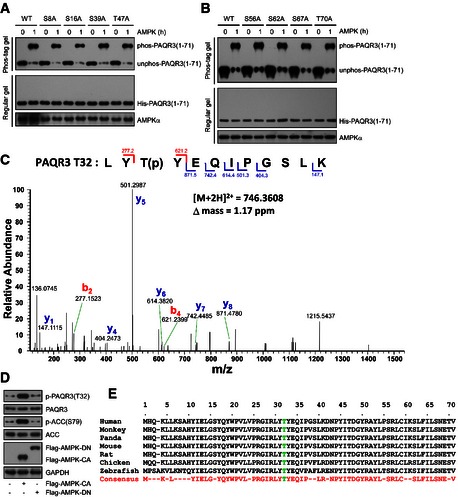
-
A, BBacterial purified His‐tagged WT or mutant NH 2‐terminal 71aa of PAQR3 was subjected to AMPK kinase assay for the indicated time, followed by Phos‐tag gel or regular SDS–PAGE analysis.
-
CBacterial purified His‐tagged NH 2‐terminal 71aa of PAQR3 was subjected to AMPK kinase assay, followed by mass spectrometry analysis. The MS/MS spectrum of a precursor ion with [M + 2H]2+ = 746.3608 was identified as PAQR3 T32‐phosphorylated tryptic peptide LYT(p)YEQIPGSLK. The labeled mass peaks indicate b and y ion series that matched the phosphopeptide.
-
DHeLa cells were transfected with constitutively activated AMPK (AMPK‐CA) or AMPK (AMPK‐DN). At 24 h after transfection, the cell lysates were harvested for immunoblotting.
-
ET32 of PAQR3 is conserved in different species. Sequence alignment of PAQR3 T32 and surrounding amino acids in a few representative species are depicted.
We next analyzed whether PAQR3 phosphorylation could affect ATG14L‐linked lipid kinase activity (Fig 6E and Appendix Fig S4A). As expected, reconstitution of wild‐type PAQR3 into PAQR3‐deficient cells could significantly augment both the basal ATG14L‐associated VPS34 activity and that stimulated by glucose starvation or a constitutive active AMPK. However, such augmentation was completely abolished by T32A mutation. In contrast, T32E mutation that mimicked the phosphorylation could greatly elevate the basal kinase activity. These results indicated that PAQR3 T32 phosphorylation is required for the activation of the ATG14‐associated VPS34 complex by AMPK upon glucose starvation.
To further address the functional importance of T32 phosphorylation in autophagy, we stably transfected wild‐type and T32A mutant of PAQR3 into PAQR3‐deleted HeLa cells, in which the expression level of these exogenous PAQR3 was comparable to that in wild‐type cells (Fig 6F). It was reported that two different regulatory mechanisms controls class III PI3K activity upon glucose starvation: (i) formation of different VPS34 complexes and (ii) posttranslational modification of its core component (Kim et al, 2013). As T32 phosphorylation of PAQR3 could not alter its interaction with ATG14L‐linked VPS34 complex (Fig 6D), we focused on the second possibility. It was worth noting that human Beclin1 phosphorylation at S93/S96 (S91/94 in mouse) is required for ATG14L‐associated VPS34 complex activation upon glucose starvation (Kim et al, 2013). Therefore, we investigated whether T32 phosphorylation of PAQR3 is required for human Beclin1 S93 phosphorylation. As shown in Appendix Fig S4B, in PAQR3‐deficient cells, phosphorylation of Beclin1 at S93 was markedly reduced no matter treated with or without glucose starvation and an AMPK activator 2‐DG. However, reconstitution of wild‐type PAQR3, rather than the T32A mutant, could significantly rescue the ameliorated Beclin1 phosphorylation (Appendix Fig S4B). These data provided one possible mechanism in which PAQR3 T32 phosphorylation might regulate ATG14L‐linked class III PI3K activity through affecting Beclin1 S93 phosphorylation.
In addition, glucose starvation‐induced autophagy was dramatically suppressed in PAQR3‐deleted cells, represented by reduced LC3‐II accumulation or increased p62 accumulations as compared to the wild‐type cells (Fig 6G and Appendix Fig S4C). However, changes of these autophagic markers were significantly rescued by reconstitution with wild‐type PAQR3, but not its T32A mutation (Fig 6G and Appendix Fig S4C), further supporting the functional importance of PAQR3 T32 phosphorylation in autophagy in response to glucose starvation.
To examine whether PAQR3 T32 phosphorylation was specifically involved in glucose starvation‐induced autophagy, we examined its effect on amino acid depletion‐ or rapamycin‐induced autophagy. As shown in Appendix Fig S4D, both amino acid depletion and rapamycin‐induced autophagy were significantly ameliorated in PAQR3 knockout cells. However, the reduced autophagic activity could be rescued by reconstitution of both wild‐type PAQR3 and its T32A mutant. Similarly, upon amino acid depletion or rapamycin treatment, both WT and T32A PAQR3 could significantly augment the impaired ATG14L‐linked VPS34 activity of PAQR3 knockout cells (Appendix Fig S4E). In summary, these data together with the results shown in Fig 6E and G indicated that PAQR3 T32 phosphorylation is specifically involved in autophagy initiation in response to glucose starvation, but not in autophagy induced by amino acid depletion or rapamycin.
As PAQR3 was redistributed into autophagosomes upon glucose starvation, during which T32 of PAQR3 was phosphorylated by active AMPK. Therefore, we suspected that PAQR3 T32 phosphorylation is implicated in its punctiform distribution. Consistent with our hypothesis, PAQR3 T32A was always localized in the Golgi apparatus regardless of glucose starvation (Appendix Fig S5A), different from the punctiform localization of wild‐type PAQR3 after glucose starvation (Fig EV1A). However, PAQR3 T32E, which mimicked its phosphorylation by AMPK, exhibited as punctiform structures under both normal medium and glucose starvation (Appendix Fig S5B and C). These data suggested that T32 phosphorylation is a switch to initiate the punctiform distribution of PAQR3 upon glucose starvation.
PAQR3‐deleted mice have deficiency in exercise‐induced autophagy and behavioral disorders
The studies above revealed the significance of PAQR3 in modulating autophagy at the cellular level. We next explored the potential function of PAQR3 in autophagy at the animal level. As exercise is a well‐recognized stimulus that induces autophagy in vivo (He et al, 2012), we applied uphill treadmill exercise to wild‐type and PAQR3‐deleted mice (Feng et al, 2007). It was previously reported that the plateau of autophagy can be reached after approximately 80 min of running (~10 m/min) (He et al, 2012), so these mice were set to run for a fixed time and fixed distance (80 min, ~900 m). Compared to the wild‐type mice, PAQR3‐deleted mice had an apparent reduction of LC3 immunohistochemical staining in both skeletal muscle and liver (Fig 7A and B). In addition, immunoblotting illustrated that both LC3‐II accumulation and p62 degradation stimulated by exercise were impeded in these PAQR3‐deficient tissues (Fig 7C and D). Collectively, these data suggested that PAQR3 is required for autophagy in vivo.
Figure 7. PAQR3‐deleted mice have deficiency in exercise‐induced autophagy and behavioral disorders.
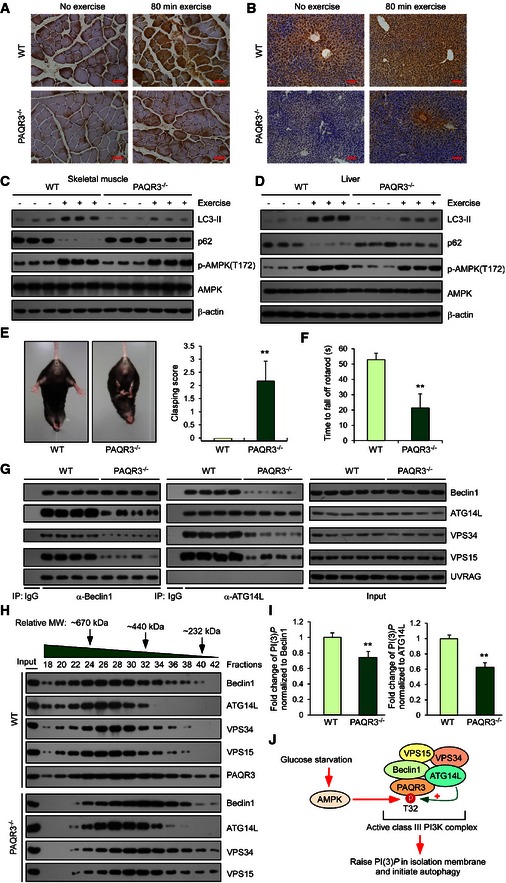
-
A, BRepresentative immunohistochemical images of LC3 staining in skeletal muscle (A) and liver (B) after 80‐min exercise in WT and PAQR3‐deleted mice. Scale bar: 20 μm (A) and 50 μm (B).
-
C, DImmunoblotting analysis of skeletal muscle (C) and liver (D) from WT or PAQR3 knockout mice at rest or after 80‐min exercise.
-
EAbnormal limb clasping of PAQR3 knockout mice when suspended by its tail. Quantification scoring of the limb clasping phenotype is shown on the right.
-
FMotor performance was measured using an accelerating rotarod apparatus, and the time of each mouse spent on the rotating rod until it fell off was recorded. Average time on the rod for each group is shown.
-
GCo‐immunoprecipitation of Beclin1 or ATG14L with their binding partners in liver tissues from mice of indicated genotype. Immunoprecipitation (IP) and immunoblotting (IB) were performed using the antibodies as indicated.
-
HThe supernatants of liver lysates form WT or PAQR3 knockout mice were subjected to gel filtration analysis, followed by IB.
-
IVPS34 complexes immunopurified with Beclin1 or ATG14L antibody in mouse liver were subjected to a quantitative PI(3)P ELISA.
-
JA schematic model of PAQR3 regulation on autophagy in response to glucose starvation.
As autophagy deficiency is associated with neurodegenerative disorders (Hara et al, 2006; Nixon, 2013), we investigated whether PAQR3 was involved in the progression of neurodegenerative phenotypes in vivo. Therefore, we evaluated the motor and behavioral characteristics of PAQR3 knockout mice by examining their stability on the accelerating rotarod, limb clasping, paw print intensity, and grip strength. There was no difference in the body weight between PAQR3 knockout mice and the wild‐type littermates at the age of 11 months (D. Xu and Y. Chen, unpublished observations). However, PAQR3‐deleted mice displayed more limb clasping and were less capable on the rotating rod than wild‐type mice (Fig 7E and F). Furthermore, abnormal paw print intensity and dispersion also indicated an ataxic walking pattern in PAQR3 knockout mice (Fig EV5A). Consistent with these results, the limb grip strength of PAQR3 knockout mice was significantly weaker than that of wild‐type mice (Fig EV5B). Thus, PAQR3 deletion is indeed associated with motor and behavioral abnormalities in the aged mice.
Figure EV5. PAQR3‐deleted mice exhibit ataxic walking pattern and ameliorated grip strength.
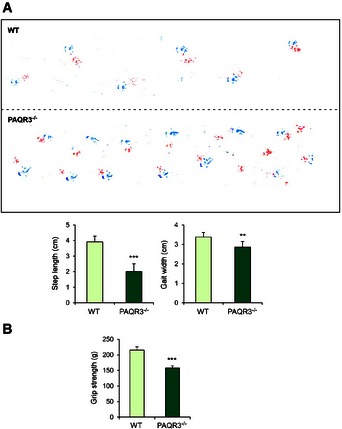
- Gait abnormalities in the PAQR3 knockout mice were evaluated by footprint analysis during walking (red, forelimbs; blue, hindlimbs). Quantification analyses of stride length and gait width of paws print were shown in the bottom panels.
- Grip strength analysis of all limbs in PAQR3 knockout mice and their WT littermates.
As we had demonstrated that PAQR3 could facilitate the formation of the ATG14L‐linked VPS34 complex at the cellular level (Fig 3), we next examined whether the constitution of this complex was affected by PAQR3 deletion in the mouse tissues. As shown in Fig 7G, the protein levels of each component of this complex were not altered by PAQR3 deletion. However, Beclin1 interacted with strikingly less ATG14L, VPS34, and VPS15 in the livers of PAQR3 knockout mice than those of the wild‐type mice when an anti‐Beclin1 antibody was used to enrich the VPS34 complex (Fig 7G). Likewise, the association of ATG14L with other components of the Beclin1–VPS34–VPS15 complex was reduced by PAQR3 deletion in the mouse liver (Fig 7G). In contrast, the association of UVRAG with Beclin1 was not altered by PAQR3 deletion (Fig 7G). These data demonstrated that PAQR3 could specifically modulate the formation of the ATG14‐linked VPS34 complex in vivo. Furthermore, if this were the case, VPS34 complex was supposed to form a larger complex in the wild‐type mice than in the PAQR3‐deleted mice. To address this hypothesis, we did a gel filtration analysis using liver tissues of the wild‐type and PAQR3 knockout mice, respectively (Fig 7H). Consistent with our hypothesis, the VPS34 complex components with the molecular weight higher than 670 kDa (fractions 18 and 20) were hardly detected in the PAQR3 knockout mice. Additionally, each “peak” of these components from PAQR3‐deleted mice was apparently shifted to fractions with lower molecular weight (Appendix Fig S6), supporting the central role of PAQR3 for ATG14L‐linked VPS34 complex constitution.
To further examine whether the activity of ATG14L‐associated class III PI3K activity was affected by PAQR3 in vivo, we analyzed PI(3)P production of the VPS34 complex associated with either Beclin1 or ATG14L. As expected, both Beclin1‐ and ATG14L‐associated lipid kinase activities were significantly reduced by PAQR3 deletion in the liver (Fig 7I). Collectively, these data provided additional evidence that the formation and activity of ATG14L‐associated class III PI3K complex are compromised by PAQR3 deletion in vivo.
Discussion
In this study, we provided strong evidence that PAQR3 is pivotal for autophagosome formation. Since neither AMPK nor mTOR activity can be modulated by PAQR3 under nutrient‐rich and starvation conditions, the candidate target for PAQR3 to regulate autophagy was narrowed down to class III PI3K. Exhilaratingly, we identified that PAQR3 can not only regulate VPS34 activity and PI(3)P production, but also modulates the relative abundance of the class III PI3K complex both in vitro and in vivo. As PAQR3 specifically promotes the formation of ATG14L–Beclin1–VPS34 complex, but not UVRAG‐associated complex, it mainly modulates autophagy initiation, rather than autophagosome maturation. Remarkably, although PAQR3 associates with the ATG14L‐linked VPS34 complex independently of autophagy‐stimulating signals, PAQR3 phosphorylation by AMPK can integrate glucose starvation to autophagy initiation. Upon glucose starvation, PAQR3 T32 can be directly phosphorylated by AMPK, which is crucial for glucose starvation‐induced autophagy and ATG14L‐associated class III PI3K activation. Additionally, PAQR3 T32 phosphorylation might be involved in the modulation of ATG14L‐linked class III PI3K activity by affecting Beclin1 S93 phosphorylation.
The mammalian VPS34 exists in different complexes and participates in a variety of cellular functions. For example, ATG14L‐associated VPS34 complex is mainly responsible for autophagy initiation, while UVRAG‐associated complex is necessary for autophagosome maturation and endosome fusion (Liang et al, 2008; Matsunaga et al, 2009; Zhong et al, 2009). Therefore, the interaction of VPS34 complex with these two partners is supposed to occur in a temporal order to guarantee sequential execution of the autophagy program. Based on our study, we propose that PAQR3 plays a crucial role in autophagy initiation by the preferential activation of the ATG14L‐linked VPS34 complex, thus coordinating timely activation of VPS34 complex during autophagy initiation. Coincidently, the functional role for PAQR3 in regulating VPS34 component assembly and kinase activity resembles largely another autophagy‐related protein NRBF2, which is a mammalian homolog of yeast Atg38 (Araki et al, 2013; Lu et al, 2014). Therefore, we also investigated the relationship between these two molecules. We found that both PAQR3 and NRBF2 are necessary to coordinately regulate ATG14L‐linked class III PI3K and autophagy activity (Appendix Figs S7 and S8). Besides, PAQR3 and NRBF2 participate in ATG14L‐associated VPS34 assembly in different manners (Appendix Fig S9).
On the whole, PAQR3 is involved in autophagy initiation via two layers of regulation (Fig 7J). In the first level, PAQR3 drives the effective assembly and stable formation of ATG14L–Beclin1–VPS34 complex, so that a higher PI(3)P generation capacity is attained ahead of starvation signal. This regulation process might be a cellular self‐protecting mechanism to quickly respond to possible incoming stress conditions. For the second level of regulation, AMPK phosphorylates PAQR3 at T32, which is a switch to initiate a powerful PI(3)P production quickly after starvation. Such regulatory properties of PAQR3 not only furnish sufficient pre‐autophagy VPS34 complexes in reserve, but also ensure a prompt and effective autophagy initiation upon starvation.
In addition, it is necessary to point out that PAQR3 is a multifunctional protein implicated in multiple biological processes. In the past, it had been assumed that PAQR3 mainly acts as a suppressor to ameliorate multiple signaling pathways, such as Ras‐Raf‐Mek‐Erk and PI3K‐AKT cascades, by sequestrating critical signaling molecules to the Golgi apparatus (Feng et al, 2007; Wang et al, 2013). However, the study here uncovers two novel concepts for PAQR3. Firstly, PAQR3 can be a positive regulator for a specific biological pathway. Secondly, PAQR3 can serve as a crucial mediator to transduce an upstream kinase signal to its downstream events through its phosphorylation.
To our knowledge, PAQR3 is the first reported molecule that acts a dual modulator for both class I PI3K and class III PI3K, although the regulation patterns are completely different. As reported previously, PAQR3 ameliorates class I PI3K activity by blocking the interaction between p110α and p85 (Wang et al, 2013). However, the formation and activity of ATG14L‐linked class III PI3K are promoted by PAQR3. Why do the cells need such an opposite modulation patterns on two types of PI3K? One possibility is that PAQR3 can protect class I PI3K signaling from overactivation upon stimulation of growth factors, so that abnormal proliferation of the cells can be curbed to certain degree. On the other hand, PAQR3 can effectively drive autophagosome formation to maintain cell survival and homeostasis under stress conditions. Therefore, PAQR3 is able to balance the cell growth and homeostasis via different regulation on two classes of PI3K.
Interestingly, the promoting function of PAQR3 on autophagy is clearly associated with its tumor suppressor activity. Autophagy is key to maintain cellular homeostasis and whereby acts as a barrier against cancer cell malignant transformation (Galluzzi et al, 2015). Although the role of autophagy in cancer development is under debate, it is generally accepted that autophagy can limit cancer initiation in a context‐dependent manner (Kimmelman, 2011; White, 2012). As a tumor suppressor, PAQR3 can not only ameliorate angiogenesis via negatively regulating the autocrine function of VEGF (Zhang et al, 2010), but also reduce EMT and tumor migration via functional cooperation with p53 (Jiang et al, 2011). In humans, the expression level of PAQR3 is decreased in many types of cancers (Ling et al, 2014; Yu et al, 2015). As PAQR3 positively modulates autophagy, it can be postulated that downregulation of PAQR3 in tumor cells is associated with reduction of autophagy under stress conditions, leading to a loss of the barrier function of autophagy in tumor initiation. Therefore, it will be important to elucidate how and to what degree the changes of autophagy contribute to the tumor suppressive activity of PAQR3 in the future.
Materials and Methods
Please refer to Appendix for more detailed Appendix Supplementary Materials and Methods.
In vitro PI(3)P ELISA
Total amount of PI(3)P was examined in a quantitative and competitive ELISA format assay according to the manufacture (Echelon Biosciences K‐3000). Briefly, different VPS34 complexes were enriched by various antibodies and then mixed with phosphatidylinositol (PI) substrates in an appropriate reaction buffer. After the PI3K reactions were complete and quenched, reaction products were diluted and added to the PI(3)P‐coated microplate for competitive binding to a PI(3)P detector protein. The amount of PI(3)P detector protein bound to the plate was determined through colorimetric detection.
Electron microscopy
After glucose starvation for 3 h, WT and PAQR3 knockout MEFs were harvested by trypsin digestion and fixed with 2.5% glutaraldehyde on ice for 2 h, followed by treatment with 1% osmium tetroxide in 0.1 M cacodylate buffer for another 1 h. Next, the fixed cells were dehydrated with sequential gradient washes from 50% to 100% ethanol and then immersed in epoxy resin after. The ultrathin sections were placed on carbon‐coated copper grids and counterstained with uranyl acetate and lead citrate. Images were taken with an FEI Tecnai G2 Spirit transmission electron microscope.
Protein purification and AMPK kinase assay
Briefly, the bacterial expression plasmids coding NH2‐terminal 71 amino acids of PAQR3 (or multiple mutations) were transformed into BL21 codon plus (Stratagene). Protein overexpression was induced under 0.5 mM IPTG at 18°C. Cells were resuspended in PBS containing 0.5% Triton X‐100, 5 mM β‐mercaptoethanol, 2 mM EDTA, and 1 mM PMSF, followed by ultrasonication. The proteins were obtained by Ni sepharose purification according to the manufacturer's protocol (GE Healthcare). After elution by imidazole, the purified proteins were dialyzed against 100 mM KCl, 0.2 mM EDTA, 20 mM HEPES‐KOH pH 8.0, and 20% glycerol. About 0.3 mg of bacterial purified proteins was used for AMPK assay. The AMPK kinase assay was conducted as previously described (Kim et al, 2001). Full‐length Atg14L was purified as previously reported (Fan et al, 2011; Kim et al, 2013).
Phos‐tag gel and immunoblotting
For analysis of phosphorylation of PAQR3, Phos‐tag (Wako) and MnCl2 were added to regular 10% SDS–PAGE gels at the levels recommended by the manufacturer. Western blotting was performed normally after soaking the gels in 1 mM EDTA for 10 min to remove Mn2+. All other steps in this analysis were identical to normal SDS–PAGE and immunoblotting protocols.
Mouse exercise studies
All animals were maintained and used in accordance with the guidelines of the Institutional Animal Care and Use Committee of the Institute for Nutritional Sciences. All of the experimental procedures were carried out in accordance with the CAS ethics commission with an approval number 2010‐AN‐8. PAQR3 knockout mice were described previously (Feng et al, 2007). The acute exercise studies were performed as previously reported (He et al, 2012). Briefly, 8‐week‐old WT and PAQR3 knockout male mice were randomly chosen and accommodated to a 10° uphill treadmill (Columbus Instruments) for 2 days in advance. On the third day, the mice were grouped randomly and subjected to a single bout of running at the initial speed of 10 m/min for 40 min. Then, the treadmill was accelerated at a rate of 1 m/min and the mice ran for another 40 min. Then, the mice were sacrificed immediately for a series of autophagy detection.
Statistical analysis
The unpaired and two‐tailed Student's t‐test was used for all the statistical analyses. Results were presented as average ± SD (standard deviation) from triplicates. *, **, and *** represent P < 0.05, P < 0.01, and P < 0.001, respectively. The symbol “ns” stands for not significant.
Author contributions
DQX, ZW, and YC conceived and designed the experiments. DQX, ZW, HDW, ZLZ, JG, YXZ, and ZGL performed the experiments and analyzed the data. ZFW, ZJK, KYM, LJL, and YP provided experimental materials and helped with computational analysis. DQX, ZW, and YC wrote the manuscript. CYW, DYZ, and ZXL commented on the manuscript. YC supervised the study.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
This work was supported by research grants from National Natural Science Foundation of China (81390350, 81130077, and 81321062 to YC), Ministry of Science and Technology of China (2012CB524900 to YC), and China Postdoctoral Science Foundation (2015M580360 to ZW).
The EMBO Journal (2016) 35: 496–514
References
- Araki Y, Ku WC, Akioka M, May AI, Hayashi Y, Arisaka F, Ishihama Y, Ohsumi Y (2013) Atg38 is required for autophagy‐specific phosphatidylinositol 3‐kinase complex integrity. J Cell Biol 203: 299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M (2010) Making membrane proteins for structures: a trillion tiny tweaks. Nat Methods 7: 429–434 [DOI] [PubMed] [Google Scholar]
- Bartz R, Sun LP, Bisel B, Wei JH, Seemann J (2008) Spatial separation of Golgi and ER during mitosis protects SREBP from unregulated activation. EMBO J 27: 948–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter EP, Beis K, Cameron AD, Iwata S (2008) Overcoming the challenges of membrane protein crystallography. Curr Opin Struct Biol 18: 581–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi AM, Ryter SW, Levine B (2013) Autophagy in human health and disease. N Engl J Med 368: 1845–1846 [DOI] [PubMed] [Google Scholar]
- Davies SP, Sim AT, Hardie DG (1990) Location and function of three sites phosphorylated on rat acetyl‐CoA carboxylase by the AMP‐activated protein kinase. Eur J Biochem 187: 183–190 [DOI] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ (2011) Phosphorylation of ULK1 (hATG1) by AMP‐activated protein kinase connects energy sensing to mitophagy. Science 331: 456–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W, Nassiri A, Zhong Q (2011) Autophagosome targeting and membrane curvature sensing by Barkor/Atg14(L). Proc Natl Acad Sci USA 108: 7769–7774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Xie X, Ding Q, Luo X, He J, Fan F, Liu W, Wang Z, Chen Y (2007) Spatial regulation of Raf kinase signaling by RKTG. Proc Natl Acad Sci USA 104: 14348–14353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, Gruss P, Piacentini M, Chowdhury K, Cecconi F (2007) Ambra1 regulates autophagy and development of the nervous system. Nature 447: 1121–1125 [DOI] [PubMed] [Google Scholar]
- Funderburk SF, Wang QJ, Yue Z (2010) The Beclin 1‐VPS34 complex–at the crossroads of autophagy and beyond. Trends Cell Biol 20: 355–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Pietrocola F, Levine B, Kroemer G (2014) Metabolic control of autophagy. Cell 159: 1263–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Pietrocola F, Bravo‐San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J, Gewirtz DA, Karantza V, Kimmelman A, Kumar S, Levine B, Maiuri MC, Martin SJ, Penninger J, Piacentini M, Rubinsztein DC, Simon HU, Simonsen A et al (2015) Autophagy in malignant transformation and cancer progression. EMBO J 34: 856–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki‐Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441: 885–889 [DOI] [PubMed] [Google Scholar]
- Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG (1996) Characterization of the AMP‐activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP‐activated protein kinase. J Biol Chem 271: 27879–27887 [DOI] [PubMed] [Google Scholar]
- He C, Klionsky DJ (2009) Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43: 67–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, Korsmeyer S, Packer M, May HI, Hill JA, Virgin HW, Gilpin C, Xiao G, Bassel‐Duby R, Scherer PE, Levine B (2012) Exercise‐induced BCL2‐regulated autophagy is required for muscle glucose homeostasis. Nature 481: 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, Guan JL, Oshiro N, Mizushima N (2009) Nutrient‐dependent mTORC1 association with the ULK1‐Atg13‐FIP200 complex required for autophagy. Mol Biol Cell 20: 1981–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Choi W, Hu W, Mi N, Guo Q, Ma M, Liu M, Tian Y, Lu P, Wang FL, Deng H, Liu L, Gao N, Yu L, Shi Y (2012) Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Res 22: 473–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL (2003) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115: 577–590 [DOI] [PubMed] [Google Scholar]
- Itakura E, Kishi C, Inoue K, Mizushima N (2008) Beclin 1 forms two distinct phosphatidylinositol 3‐kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 19: 5360–5372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Xie X, Li Z, Wang Z, Zhang Y, Ling ZQ, Pan Y, Wang Z, Chen Y (2011) Functional cooperation of RKTG with p53 in tumorigenesis and epithelial‐mesenchymal transition. Cancer Res 71: 2959–2968 [DOI] [PubMed] [Google Scholar]
- Jin T, Ding Q, Huang H, Xu D, Jiang Y, Zhou B, Li Z, Jiang X, He J, Liu W, Zhang Y, Pan Y, Wang Z, Thomas WG, Chen Y (2012) PAQR10 and PAQR11 mediate Ras signaling in the Golgi apparatus. Cell Res 22: 661–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH (2009) ULK‐Atg13‐FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 20: 1992–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Yoon MY, Choi SL, Kang I, Kim SS, Kim YS, Choi YK, Ha J (2001) Effects of stimulation of AMP‐activated protein kinase on insulin‐like growth factor 1‐ and epidermal growth factor‐dependent extracellular signal‐regulated kinase pathway. J Biol Chem 276: 19102–19110 [DOI] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL (2013) Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152: 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmelman AC (2011) The dynamic nature of autophagy in cancer. Genes Dev 25: 1999–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita E, Kinoshita‐Kikuta E, Takiyama K, Koike T (2006) Phosphate‐binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics 5: 749–757 [DOI] [PubMed] [Google Scholar]
- Kinoshita E, Kinoshita‐Kikuta E, Koike T (2009) Separation and detection of large phosphoproteins using Phos‐tag SDS‐PAGE. Nat Protoc 4: 1513–1521 [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo‐Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre‐Ghiso JA, Ahn HJ, Ait‐Mohamed O, Ait‐Si‐Ali S, Akematsu T, Akira S, Al‐Younes HM, Al‐Zeer MA, Albert ML, Albin RL, Alegre‐Abarrategui J et al (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8: 445–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama‐Honda I, Itakura E, Fujiwara TK, Mizushima N (2013) Temporal analysis of recruitment of mammalian ATG proteins to the autophagosome formation site. Autophagy 9: 1491–1499 [DOI] [PubMed] [Google Scholar]
- Lamb CA, Yoshimori T, Tooze SA (2013) The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol 14: 759–774 [DOI] [PubMed] [Google Scholar]
- Li X, He L, Che KH, Funderburk SF, Pan L, Pan N, Zhang M, Yue Z, Zhao Y (2012) Imperfect interface of Beclin1 coiled‐coil domain regulates homodimer and heterodimer formation with Atg14L and UVRAG. Nat Commun 3: 662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C, Jung JU (2008) Beclin1‐binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol 10: 776–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling ZQ, Guo W, Lu XX, Zhu X, Hong LL, Wang Z, Wang Z, Chen Y (2014) A Golgi‐specific protein PAQR3 is closely associated with the progression, metastasis and prognosis of human gastric cancers. Ann Onco 25: 1363–1372 [DOI] [PubMed] [Google Scholar]
- Lu J, He L, Behrends C, Araki M, Araki K, Jun Wang Q, Catanzaro JM, Friedman SL, Zong WX, Fiel MI, Li M, Yue Z (2014) NRBF2 regulates autophagy and prevents liver injury by modulating Atg14L‐linked phosphatidylinositol‐3 kinase III activity. Nature Commun 5: 3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Feng L, Jiang X, Xiao F, Wang Z, Feng GS, Chen Y (2008) Characterization of the topology and functional domains of RKTG. Biochem J 414: 399–406 [DOI] [PubMed] [Google Scholar]
- Maiuri MC, Zalckvar E, Kimchi A, Kroemer G (2007) Self‐eating and self‐killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 8: 741–752 [DOI] [PubMed] [Google Scholar]
- Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama‐Noda K, Ichimura T, Isobe T, Akira S, Noda T, Yoshimori T (2009) Two Beclin 1‐binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 11: 385–396 [DOI] [PubMed] [Google Scholar]
- Nixon RA (2013) The role of autophagy in neurodegenerative disease. Nat Med 19: 983–997 [DOI] [PubMed] [Google Scholar]
- Nojima H, Tokunaga C, Eguchi S, Oshiro N, Hidayat S, Yoshino K, Hara K, Tanaka N, Avruch J, Yonezawa K (2003) The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E‐BP1 through their TOR signaling (TOS) motif. J Biol Chem 278: 15461–15464 [DOI] [PubMed] [Google Scholar]
- Polson HE, de Lartigue J, Rigden DJ, Reedijk M, Urbe S, Clague MJ, Tooze SA (2010) Mammalian Atg18 (WIPI2) localizes to omegasome‐anchored phagophores and positively regulates LC3 lipidation. Autophagy 6: 506–522 [DOI] [PubMed] [Google Scholar]
- Ropolo A, Grasso D, Pardo R, Sacchetti ML, Archange C, Lo Re A, Seux M, Nowak J, Gonzalez CD, Iovanna JL, Vaccaro MI (2007) The pancreatitis‐induced vacuole membrane protein 1 triggers autophagy in mammalian cells. J Biol Chem 282: 37124–37133 [DOI] [PubMed] [Google Scholar]
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL (2013) ULK1 induces autophagy by phosphorylating Beclin‐1 and activating VPS34 lipid kinase. Nat Cell Biol 15: 741–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalm SS, Fingar DC, Sabatini DM, Blenis J (2003) TOS motif‐mediated raptor binding regulates 4E‐BP1 multisite phosphorylation and function. Curr Biol 13: 797–806 [DOI] [PubMed] [Google Scholar]
- Schu PV, Takegawa K, Fry MJ, Stack JH, Waterfield MD, Emr SD (1993) Phosphatidylinositol 3‐kinase encoded by yeast VPS34 gene essential for protein sorting. Science 260: 88–91 [DOI] [PubMed] [Google Scholar]
- Simonsen A, Tooze SA (2009) Coordination of membrane events during autophagy by multiple class III PI3‐kinase complexes. J Cell Biol 186: 773–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mule JJ, Pledger WJ, Wang HG (2007) Bif‐1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 9: 1142–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J, Levine B (2012) Akt‐mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 338: 956–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang L, Zhu L, Pan Y, Xiao F, Liu W, Wang Z, Guo F, Liu Y, Thomas WG, Chen Y (2013) PAQR3 modulates insulin signaling by shunting phosphoinositide 3‐kinase p110alpha to the Golgi apparatus. Diabetes 62: 444–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei JH, Seemann J (2009) The mitotic spindle mediates inheritance of the Golgi ribbon structure. J Cell Biol 184: 391–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, Kinch L, Koduru P, Christudass CS, Veltri RW, Grishin NV, Peyton M, Minna J, Bhagat G, Levine B (2013) EGFR‐mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 154: 1269–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E (2012) Deconvoluting the context‐dependent role for autophagy in cancer. Nat Rev Cancer 12: 401–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Klionsky DJ (2010) Eaten alive: a history of macroautophagy. Nat Cell Biol 12: 814–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Li Z, Chan MT, Wu WK (2015) PAQR3: a novel tumor suppressor gene. Am J Cancer Res 5: 2562–2568 [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Jiang X, Qin X, Ye D, Yi Z, Liu M, Bai O, Liu W, Xie X, Wang Z, Fang J, Chen Y (2010) RKTG inhibits angiogenesis by suppressing MAPK‐mediated autocrine VEGF signaling and is downregulated in clear‐cell renal cell carcinoma. Oncogene 29: 5404–5415 [DOI] [PubMed] [Google Scholar]
- Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z (2009) Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1‐phosphatidylinositol‐3‐kinase complex. Nat Cell Biol 11: 468–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File