

One DNA sequence can code for multiple different mRNAs, and therefore many different proteins. Conversely, a variant identified at the protein or transcript level may have non-unique genomic origins. For example, EGFR:p.L747S, which mediates acquired resistance of non-small cell lung cancer to tyrosine kinase inhibitors1, can be translated from multiple genomic variants such as chr7:g.55249076_55249077delinsAG and chr7:g.55242470T>C on different isoforms defined on the human reference assembly GRCh37. One-to-many, many-to-one and many-to-many relationships among sequence variants at the genomic level and those at transcript and protein levels introduce frequent inconsistencies in current practice when vital information about the annotation process (e.g., transcript or isoform IDs) is omitted from variant identifiers.
To facilitate standardization and reveal inconsistency in existing variant annotations, we have designed a novel variant annotator, TransVar, to perform three main functions supporting diverse reference genomes and transcript databases (Fig. 1a): (i) “forward annotation”, which annotates all potential effects of a genomic variant on mRNAs and proteins; (ii) “reverse annotation”, which traces an mRNA or protein variant to all potential genomic origins; and (iii) “equivalence annotation”, which, for a given protein variant, searches for alternative protein variants that have identical genomic origin but are represented based on different isoforms.
Figure 1.
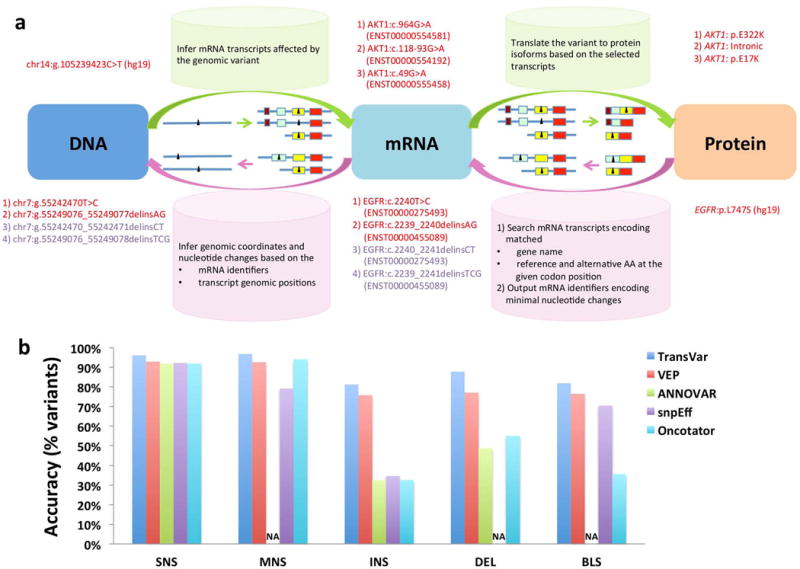
Schematic overview of TransVar and comparison of TransVar with other tools. (a) TransVar performs forward (green arrows) and reverse annotation (pink arrows) and considers all possible mRNA transcripts or protein isoforms available in user-specified reference genome and transcript databases (colored boxes representing exons in various transcripts or isoforms of a gene). Given a variant (black triangle) at any of the genomic, mRNA or protein levels, TransVar is able to infer the associated variants at the other two levels. In reverse annotation, TransVar searches all potential transcripts and reports one variant on each transcript. When there are multiple variants on the same transcript, TransVar reports the variant with minimal nucleotide changes (red text) instead of other alternatives (purple text). (b) Comparison of forward annotation consistency among TransVar, VEP, ANNOVAR, snpEff and Oncotator. Plotted are percentages of variants (Y axis) that had matched protein annotations in COSMIC v67 based on 964,132 unique SNSs, 3,715 MNSs, 11,761 INSs, 24,595 DELs and 166 BLSs (X axis). NA: Protein level annotations not available.
We annotated 964,132 unique single-nucleotide substitutions (SNS), 3,715 multi-nucleotide substitutions (MNS), 11,761 insertions (INS), 24,595 deletions (DEL) and 166 block substitutions (BLS) in the Catalogue of Somatic Mutations in Cancer (COSMIC v67) using TransVar, ANNOVAR2, VEP3, snpEff4, and Oncotator5, and asked whether the resulting protein identifiers (gene name, protein coordinates, and reference amino acid (AA)) match those in COSMIC. We observed comparable consistency in SNS and MNS but variable consistency in INS, DEL and BLS from different annotators (Fig. 1b, Supplementary Table 1 and Supplementary Notes). That finding can largely be attributed to a lack of standardization among variant annotations (codon or AA positions of variants) submitted to COSMIC and among conventions implemented in various annotators. Inconsistency in annotations blurred the lines of evidence for variant frequency estimation and led to inaccurate determination of variant function. TransVar revealed hidden inconsistency in these variant annotations by comprehensively outputting alternative annotations in all available transcripts in standard HGVS nomenclature, and thus resulted in greater consistency in this experiment.
TransVar’s novel reverse annotation can be used to ascertain if two protein variants have identical genomic origin, thus reducing inconsistency in annotation data. It can also reveal whether or not a protein variant has non-unique genomic origins and requires caution in genetic and clinical interpretation. We reverse-annotated the protein level variants in COSMIC and found that even under the constraints imposed by the reference base or AA identity, a sizeable fraction (e.g., 11.9% of single-AA substitutions) were associated with multiple genomic variants (Supplementary Table 2), if transcripts were not specified. Among the 537 variants that were cited as clinically actionable at PersonalizedCancerTherapy.org, 78 (14.5%) (e.g., CDKN2A:p.R87P and ERBB2:p.L755_T759del) could be mapped to multiple genomic locations (Supplementary Table 3). The reverse-annotation functionality also enabled systematic genomic characterization of variants directly identified from proteomic or RNA-seq data. For example, we were able to identify in just a few minutes of compute-time the putative genomic origins of 187,464 (97.69%) protein phosphorylation sites (e.g., p.Y308/p.S473 in AKT1 and p.Y1068/p.Y1172 in EGFR) in human proteins6.
Our investigation revealed frequent inconsistencies in current databases and tools and highlighted the importance of standardization. With both forward and reverse annotation enabled in TransVar, we can reveal hidden inconsistency and improve the precision of translational and clinical genomics. The source code and detailed instructions of TransVar is available at https://bitbucket.org/wanding/transvar and a web interface is at http://www.transvar.net.
Supplementary Material
Supplementary Table 1 Comparing the annotation accuracy of different mutation types using TransVar, ANNOVAR, snpEff, VEP and Oncotator
Supplementary Table 2 Reverse annotation accuracy of 1,080,304 COSMIC single-amino acid substitutions via different transcript databases
Supplementary Table 3 Clinically actionable cancer mutations with non-unique genomic origins
Acknowledgments
We thank P. Ng and K. Shaw for critical feedback, A. Johnson, A. Bailey, V. Holla, B. Litzenburger, J. Zhang and A. Chang for assistance. This work was supported in part by the National Institutes of Health [grant numbers CA172652, CA168394, CA083639, CA143883, UL1 TR000371, P50 CA083639, U54 CA112970 and P50 CA098258], the MD Anderson Cancer Center Sheikh Khalifa Ben Zayed Al Nahyan Institute of Personalized Cancer Therapy, the Bosarge Family Foundation, the Mary K. Chapman Foundation, the Michael & Susan Dell Foundation (honoring Lorraine Dell) and the National Cancer Institute Cancer Center Support Grant [P30 CA016672].
Footnotes
Competing interests
The authors declare that they have no competing interests.
Author’s contributions
KC conceived the project, TC, WZ and KC designed the studies, WZ and TC developed the tool and performed the analysis, ZC prepared the databases, WZ, TC, MR, JM and CW set up the web application interface, JZ and FMB detected clinical actionable mutations and informed clinical impact, TC, WZ, JW, GBM and KC interpreted the results and wrote the manuscript.
References
- 1.Yamaguchi F, et al. Acquired resistance L747S mutation in an epidermal growth factor receptor-tyrosine kinase inhibitor-naive patient: A report of three cases. Oncol Lett. 2014;7:357–360. doi: 10.3892/ol.2013.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McLaren W, et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cingolani P, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramos AH, et al. Oncotator: Cancer Variant Annotation Tool. Human mutation. 2015;36:E2423–E2429. doi: 10.1002/humu.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hornbeck PV, et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic acids research. 2012;40:D261–270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1 Comparing the annotation accuracy of different mutation types using TransVar, ANNOVAR, snpEff, VEP and Oncotator
Supplementary Table 2 Reverse annotation accuracy of 1,080,304 COSMIC single-amino acid substitutions via different transcript databases
Supplementary Table 3 Clinically actionable cancer mutations with non-unique genomic origins