

Abstract
Non-steroidal anti-inflammatory drug (NSAID)-activated gene-1 (NAG-1) and COX-2 are involved in cellular processes such as inflammation, apoptosis, and tumorigenesis. To address the relationship between COX-2 and NAG-1 expression, we investigated the expression of NAG-1 and COX-2 in normal and tumor tissue from human patients, ApcMin/+ mice, and COX-2(−/−) mice. While COX-2 expression is highly induced in tumor tissue, NAG-1 expression is reduced. Furthermore, PGE2 reduces NAG-1 while celebrex induces NAG-1 expression. The results suggest that a possible inverse relationship exists between the expression of NAG-1 and COX-2 in tumor formation of colon tissue.
Keywords: GDF-15, NAG-1, COX-2, Colorectal cancer
1. Introduction
Colorectal cancer (CRC) represents the third leading cause of cancer-related deaths in both men and women in the United States [1]. Epidemiological studies have shown that patients who use non-steroidal anti-inflammatory drugs (NSAIDs) for 10–15 years have a reduced risk of developing colorectal cancer [2]. NSAIDs inhibit two enzymes involved in prostaglandin biosynthesis: cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2), reduce the number and size of adenomas in patients with familial adenomatous polyposis (FAP), and prevent the development of colon cancer in ApcMin/+ mice [3]. The chemopreventive and anti-tumorigenic activities of NSAIDs against CRC and other cancers are well established. Emerging clinical and experimental evidence now supports an anti-tumorigenic efficacy of NSAIDs in CRC [4,5] and implicates the contribution of COX-2 and one of its metabolites, prostaglandin E2 (PGE2), in CRC development [6,7]. Celecoxib, a COX-2 specific inhibitor, reduces polyp burden in FAP patients [8]. However, other evidence suggests that COX-2 may not be the only target for chemoprevention by NSAIDs as NSAIDs also inhibit tumor growth through COX-independent pathways [2].
Previously, we showed that NSAIDs modulate the expression of several genes, including non-steroidal anti-inflammatory drug-activated gene (NAG-1), also known as MIC-1, GDF-15, PTGFB, PLAB, and PDF [9]. NAG-1 is a member of the transforming growth factor (TGF-β) super-family and is involved in cellular processes such as inflammation, apoptosis, and anti-tumorigenesis [9]. NAG-1 expression is up-regulated by several NSAIDs independently of prostaglandin formation in human colorectal cancer cells [9]. In addition, anti-tumorigenic compounds, such as resveratrol, genistein, and a peroxisome proliferator-activated receptor (PPAR-γ) ligand, troglitazone, induce NAG-1 expression [10]. NAG-1 is regulated by p53, Egr-1, and AKT, and may have tumor suppressor activity [11]. Indeed, NAG-1 transgenic mice that ubiquitously express the human protein are less susceptible to both chemical and genetically induced intestinal cancer [12].
NAG-1 is expressed in mature intestinal epithelial cells; however its expression is significantly reduced in human colorectal carcinoma samples [13]. A similar finding of reduced NAG-1 expression was observed in neoplastic intestinal polyps of ApcMin/+ mice [13]. However, COX-2 is up-regulated in human colorectal tumors and is also expressed at higher levels in the neoplastic intestinal polyps of ApcMin/+ mice [14–16]. These results suggest a possible inverse relationship between the expression of NAG-1 and COX-2 in intestinal tumor tissue, implying that the expression of one protein could cause the suppression of the other. In this study, we demonstrate that NAG-1 expression is greater in COX-2(−/−) mice compared to wild type littermates, but its expression is decreased in COX-2(−/−) mice treated with PGE2. Furthermore, treatment of COX-2 mice with doses of celecoxib that inhibit tumor growth induce NAG-1 expression.
2. Materials and methods
2.1. Animals
The development of COX-1 and COX-2 deficient mice has been previously reported [17,18]. These mice carry mutations on the genes encoding COX-1 and COX-2 and have been maintained on the 129/Ola/C57BL/6 background for more than 40 generations. COX-2(−/−) mice were given PBS (vehicle) or 0,150, or 300 μg/mouse of PGE2 twice daily by oral gavage for 7 days. Celecoxib was administered in the food for 4 weeks at 0, 1000, and 1500 ppm. The mice were then euthanized and the intestines removed for analysis.
ApcMin/+ mice were purchased from Jackson laboratories (West grove, PA). The mice were euthanized by CO2 inhalation after 4 h of fasting and the entire intestinal tract was removed, flushed with cold saline, and opened longitudinally. Both polyps and normal adjacent normal tissue were stored in RNAlater at 4 °C until RT-PCR analysis. Human NAG-1 transgenic (NAG-1Tg/Lox) mice were crossed with ApcMin/+ mice to yield mice (ApcMin/+/NAG-1Tg/Lox) that have mutant Apc and express human NAG-1 in the intestinal tract [12].
2.2. Human colorectal tissue samples
Human colorectal tumor specimens were obtained from surgical resections by the Southern Division of the Cooperative Human Tissue Network. For each tumor sample, matched adjacent normal mucosa was collected for comparison. All samples were snap-frozen and stored in liquid nitrogen.
2.3. RNA isolation and reverse transcription
RNA was isolated from tissue using the RNeasy Mini Kit from Qiagen (Valencia, CA) according to the manufacturer’s protocol. RNA was stored in a −80° freezer until use. RNA was treated with 1 unit of amplification grade Deoxyribonuclease I (Life Technologies) per μg of RNA at room temperature for 15 min to remove genomic DNA The addition of 2.5 mM EDTA and incubation at 65 °C inactivated the Deoxyribonuclease I reaction. Reverse transcription (RT) was performed using Qiagen’s Omniscript reverse transcription kit according to the manufacturer’s protocol. A negative control containing all of the RT reagents in the absence of RT enzyme was routinely performed. After RT, cDNA was treated with 1 unit RNase H (Life Technologies) per μg RNA at 37 °C for 20 min.
2.4. Real-time PCR
Real-time RT-PCR was performed in triplicate two or more times for each gene tested. Primer design (Table 1), deoxyribonuclease treatment, reverse transcription, and real-time RT-PCR assay using ABI Prism 7700 (Applied Bio-systems, Foster City, CA) were performed as previously described.
Table 1.
The list of primers.
| Primer | Forward Sequence | Reverse Sequence |
|---|---|---|
| Mouse NAG-1 | 5′ TTCTGTGGGGACGGTCAG 3′ | 5′ CGGGTGACCAGGCTAATTC 3′ |
| Mouse Actin | 5′ TGACAGGATGCAGAAGGAGA 3′ | 5′ CGCTCAGGAGGAGCAATG 3′ |
| Human NAG-1 | 5′ TGCCCGCCAGCTACAATC 3′ | 5′ TCTTTGGCTAACAAGTCATCATAGGT 3′ |
| Human Actin | 5′ CCTGGCACCCAGCACAAT 3′ | 5′ GCGGATCCACACGGAGTACT 3′ |
| Mouse COX-1 | 5′ CCTCTTTCCAGGAGCTCACA 3′ | 5′ TCGATGTCACCCGTACAGCTC 3′ |
| Mouse COX-2 | 5′ GCTCTTCCGAGCTGTGCT 3′ | 5′ GATTGGAACAGCAAGGATTTG 3′ |
2.5. Western blotting
Proteins from mouse tissue and tissue culture cells were isolated as previously described [13], at 50 μg and 20 μg protein, respectively, separated by SDS–PAGE, and transferred onto nitrocellulose membranes. Blots were blocked overnight with 10% skim milk in TBS containing 0.1% Tween-20 (TBS-T), probed for 1 h at room temperature in 5% milk with TBS-T containing anti-NAG-1 (generated in this laboratory), anti-COX-2 (Cayman), or anti-actin (Santa Cruz Biotechnologies, Santa Cruz, CA), and washed in TBS-T. Anti-mouse NAG-1 was generated in this laboratory in rabbit from a specific peptide of the mouse NAG-1 C-terminus (DSGVSLQTYDDLVARG). The antibody recognized both the precursor and secreted forms of NAG-1. The membranes were treated with the appropriate horseradish peroxidase-conjugated secondary antibody (Santa Crux Biotechnologies) in 5% milk with TBS-T for 1 h at room temperature, and then washed several times with TBS-T and the signal was detected by enhanced chemiluminescence (Amersham Biosciences) followed by autoradiography. When necessary, membranes were stripped by sealing in a plastic bag containing 62.5 mM Tris–HCl (pH 6.8), 2% (w/v) SDS, and 100 mM β-mercaptoethanol for 30 min with constant agitation at 50 °C and washed with TBS-T prior to reprobing.
2.6. Statistical analysis
Statistical significance was determined according to a one-sided t test with a 0.05 level of significance on Ct values following adjustment for β-actin.
3. Results
3.1. NAG-1 expression is inversely associated with COX-2 expression in human colorectal cancer tissue and ApcMin/+ polyps
We previously reported that the expression of NAG-1 on the surface epithelium of intestinal villi overlaps with regions that are undergoing apoptosis and is down-regulated in colon tumors as compared to normal adjacent tissue [13]. A comparison of human colorectal cancer and adjacent normal tissue revealed a reduction in NAG-1 expression in malignant tissue compared to normal tissue (Fig. 1). Real-time PCR analysis indicated that NAG-1 expression was reduced by approximately 65% in the colon cancer tissue as compared to the corresponding adjacent normal tissue (Fig. 1A). Because COX-2 expression is frequently up-regulated in colon cancer tissue [19,20], we measured COX-2 expression in colon cancer tissue and compared its expression to the adjacent normal tissue. RT-PCR analysis indicated that COX-2 expression was induced about eightfold in cancer tissue compared to normal tissue (Fig. 1B).
Fig. 1.
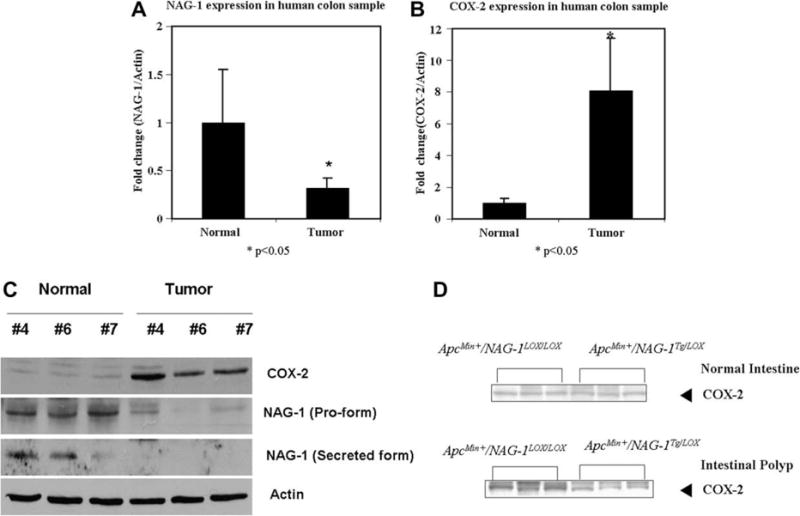
The protein and mRNA level of NAG-1 was up-regulated in normal tissue while COX-2 expression was up-regulated in neoplastic tissue. (A) and (B) NAG-1 and COX-2 mRNA expressions in normal and neoplastic tissue from human colon samples. Values are means (±SEM) and are expressed as fold induction relative to control. Five pairs of samples were analyzed in five replicates and differed at P < 0.05 compared with control group. (C) Western blot of COX-2 and NAG-1 proteins in normal and tumor tissue of ApcMin/+ mouse. (D) Expression of COX-2 in normal and tumor tissue of ApcMin/+/NAG-1Tg/Lox.
The findings of a possible reciprocal relationship between COX-2 and NAG-1 in human samples led us to examine if this relationship also existed in the ApcMin/+ mouse, a mouse model for intestinal cancer. Human colorectal polyps contain inactivating mutations in the Apc gene and ApcMin/+ mice, which have a mutation in the Apc gene, show increased formation of polyps in the small intestine. As shown in Fig. 1C, the expression of the NAG-1 pro-form and secreted form are reduced in ApcMin/+ polyps compared to normal tissue, whereas Apc-Min/+ polyps had increased expression of COX-2 compared to normal tissue. Thus, there appears to be an inverse relationship between COX-2 and NAG-1 expression in both normal murine intestinal tissue and in tumors.
3.2. COX-2 expression was reduced in the intestinal polyps from ApcMin/+/NAG-1Tg/Lox mice
To further investigate the expression of COX-2 and NAG-1 in normal and adenoma tissue, ApcMin/+ mice were mated with transgenic mice (NAG-1Tg/Lox) expressing human NAG-1. Previously we reported that ApcMin/+/NAG-1Tg/Lox mice had about a 40% reduction in both small intestine polyp numbers and tumor load [12] compared to littermate ApcMin/+ mice. These data indicate a role for NAG-1 in suppression of polyp formation in ApcMin/+ mice, and the effect of hNAG-1 expression on the expression of COX-2 was examined in the polyps of ApcMin/+/NAG-1Tg/Lox mice. Fig. 1D shows a low level of COX-2 expression in the normal intestinal tissue from both Apc-Min/+/NAG-1Lox/Lox and ApcMin/+/NAG-1Tg/Lox mice. In contrast, the intestinal polyps from ApcMin/+/NAG-1Lox/Lox mice had higher expression of COX-2 compared to ApcMin/+/NAG-1Tg/Lox mouse polyps. These findings suggest that hNAG-1 may suppress polyp formation in ApcMin/+ mice, in part, by suppressing COX-2 expression, or that the reduced expression of COX-2 results in tumor growth inhibition, both suggesting an inverse relationship.
3.3. NAG-1 expression was up-regulated in COX-2(−/−) mice, but not in COX-1(−/−) mice
COX-1(−/−) and COX-2(−/−) mice, when crossed with ApcMin/+ mice, have reduced intestinal polyps [14] and therefore, mNAG-1 expression in COX-1(−/−) and COX-2(−/−) was measured. As shown in Fig. 2, mNAG-1 mRNA expression was higher in COX-2(−/−) mice as compared to COX-2(+/+) littermate mice. However, there was no difference in the expression of mNAG-1 in COX-1(+/+) and COX-1(−/−) mice. These results suggest that COX-2 may act to suppress NAG-1 expression.
Fig. 2.
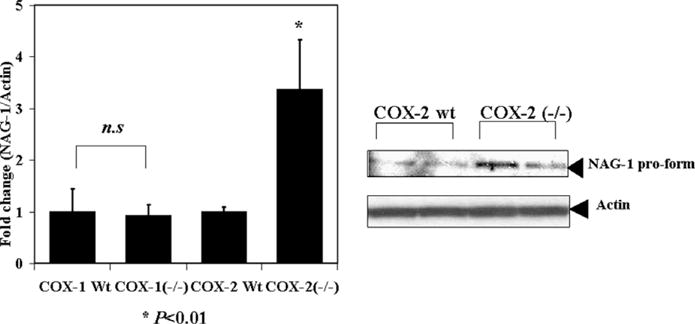
The protein and mRNA levels of NAG-1 were up-regulated in COX-2(−/−) mice, but no change was observed in COX-1 (−/−) mice. NAG-1 expression in COX-1/COX-2(+/+) and (−/−) mice as measured by Western blot and real-time RT-PCR. Values are means (±SEM) and are expressed as fold induction relative to control. Each group consists of at least five mice. (n.s. not significant, *P< 0.01 compared with control group).
3.4. PGE2 treatment suppressed NAG-1 expression in COX-2(−/−) mice
NSAIDs inhibit cyclooxygenase activity and lower the level of prostaglandins such as PGE2. Both COX-2 and PGE2 are increased in intestinal tumor tissue, compared to normal tissue [21]. Furthermore, treatment of ApcMin/+ mice with PGE2 leads to enhanced development of intestinal adenoma growth [22]. To determine the effect of PGE2 treatment on NAG-1 expression, COX-2(−/−) mice were treated with 0, 150, or 300 μg/mouse of PGE2 twice daily for 7 days. These doses are reported to enhance polyp formation in mice [22]. PGE2 treatment of the COX-2(−/−) mice resulted in a dose dependent suppression of NAG-1 mRNA and protein expression (Fig. 3). These results indicate that PGE2 causes NAG-1 suppression and also further supports an inverse association between NAG-1 and COX-2.
Fig. 3.
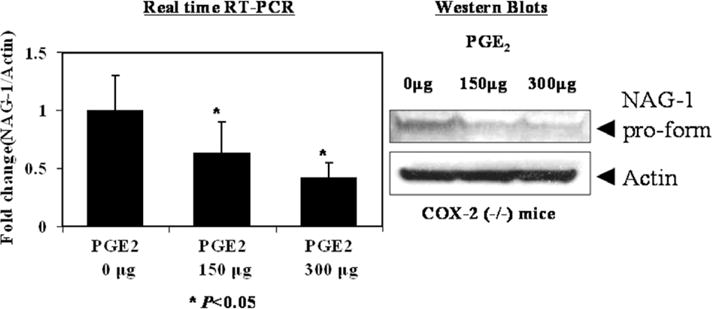
PGE2 suppressed NAG-1 expression in COX-2(−/−) mice. NAG-1 mRNA expression was quantified by real time RT-PCR in COX-2(−/−) mice treated with vehicle or 0,150, and 300 μg of PGE2 in 100 μl sterile PBS by twice daily gavage for 7 days. Values are means (±SEM) for four mice in each group and are expressed as fold induction relative to control. * P< 0.05 compared with control group.
3.5. NAG-1 expression in celecoxib-treated wild type and COX-2(−/−) mice
Celecoxib, a selective COX-2 inhibitor, has been used extensively to inhibit tumor growth [23–25] and induces NAG-1 expression in cancer cells. NAG-1 expression induced by celebrex appears to be mediated by blocking the activation of the AKT pathway [26], a pathway activated by PGE2. In addition, ATF-3 is also induced by NSAIDs and its expression is repressed in colorectal cancer tissue compared to normal tissue [27]. NAG-1 and ATF-3 are known downstream targets of GSK-3β [26,28] and an increase in ATF-3 expression causes apoptosis and altered growth of cells in a xenograft mouse model [27]. However, there are no reports in the literature investigating the effect of celecoxib on NAG-1 or ATF-3 expression in vivo. Therefore, we administered celecoxib to mice to determine its effects on NAG-1 and ATF-3 expression to access if prostaglandin formation altered the expression and if celebrex increased expression in vivo. COX-2(+/+) and COX-2(−/−) littermate mice were treated with 0, 1000, and 1500 ppm celecoxib for 4 weeks, at doses reported to suppress tumor growth [29]. Celecoxib treatment increased NAG-1 expression in a dose dependent manner in COX-2(−/−) mice and ATF-3 in wild type mice (Fig. 4). Thus, celebrex not only inhibits COX-2 and lowers PGE2 levels, but also induces the expression of NAG-1 and ATF-3, suggesting both targets may play a role in the suppression tumor growth by this NSAID.
Fig. 4.
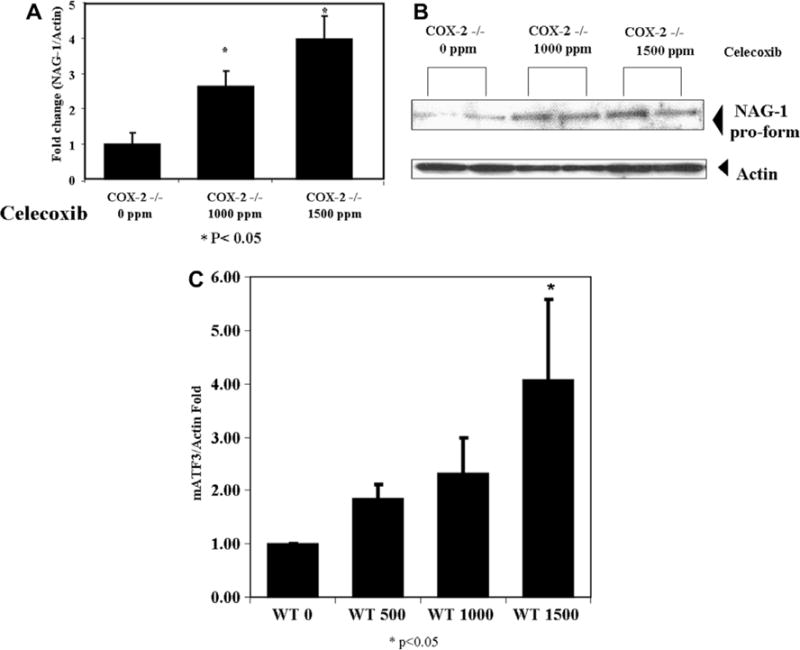
The expression of NAG-1 in Celecoxib treated COX-2(−/−) mice. (A) and (B) NAG-1 mRNA and protein expression in COX-2 knockout mice and (C) ATF-3 mRNA expression in COX-2 wild type mice treated with 0,1000, and 1500 ppm celecoxib for 4 weeks. Western blot and real time RT-PCR analyzes are relative to 0 ppm treated mice and adjusted for β-actin. Values are means (±SEM) for three mice in each group and are expressed as fold induction relative to control (* P < 0.05 compared with control group).
4. Discussion
NAG-1 was identified as a divergent member of the TGF-β superfamily from an NSAID-induced library in COX-2 negative colorectal cancer cells by a PCR-based subtractive hybridization [30] Many chemopreventive drugs, including COX inhibitors, up-regulate NAG-1 expression in human colorectal cells in culture and the increased NAG-1 expression appears to mediate NSAID-induced increases in apoptosis. The up-regulation of mNAG-1 was observed in the intestinal tract and liver of mice fed tumor suppressive doses of sulindac [13]. Recently, we generated NAG-1 transgenic mice (Cre/NAG-1Tg/Lox) which express human NAG-1, and the anti-tumorigenic activity in mice expressing hNAG-1 was evaluated with the known colon carcinogen azoxymethane (AOM). NAG-1Tg/Lox mice had smaller numbers of aberrant crypt foci (ACF) in their colons compared to littermate controls. Furthermore, ApcMin/+/NAG-1Tg/Lox mice showed ~40% reduction in small intestine polyp numbers and in tumor load [12]. These findings suggest that NAG-1 may play a role in the development of intestinal polyps in mice and CRC in humans.
COX-2 expression in mouse models of intestinal cancer and in human CRC is well documented. Higher COX-2 expression is noted in human CRC and in several mouse models for intestinal cancer. Deletion of the COX-2 gene results in decreased tumor formation in the small intestine and colon of ApcMin/+ and ApcΔ716 mice [14,31]. Hansen-Petrik et al. [21] reported that PGE2 reversed NSAID-in-duced adenoma repression in ApcMin/+ mice. Wang et al. [22] also reported that PGE2 accelerated intestinal adenoma growth in ApcMin/+ mice. PGE2 exerts its effects by binding to its cognate receptors, EP1–4, which belong to the seven transmembrane G-protein coupled receptor family. PGE2, via the EP2/4 receptors, activates the PI3 Kinase/AKT pathway and interacts with the EGFR pathway [32,33]. COX-2 expression causes transactivation of EGFR by enhancing phosphorylation of EGFR, leading to tumor growth. The evidence clearly indicates the importance of COX-2 and the EP receptors in the development of intestinal cancer.
Because COXs and NAG-1/ATF3 are targets for COX inhibitors and COX-2 is reported to be highly expressed in intestinal tumors, we decided to further investigate a possible relationship between the expressions of these two proteins. We first confirmed in human colorectal tissue samples the high expression of COX-2 and a corresponding lower expression of NAG-1. A similar pattern of expression was observed in normal tissue and polyps obtained from ApcMin/+ mice, suggesting an inverse relationship between these two proteins in intestinal cancer. The expression of hNAG-1 in the ApcMin/+ not only inhibited polyp formation [12], but also reduced COX-2 expression was observed in the polyps. NAG-1 could directly inhibit the expression of COX-2, but more likely the decrease in COX-2 is the result of suppression of tumor development by NAG-1. However, we do not know if the expression of COX-2 in normal cells would suppress NAG-1 expression. Higher mNAG-1 expression was observed in the intestinal tract of COX-2(−/−), but not in the COX-1(−/−) as compared to littermate controls. Furthermore, treatment of COX-2(−/−) mice with PGE2 at doses that increase polyp formation and activate the PI3 K/AKT/GSK-3β pathway caused a dose dependent decrease in the expression of mNAG-1. Thus, based on these data, we proposed that the increase in COX-2 expression in the tumor suppresses the expression of NAG-1.
NAG-1 is an important downstream target of the tumor suppressor gene p53 and Egr-1 [11] and is up-regulated following inhibition of PI3 K/AKT/GSK-3β pathway [26]. Activation of the PI3 K/AKT/GSK-3β pathway by PGE2 formed by COX-2 would result in lower expression of NAG-1 in tumors expressing COX-2. We have previously shown that some NSAIDs increase NAG-1 expression in vitro [9]. Sulindac, administered in drinking water at doses that reduce tumor formation in mice, induces mNAG-1 expression in the liver and colon [12]. Celebrex increases the expression of NAG-1 in a number of cells in culture by inhibiting the phosphorylation, and hence activation, of AKT [24]. Treatment of COX-2(+/+) and COX-2(−/−) mice with celebrex caused a dose dependent increase in the expression of NAG-1 and another down steam target of AKT, ATF-3, in intestinal tissue. Modulation of AKT activity either by inhibition with celebrex or other chemicals, or by activation via the EP2/4 receptors via increases in COX-2, can increase or decrease, respectively, the expression of NAG-1 in intestinal tissue, thereby altering the development of intestinal tumors.
We suspect that many COX-2 inhibitors have two cellular targets that contribute to the inhibition of tumor growth. One is COX-2, the inhibition of which reduces the levels of prostaglandins. A second is NAG-1, whose induced expression is mediated by inhibition of AKT activation. Thus we suspect both of these targets may contribute to the inhibition of intestinal cancer by the long term use of COX inhibitor, celebrex.
COX-2 selective inhibitors have been touted for their possible use as chemoprevention agents. However, recent concerns over the safety of COX-2 inhibitors have prompted researchers to develop more effect agents with minimal toxicity. Understanding the molecular mechanism for the inhibition of tumor development will help to identify specific safer target. NAG-1 may play an important role as a potential candidate.
Acknowledgments
We wish to thank Chris Lee and Colleen Anna for their support. This work was supported, in part, by the National Institute of Environmental Health Sciences, National Institutes of Health, Intramural program.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer Statistics. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Wang D, DuBois RN. Prostaglandins and cancer. Gut. 2006;55:115–122. doi: 10.1136/gut.2004.047100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Torrance CJ, Jackson PE, Montgomery E, Kinzler KW, Vogelstein B, Wissner A, Nunes M, Frost P, Discafani CM. Combinatorial chemoprevention of intestinal neoplasia. Nat Med. 2000;6:1024–1028. doi: 10.1038/79534. [DOI] [PubMed] [Google Scholar]
- 4.Thun MJ, Namboodiri MM, Calle EE, Flanders WD, Heath CW., Jr Aspirin use and risk of fatal cancer. Cancer Res. 1993;53:1322–1327. [PubMed] [Google Scholar]
- 5.Saha D, Roman C, Beauchamp RD. New strategies for colorectal cancer prevention and treatment. World J Surg. 2002;26:762–766. doi: 10.1007/s00268-002-4049-1. [DOI] [PubMed] [Google Scholar]
- 6.Wang D, Buchanan FG, Wang H, Dey SK, DuBois RN. Prostaglandin E2 enhances intestinal adenoma growth via activation of the Ras-mitogen-activated protein kinase cascade. Cancer Res. 2005;65:1822–1829. doi: 10.1158/0008-5472.CAN-04-3671. [DOI] [PubMed] [Google Scholar]
- 7.Castellone MD, Teramoto H, Gutkind JS. Cyclooxygenase-2 and colorectal cancer chemoprevention: the beta-catenin connection. Cancer Res. 2006;66:11085–11088. doi: 10.1158/0008-5472.CAN-06-2233. [DOI] [PubMed] [Google Scholar]
- 8.Phillips RK, Wallace MH, Lynch PM, Hawk E, Gordon GB, Saunders BP, Wakabayashi N, Shen Y, Zimmerman S, Godio L, Rodrigues-Bigas M, Su LK, Sherman J, Kellof G, Levin B, Steinbach G, FAP Study Group A randomized, double blind, placebo controlled study of celecoxib, a selective cyclooxygenase-2 inhibitor, on duodenal polyposis in familial adenomatous polyposis. Gut. 2002;50:857–860. doi: 10.1136/gut.50.6.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baek SJ, Kim KS, Nixon JB, Wilson LC, Eling TE. Cyclooxygenase inhibitors regulate the expression of a TGF-beta superfamily member that has proapoptotic and anti-tumorigenic activities. Mol Pharmacol. 2001;59:901–908. [PubMed] [Google Scholar]
- 10.Baek SJ, Kim J, Nixon JB, DiAugustine RP, Eling TE. Expression of NAG-1, a transforming growth factor-β superfamily member, by troglitazone requires the early growth response gene EGR-1. J Biol Chem. 2004;279:6883–6892. doi: 10.1074/jbc.M305295200. [DOI] [PubMed] [Google Scholar]
- 11.Baek SJ, Eling TE. Changes in gene expression contribute to cancer prevention by COX inhibitors. Prog Lipid Res. 2006;45:1–16. doi: 10.1016/j.plipres.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 12.Baek SJ, Okazaki R, Lee SH, Martinez J, Kim JS, Yamaguchi K, Mishina Y, Martin DW, Shoieb A, McEntee MF, Eling TE. Nonsteroidal anti-inflammatory drug-activated gene-1 over expression in transgenic mice suppresses intestinal neoplasia. Gastroenterology. 2006;131:1553–1560. doi: 10.1053/j.gastro.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 13.Kim KS, Baek SJ, Flake GP, Loftin CD, Calvo BF, Eling TE. Expression and regulation of nonsteroidal anti-inflammatory drug-activated gene (NAG-1) in human and mouse tissue. Gastroenterology. 2002;122:1388–1398. doi: 10.1053/gast.2002.32972. [DOI] [PubMed] [Google Scholar]
- 14.Chulada PC, Thompson MB, Mahler JF, Doyle CM, Gaul BW, Lee C, Tiano HF, Morham SG, Smithies O, Langenbach R. Genetic disruption of Ptgs-1, as well as Ptgs-2, reduces intestinal tumorigenesis in min mice. Cancer Res. 2000;60:4705–4708. [PubMed] [Google Scholar]
- 15.Hull MA, Booth JK, Scott N, Bonifer C, Markham AF, Coletta PL. Cyclooxygenase-2 is up-regulated and localized to macrophages in the intestine of min mice. Brit J Cancer. 1999;79:1399–1405. doi: 10.1038/sj.bjc.6690224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams CS, Luongo C, Radhika A, Zhang T, Lamps LW, Nanney LB, Beauchamp RD, DuBois RN. Elevated cyclooxygenase-2 levels in min mouse adenomas. Gastroenterology. 1996;111:1134–1140. doi: 10.1016/s0016-5085(96)70083-5. [DOI] [PubMed] [Google Scholar]
- 17.Langenbach R, Morham SG, Tiano HF, Loftin CD, Ghanayem BI, Chulada PC, Mahler JF, Lee CA, Goulding EH, Kluckman KD, Kim HS, Smithies O. Prostaglandin synthase-1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell. 1995;83:483–492. doi: 10.1016/0092-8674(95)90126-4. [DOI] [PubMed] [Google Scholar]
- 18.Morham SG, Langenbach R, Mahler J, Smithies O. Characterization of prostaglandin H synthase 2 deficient mice and implications for mechanisms of NSAID action. Adv Exp Med Biol. 1997;407:131–138. doi: 10.1007/978-1-4899-1813-0_20. [DOI] [PubMed] [Google Scholar]
- 19.Kargman SL, O’Neill GP, Vickers PJ, Evans JF, Mancini JA, Jothy S. Expression of prostaglandin G/H synthase-1 and -2 protein in human colon cancer. Cancer Res. 1995;55:2556–2559. [PubMed] [Google Scholar]
- 20.Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Up-regulation of cyclooxygenase-2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107:1183–1188. doi: 10.1016/0016-5085(94)90246-1. [DOI] [PubMed] [Google Scholar]
- 21.Hansen-Petrik MB, McEntee MF, Jull B, Shi H, Zemel B, Whelan J. Prostaglandin E2 protects intestinal tumors from nonsteroidal anti-inflammatory drug-induced regression in ApcMin/+ mice. Cancer Res. 2002;62:403–408. [PubMed] [Google Scholar]
- 22.Wang D, Wang H, Shi Q, Katkuri S, Walhi W, Desvergne B, Das SK, Dey SK, DuBois RN. Prostaglandin E2 promotes colorectal adenoma growth via transactivation of the nuclear peroxisome proliferator-activated receptor δ. Cancer Cell. 2004;6:285–295. doi: 10.1016/j.ccr.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 23.Maier TJ, Janssen A, Schmidt R, Geisslinger G, Grösch S. Targeting the beta-catenin/APC pathway: a novel mechanism to explain the cyclooxygenase-2-independent anticarcinogenic effects of celecoxib in human colon carcinoma cells. FASEB J. 2005;19:1353–1355. doi: 10.1096/fj.04-3274fje. [DOI] [PubMed] [Google Scholar]
- 24.Pang RP, Zhou JG, Zeng ZR, Li XY, Chen W, Chen MH, Hu PJ. Celecoxib induces apoptosis in COX-2 deficient human gastric cancer cells through AKT/GSK3beta/NAG-1 pathway. Cancer Lett. 2007;251:268–277. doi: 10.1016/j.canlet.2006.11.032. [DOI] [PubMed] [Google Scholar]
- 25.Hsu AL, Ching TT, Wang DS, Song X, Rangnekar VM, Chen CS. The cyclooxygenase-2 inhibitor celecoxib induces apoptosis by blocking AKT activation in human prostate cancer cells independently of Bcl-2. J Biol Chem. 2000;275:11397–11403. doi: 10.1074/jbc.275.15.11397. [DOI] [PubMed] [Google Scholar]
- 26.Yamaguchi K, Lee SH, Eling TE, Baek SJ. Identification of nonsteroidal anti-inflammatory drug-activated gene (NAG-1) as a novel downstream target of phosphatidylinositol 3-kinase/AKT/GSK-3β pathway. J Biol Chem. 2004;279:49617–49623. doi: 10.1074/jbc.M408796200. [DOI] [PubMed] [Google Scholar]
- 27.Bottone FR, Jr, Moon Y, Kim JS, Alston-Mills B, Ishibashi M, Eling TE. The anti-invasive activity of cyclooxygenase inhibitors is regulated by the transcription factor ATF3 (activating transcription factor 3) Mol Cancer Ther. 2005;4:693–703. doi: 10.1158/1535-7163.MCT-04-0337. [DOI] [PubMed] [Google Scholar]
- 28.Lee SH, Yamaguchi K, Kim JS, Eling TE, Safe S, Park Y, Baek SJ. Conjugated linoleic acid stimulates an anti-tumorigenic protein NAG-1 in an isomer specific manner. Carcinogenesis. 2006;27:972–981. doi: 10.1093/carcin/bgi268. [DOI] [PubMed] [Google Scholar]
- 29.Buchanan FG, Holla V, Katkuri S, Matta P. Targeting cyclooxygenase-2 and epidermal growth factor receptor for the prevention and treatment of intestinal cancer. Cancer Res. 2007;67:9380–9388. doi: 10.1158/0008-5472.CAN-07-0710. [DOI] [PubMed] [Google Scholar]
- 30.Baek SJ, Horowitz JM, Eling TE. Molecular cloning and characterization of human nonsteroidal anti-inflammatory drug-activated gene promoter. Basal transcription is mediated by Sp1 and Sp3. J Biol Chem. 2001;276:33384–33392. doi: 10.1074/jbc.M101814200. [DOI] [PubMed] [Google Scholar]
- 31.Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, Trzaskos JM, Evans JF, Taketo MM. Suppression of intestinal polyposis in ApcΔ716 knockout mice by inhibition of cyclooxygenase-2 (COX-2) Cell. 1996;87:803–809. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- 32.Fujino H, Xu W, Regan JW. Prostaglandin E2 induced functional expression of early growth response factor-1 by EP4, but not EP2, prostanoid receptors via the phophatydlinositol-3 kinase and extracellular signal-related kinases. J Biol Chem. 2003;278:12151–12156. doi: 10.1074/jbc.M212665200. [DOI] [PubMed] [Google Scholar]
- 33.Buchanan FG, Wang D, Bargiacchi F, DuBois RN. Prostaglandin E2 regulates cell migration via the intracellular activation of the epidermal growth factor receptor. J Biol Chem. 2003;278:35451–35457. doi: 10.1074/jbc.M302474200. [DOI] [PubMed] [Google Scholar]