

Abstract
As a genome guardian, p53 maintains genome stability by arresting cells for damage repair or inducing cell apoptosis to eliminate the damaged cells in stress response. Several nucleolar proteins stabilize p53 by interfering Mdm2–p53 interaction upon cellular stress, while other mechanisms by which nucleolar proteins activate p53 remain to be determined. Here, we identify NAT10 as a novel regulator for p53 activation. NAT10 acetylates p53 at K120 and stabilizes p53 by counteracting Mdm2 action. In addition, NAT10 promotes Mdm2 degradation with its intrinsic E3 ligase activity. After DNA damage, NAT10 translocates to nucleoplasm and activates p53‐mediated cell cycle control and apoptosis. Finally, NAT10 inhibits cell proliferation and expression of NAT10 decreases in human colorectal carcinomas. Thus, our data demonstrate that NAT10 plays a critical role in p53 activation via acetylating p53 and counteracting Mdm2 action, providing a novel pathway by which nucleolar protein activates p53 as a cellular stress sensor.
Keywords: acetylation, E3 ligase, Mdm2, NAT10, p53
Subject Categories: Post-translational Modifications, Proteolysis & Proteomics; Signal Transduction
Introduction
Tumor suppressor p53 is pivotal to cell fate determination under genotoxic stress and is mutated in over 50% of human cancers 1, 2. As a transcriptional factor, p53 regulates expression of diverse sets of genes corresponding to different stimuli, functioning in cell cycle arrest, DNA repair, apoptosis, and senescence 3, 4. p53 protein exists at low levels with high turnover rate under normal conditions but becomes rapidly stabilized and activated under stress. p53 dynamics is mainly modulated by posttranslational modifications, such as ubiquitination, acetylation, and phosphorylation 5.
Mdm2 is a ubiquitin ligase which functions as an essential inhibitor of p53 6, 7, 8. Mdm2 binds to and antagonizes p53 by either targeting p53 for degradation or repressing p53 transcriptional activity, which is beneficial for cell proliferation in normal cells 9, 10. Mdm2 gene is amplified in at least 7% of all human cancers without concomitant p53 mutation, and these amplifications disrupt p53‐mediated tumor suppression pathway and facilitate tumorigenesis 11, 12, 13. Inhibition or degradation of Mdm2 mediated by multiple proteins is a crucial step and an important mechanism for p53 activation 14. In addition to its role as the workshop for ribosomal biogenesis, the nucleolus also acts as a cellular stress sensor to activate p53 15. Nucleolar protein ARF binds to and promotes degradation of Mdm2, leading to p53 stabilization and activation in response to oncogenic stress 16, 17. Ribosomal proteins (RPs), particularly L5, L11, and L23, have also been shown to interfere with Mdm2–p53 interaction and activate p53 upon ribosomal stress 18, 19, 20. Nevertheless, the signaling through ARF/RP pathway is dispensable for DNA damage response 21, 22. Other mechanisms by which nucleolar proteins contribute to p53 activation in DNA damage response remain to be determined.
Histone acetyltransferases (HATs) have been shown to activate p53 through acetylating p53. For example, CBP/p300 enhances p53‐dependent transcription by directly acetylating the lysine residues in the C‐terminus of p53 23. Acetylation of p53 is reversible with deacetylases such as HDAC1 and SIRT1, suggesting that the transition between acetylation and deacetylation is crucial for p53 activity 24, 25. C‐terminal acetylation of p53 is important for its sequence‐specific DNA binding activity and for activation of expression of p53 target genes 26. However, the C‐terminal acetylation‐deficient p53‐6KR knock‐in mice showed that p53 acetylation at its C‐terminus is not as essential as originally anticipated although it regulates multiple aspects of p53 function 27. Ensuing studies demonstrated that p53 acetylation at lysine 120 (K120) within the DNA binding domain is required for p53‐mediated apoptosis and K120 is acetylated by MYST family acetyltransferases including Tip60, hMOF, and MOZ 28, 29, 30. More importantly, K120 is a common p53 mutation site in human cancer and loss of this acetylation site completely abrogates p53‐mediated apoptosis of thymocytes in mice 31.
N‐acetyltransferase 10, NAT10 (also known as hALP), is a member of GNAT family of HATs. Truncated recombinant NAT10 (amino acids 164–834) shows the ability to acetylate calf thymus histones in vitro 32. Previous study found that NAT10 is a nucleolar protein, promoting RNA polymerase I transcription 33. It was recently demonstrated that Remodelin, a NAT10 inhibitor, ameliorates laminopathies by correcting nuclear architecture and reducing senescence 34. Additionally, NAT10 is highly expressed in some tumors 35. Although related to senescence and cancer, the roles and targets of NAT10 in tumorigenesis remain unknown.
In search for NAT10 targets, we purified the NAT10‐binding proteins from U2OS cells and identified p53 as a novel NAT10‐binding protein by mass spectrometry. NAT10 acetylates p53 at K120 and stabilizes p53 by counteracting Mdm2 action. Consequently, NAT10 activates p53 in response to DNA damage. Our findings demonstrated that NAT10 is a critical regulator for p53 homeostasis.
Results
NAT10 interacts with p53 and Mdm2
To identify the novel targets of NAT10, we purified a NAT10‐contained complex from U2OS cells (Fig 1A). Among the various NAT10 interacting proteins identified by mass spectrometry, p53 was of particular interest due to its pivotal role in tumor suppression. We performed coimmunoprecipitation experiments using the NAT10‐specific antibody to verify that NAT10 interacts with p53. As shown in Fig 1B, p53 existed in the NAT10‐specific immunoprecipitates from U2OS cell extracts, but was absent in the control IgG immunoprecipitates. By serendipity, we found that Mdm2 was also present in the NAT10‐specific immunoprecipitates (Fig 1B). Reciprocal immunoprecipitation with anti‐p53 and anti‐Mdm2 antibodies also pulled down NAT10 in U2OS cells and HCT116 p53+/+ cells (Fig 1C and D). To examine whether the interaction between NAT10 and Mdm2 depends on p53, coimmunoprecipitation assays were conducted in HCT116 p53−/− cells. As shown in Fig 1E and F, Mdm2 and NAT10 coimmunoprecipitated with each other. These results suggested that NAT10 interacts with p53 and Mdm2 in cells. In order to determine whether the interactions are direct, we generated purified GST‐p53 and GST‐Mdm2 from bacteria, and Flag‐NAT10 from Sf9 insect cells. Purified Flag‐NAT10 was able to interact with GST‐p53 and GST‐Mdm2 under cell‐free conditions, suggesting that NAT10 interacts directly with p53 and Mdm2 (Fig 1G). Furthermore, Flag‐NAT10 binds to both DNA binding domain and C‐terminal domain of GST‐p53 in vitro (Fig 1H). Mapping the region of NAT10 required for p53 and Mdm2 binding revealed that both the N‐terminus and the C‐terminus of NAT10 interact with p53, while N‐terminus is critical for the interaction between NAT10 and Mdm2 (Fig 1I). Taken together, these data demonstrated that NAT10 interacts with p53 and Mdm2 both in cells and in vitro.
Figure 1. NAT10 interacts with p53 and Mdm2.
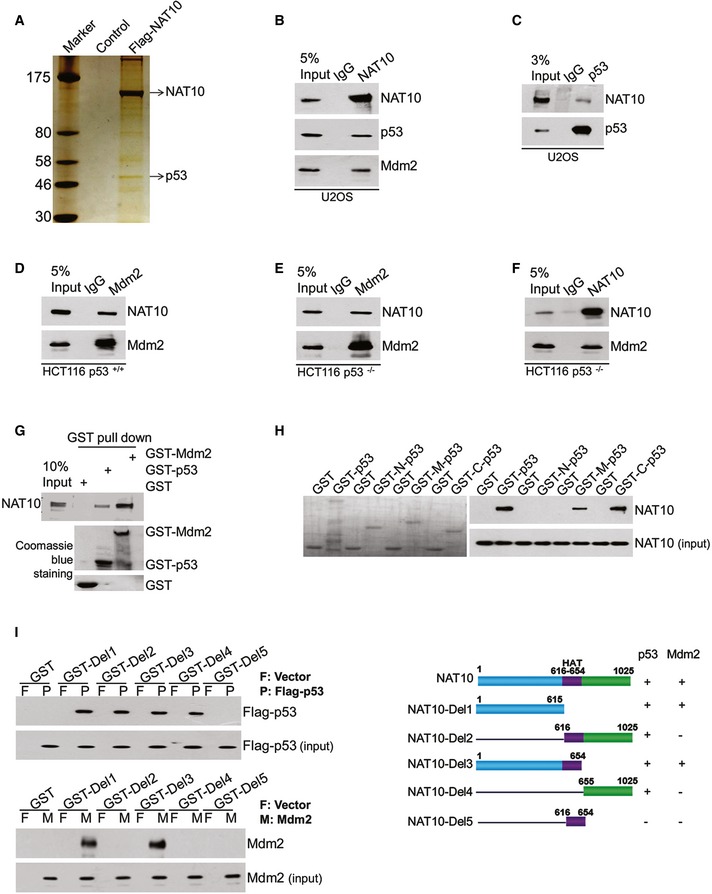
-
AU2OS cells were transfected with Flag‐NAT10 or control vectors. Forty‐eight hours later, cells were harvested and whole‐cell extracts were immunoprecipitated with Flag antibody affinity resin. The NAT10‐binding proteins were resolved by SDS–PAGE and detected by silver staining.
-
B–FU2OS, HCT116 p53+/+, or HCT116 p53−/− cell lysates were immunoprecipitated with control IgG, anti‐NAT10 (B and F), anti‐p53 (C), and anti‐Mdm2 (D and E) antibodies. The immunoprecipitates were subsequently immunoblotted with the indicated antibodies.
-
GPurified NAT10 was incubated with GST, GST‐p53, or GST‐Mdm2 proteins coupled to Glutathione Sepharose 4B. Proteins retained on the Sepharose were then analyzed by Western blot using the antibodies as indicated. The amount of GST fusion proteins are shown in the lower panel.
-
HFull‐length GST‐p53 fusion protein, its deletion mutants, or GST protein was used in pull‐down experiments with purified NAT10 protein. The levels of the GST fusion proteins are shown in the left panel.
-
IGST pull‐down assay was performed using purified GST‐NAT10 deletion mutants or GST protein and overexpressed Flag‐p53 or Mdm2 protein in HEK293T cells. Schematic diagram represents the constructs of GST‐NAT10 deletion mutants (right panel).
NAT10 acetylates p53 at K120
Previous studies indicated that p53 acetylation is indispensable for its activation 36. As NAT10 has been shown to acetylate histones with its acetyltransferase activity 32, we hypothesized that NAT10 could acetylate p53. To test this possibility, we performed in vitro acetylation assay using highly purified Flag‐NAT10 and His‐p53. As shown in Fig 2A, p53 was acetylated only in the presence of both acetyl‐CoA and NAT10. In the midst of GNAT motif of NAT10, there lies a conserved Arg/Gln‐X‐X‐Gly‐X‐Gly/Ala segment (X denotes variation), Q‐G‐M‐G‐Y‐G, which is the acetyl‐CoA binding site common for acetyltransferases. It has been shown that one or more mutations of these three conserved residues dramatically impair acetyltransferase activity of human N‐acetyltransferases 37. To further investigate the HAT activity of NAT10, we generated NAT10 GE mutant by mutating conserved glycine residue 641 to glutamate (G641E) (Fig EV1A). Purified NAT10 GE mutant dramatically lowered its ability to acetylate p53 (Fig 2B). As different acetylation sites of p53 distinctly function in regulating its activity 31, 36, we used mass spectrometric analysis to identify the acetylation sites induced by NAT10. As shown in Fig 2C, lysine 120 (K120) of p53 was acetylated by NAT10. To further confirm this result, we used anti‐Ac‐p53‐K120 antibody which specifically detects K120 acetylation of p53 to evaluate NAT10‐mediated p53 acetylation. As shown in Fig 2D, wild‐type NAT10 rather than the NAT10 GE mutant acetylated p53 at K120 and mutation of K120 (K120R) specifically abrogated NAT10‐mediated acetylation of p53 in vitro. Acetylation of p53 at K120 catalyzed by NAT10 was also determined in cells. High levels of K120‐acetylated p53 were detected in the cells cotransfected with NAT10 and wild‐type p53, but not the p53K120R mutant (Fig 2E). hMOF, another HAT which has been shown to acetylate p53 at K120 in previous study, also increased K120‐acetylated p53 (Fig 2E) 29. In addition, expression of wild‐type NAT10 still strongly increased K120‐acetylated p53 in cells when hMOF was silenced by small interfering RNA (siRNA), while the NAT10 GE mutant lost this ability (Fig EV1B). These results demonstrated that NAT10 is a novel acetyltransferase for p53.
Figure 2. NAT10 acetylates p53 at K120.
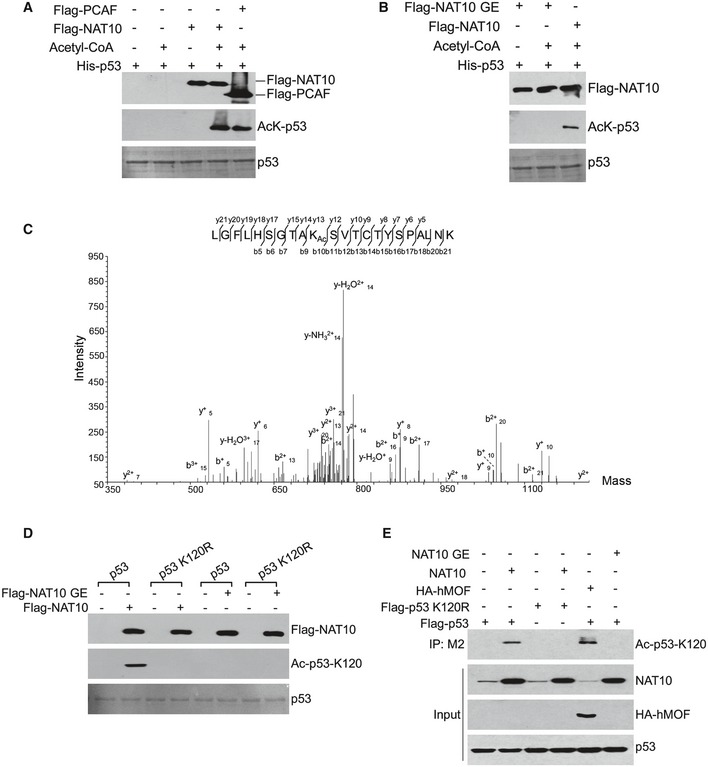
- Recombinant His‐p53 was incubated with or without purified Flag‐NAT10 as described in Materials and Methods. The reaction products were resolved by SDS–PAGE and immunoblotted using anti‐acetyl‐lysine antibodies. Flag‐PCAF was used as a positive control.
- In vitro acetylation was performed and the acetylated p53 was detected as in (A).
- In vitro acetylation assay was performed as described in (A). The reaction products were resolved by SDS–PAGE, and acetylated p53 was purified from the SDS–PAGE and subjected to mass spectrometry analysis.
- His‐p53 or His‐p53‐K120R fusion protein was incubated with purified NAT10 or NAT10 GE mutant as described in Materials and Methods. Reaction mixtures were subjected to Western blot using the site‐specific monoclonal anti‐Ac‐p53‐K120 antibody.
- H1299 cells were transfected with the indicated vectors. Total proteins and the anti‐Flag antibody M2‐specific immunoprecipitates were analyzed by Western blot using the indicated antibodies.
Figure EV1. NAT10 inhibits ubiquitination of p53.
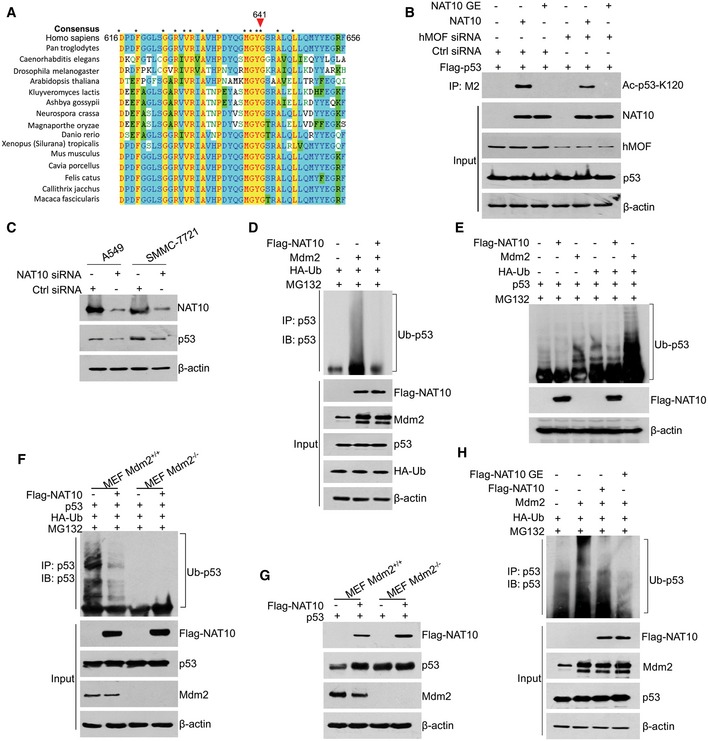
- Alignment of NAT10 GNAT (Gcn5‐related N‐acetyltransferase) domain showing the conservation of G641.
- H1299 cells treated with the indicated siRNAs were transfected with the indicated constructs. Forty‐eight hours later, cells were harvested and the acetylated p53 at K120 was analyzed.
- The A549 or SMMC‐7721 cells were transfected with the indicated siRNAs. Seventy‐two hours later, cells were harvested and the total proteins were subjected to Western blot using the indicated antibodies.
- HCT116 cells were transfected with the indicated plasmids and treated with MG132 for 4 h before harvest. Total proteins were subjected to immunoprecipitation using p53 polyclonal antibody. The ubiquitination levels of p53 were analyzed by Western blot using anti‐p53 antibody (DO‐1).
- H1299 cells were transfected with the vectors as indicated. Twenty‐four hours later, cells were treated with MG132 for 4 h before harvest and Western blot was performed for the indicated proteins.
- MEF Mdm2‐expressing or Mdm2‐null cells were transfected with the indicated plasmids and treated with MG132 for 4 h before harvest. Total proteins were subjected to immunoprecipitation using p53 polyclonal antibody. The ubiquitination levels of p53 were analyzed by Western blot using anti‐p53 antibody (DO‐1).
- MEF Mdm2‐expressing or Mdm2‐null cells were transfected with the vectors as indicated. Twenty‐four hours later, cells were harvested and Western blot was performed for the indicated proteins.
- HCT116 cells were transfected with the indicated plasmids and treated with MG132 for 4 h before harvest. Total proteins were subjected to immunoprecipitation using p53 polyclonal antibody. The ubiquitination levels of p53 were analyzed by Western blot using anti‐p53 antibody (DO‐1).
NAT10 stabilizes p53 by counteracting Mdm2 action
To further study the modulation of p53 acetylation mediated by NAT10, we depleted NAT10 using NAT10‐specific siRNAs. Surprisingly, we found that downregulation of NAT10 decreased p53 protein levels without affecting p53 mRNA levels and both protein and mRNA levels of p21 and Bax, two p53 target genes, were also decreased, while p53‐specific siRNA reduced both mRNA and protein levels of p53 (Fig 3A and B). The NAT10 knockdown‐induced decrease of p53 was confirmed in human lung cancer A549 and liver cancer SMMC‐7721 cells (Fig EV1C). In contrast, overexpression of NAT10 increased the levels of endogenous p53, p21, and Puma (Fig 3C). To corroborate that NAT10 affects p53 stability per se, we treated cells with cycloheximide (CHX) and examined p53 stability after knockdown of NAT10. p53 stability was compromised in the cells transfected with NAT10 siRNA (Fig 3D). These results suggested that NAT10 stabilizes p53 in cells.
Figure 3. NAT10 stabilizes p53 by counteracting Mdm2 action.
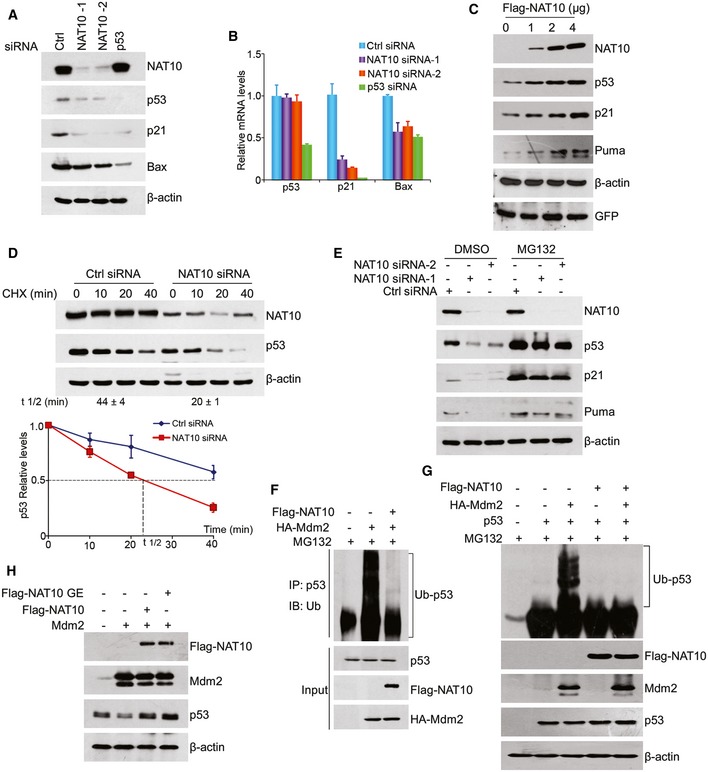
- U2OS cells were transfected with the indicated siRNAs. Seventy‐two hours later, cell lysates were prepared and proteins from lysates were subjected to Western blot using the indicated antibodies.
- U2OS cells were transfected as in (A). Seventy‐two hours later, total mRNAs were extracted and subjected to RT–qPCR for the indicated genes.
- U2OS cells were transfected with increasing amounts of plasmid DNA expressing NAT10. Proteins from whole‐cell extracts were subjected to Western blot using the indicated antibodies.
- U2OS cells were transfected with control or NAT10 siRNAs. Seventy‐two hours later, cells were treated with cycloheximide (CHX) and harvested at the indicated time points. Proteins from cell lysates were subjected to immunoblot for the evaluation of NAT10 and p53 (upper panels). Beta‐actin was evaluated as a loading control (lower panel). p53 half‐life (t1/2) obtained from three independent experiments is presented as mean ± SEM at the bottom.
- U2OS cells were treated with MG132 (10 μM) for 4 h after transfected with the indicated siRNAs. Proteins from cell extracts were detected by Western blot for the indicated proteins.
- HCT116 p53+/+ cells were transfected with the indicated constructs and treated with MG132 for 4 h before harvest. p53 was immunoprecipitated with anti‐p53 polyclonal antibody and immunoblotted with anti‐Ub antibody.
- p53 and Mdm2 double‐null MEF cells were transfected with the indicated constructs and treated with MG132 for 4 h before harvest. Proteins from whole‐cell extracts were subjected to Western blot using the indicated antibodies.
- U2OS cells were transfected with vectors as indicated. Forty‐eight hours later, cells were harvested and proteins from whole‐cell extracts were subjected to Western blot using the indicated antibodies.
The ubiquitin–proteasome system (UPS) is critical for determination of p53 stability 6, 10. To examine whether NAT10‐mediated p53 stabilization depends on UPS, we treated cells with a proteasome inhibitor MG132 after transfection of control or NAT10 siRNAs. As shown in Fig 3E, p53 reduction by NAT10 knockdown was rescued by addition of MG132, revealing that NAT10 regulates p53 levels through proteasome‐dependent pathway. To investigate if NAT10 regulates p53 ubiquitination in cells, we performed in vivo ubiquitination assay. As expected, Mdm2 induced p53 ubiquitination, while this effect was significantly diminished by coexpression of NAT10 (Figs 3F and EV1D and E). These results suggested that NAT10 stabilizes p53 through inhibiting Mdm2‐induced ubiquitination. To confirm this suggestion, in vivo ubiquitination assay was performed in Mdm2‐null MEF cells. NAT10 inhibited p53 ubiquitination only when Mdm2 was coexpressed (Fig 3G). Furthermore, NAT10 inhibited ubiquitination of p53 and increased p53 levels in Mdm2‐expressing MEF cells, while these effects were not observed in Mdm2‐null MEF cells (Fig EV1F and G). These results demonstrated that NAT10 inhibits Mdm2‐mediated ubiquitination of p53.
Acetylation and ubiquitination may compete for the same lysine residues at the C‐terminus of p53; thus, p53 acetylation at these lysines inhibits Mdm2‐mediated ubiquitination of p53 38. However, acetylation at K120 has no significant effect on p53 stability 29. Since NAT10 acetylates p53 at K120, it is likely that NAT10‐mediated p53 stabilization is independent of its acetyltransferase activity. Therefore, we examined the effect of the NAT10 GE mutant on p53 stability. As expected, NAT10 GE still counteracts Mdm2‐induced p53 degradation (Figs 3H and EV1H), suggesting that NAT10‐mediated stabilization of p53 is independent of its acetyltransferase activity.
NAT10 promotes Mdm2 degradation with its intrinsic E3 ligase activity
NAT10 stabilizes p53 protein through inhibiting Mdm2‐mediated p53 ubiquitination. As NAT10 interacts with Mdm2 in cells and in vitro, we further explored the mechanism by which NAT10 targets Mdm2. Ectopic expression of NAT10 resulted in a decrease of Mdm2 protein levels in both HCT116 p53+/+ (Fig 4A) and HCT116 p53−/− cells (Fig EV2A), without changing the mRNA levels of Mdm2 significantly (Fig EV2B). NAT10 also promoted Mdm2 degradation and increased p53 levels in A549 and SMMC‐7721 cells (Fig EV2C). Additionally, the half‐life of Mdm2 was shortened by ectopic expression of NAT10 (Fig 4B). These observations revealed that NAT10 promotes degradation of Mdm2.
Figure 4. NAT10 is a novel E3 ligase for Mdm2.
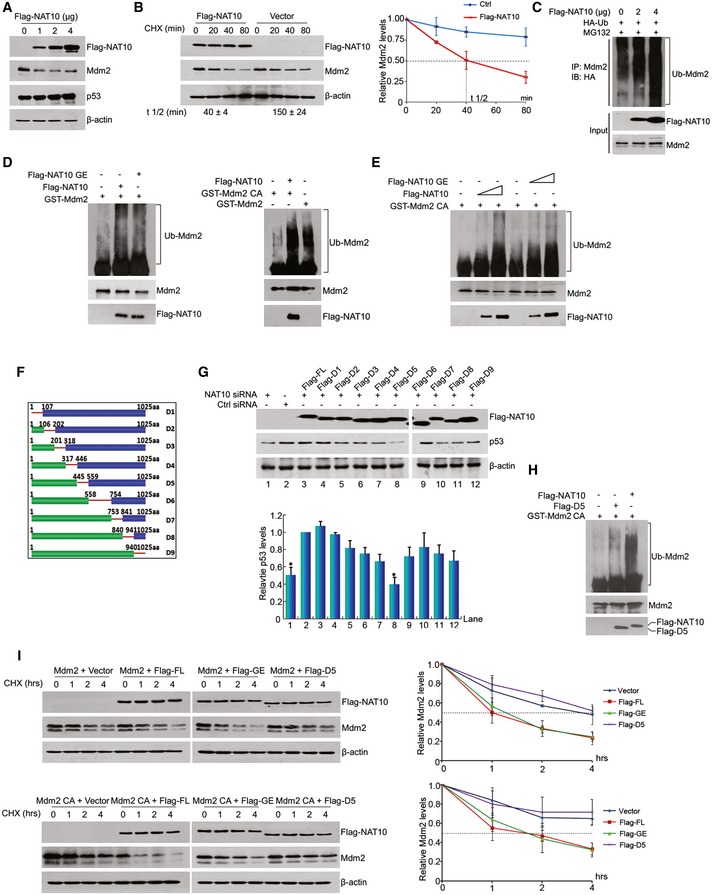
- HCT116 cells were transfected with increasing amounts of DNA expressing NAT10. Forty‐eight hours later, whole‐cell lysates were prepared and proteins from lysates were analyzed by Western blot using the indicated antibodies.
- HCT116 cells were transfected with Flag‐NAT10 and harvested at indicated time points after treatment with CHX. Proteins from cell lysates were immunoblotted with the antibodies as indicated (left panel). Relative Mdm2 protein levels standardized by β‐actin are shown in the right. Mdm2 half‐life (t 1/2) obtained from three independent experiments was presented as mean ± SEM at the bottom.
- HCT116 cells transfected with the indicated constructs were harvested after treatment with MG132 for 4 h. The Mdm2 protein was immunoprecipitated with anti‐Mdm2 antibody and immunoblotted with anti‐HA antibody.
- In vitro ubiquitination was performed with recombinant GST‐Mdm2 (left) or GST‐Mdm2 C464A mutant (right) and Flag‐NAT10 or Flag‐NAT10 GE in the presence of E1, E2, and Ub in vitro as described in Materials and Methods. The reaction products were resolved by SDS–PAGE, followed by Western blot for evaluation of the indicated proteins.
- Mdm2 is ubiquitinated by NAT10 in a dose‐dependent manner. In vitro ubiquitination assay were carried out with Mdm2 and different doses of purified Flag‐NAT10 as described in (D).
- Schematic representation of NAT10 deletion mutant constructs.
- U2OS cells transfected with NAT10 siRNA‐3 (targeting 3′‐UTR of NAT10 mRNA) or control siRNA were transfected with different NAT10 deletion mutants. Cell lysates were extracted, and proteins from lysates were subjected to Western blot using the indicated antibodies. Relative p53 protein levels standardized by β‐actin are shown in the lower panel.
- In vitro ubiquitination reaction was conducted using recombinant GST‐Mdm2 C464A and full‐length Flag‐NAT10 or Flag‐NAT10‐D5. Ubiquitinated Mdm2 was detected as in (D).
- HCT116 cells were transfected with the indicated constructs. Cells were harvested after CHX treatment for the indicated times, and cell lysates were prepared. Mdm2 protein levels were evaluated by Western blot. Relative Mdm2 protein levels standardized by β‐actin are shown in the right.
Figure EV2. NAT10 is a novel E3 ligase for Mdm2.
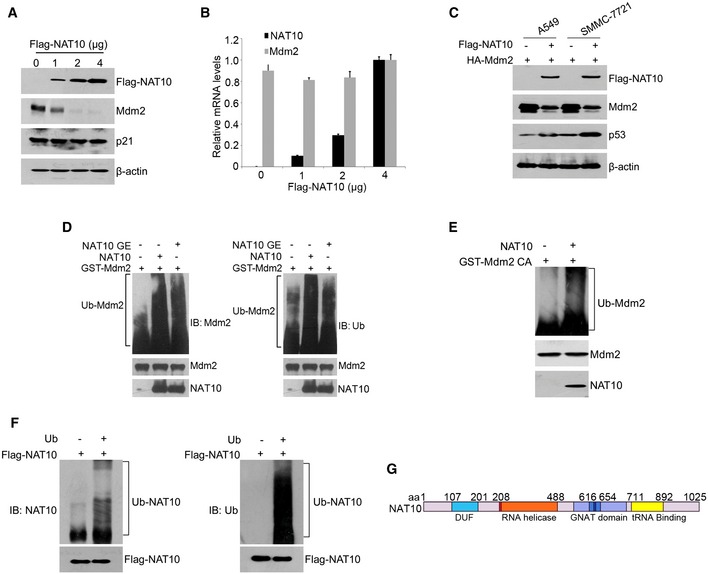
- HCT116 p53−/− cells were transfected with increasing amounts of NAT10 plasmids. Forty‐eight hours later, cells were harvested and the total proteins were subjected to Western blot using the indicated antibodies.
- HCT116 p53+/+ cells were transfected with the indicated vectors. Twenty‐four hours later, cells were harvested after treatment with MG132 for 4 h. Relative mRNA levels were analyzed by RT–qPCR for the indicated genes.
- A549 or SMMC‐7721 cells were transfected with the indicated plasmids. Forty‐eight hours later, cells were harvested and lysed. Western blot was performed for the indicated proteins.
- In vitro ubiquitination of Mdm2 by NAT10. Recombinant GST‐Mdm2 was incubated with Flag‐NAT10 or Flag‐NAT10 GE in the presence of E1, E2, and Ub in vitro as described in Materials and Methods. The reaction products were resolved by SDS–PAGE, followed by Western blot for the indicated proteins.
- In vitro ubiquitination of Mdm2 by bacterial NAT10. Recombinant GST‐Mdm2 CA was incubated with bacterial NAT10 in the absence or presence of Ub in vitro as described in Materials and Methods. The reaction products were resolved by SDS–PAGE, followed by Western blot for the indicated proteins.
- In vitro auto‐ubiquitination of NAT10 was carried out as described in Materials and Methods. Purified NAT10 were incubated with or without Ub in the presence of E1 and E2. The reaction products were resolved by SDS–PAGE, followed by Western blot for the indicated proteins.
- Representation of NAT10 with its known domains.
Given that Mdm2 is degraded by UPS 39, it is possible that NAT10 may regulate Mdm2 through ubiquitination pathway. Indeed, ectopic expression of NAT10 increased ubiquitination levels of endogenous Mdm2 in cells (Fig 4C). To further investigate whether NAT10 enhances Mdm2 ubiquitination in cell‐free system, NAT10 and NAT10 GE were expressed and purified from Sf9 insect cells and in vitro ubiquitination assay was performed with purified NAT10 and GST‐Mdm2. Surprisingly, both purified NAT10 and NAT10 GE enhanced ubiquitination of GST‐Mdm2 in vitro (Figs 4D and EV2D), indicating that NAT10 promotes Mdm2 ubiquitination independent of its acetyltransferase activity. As Mdm2 can ubiquitinate itself 40, we next performed in vitro ubiquitination assay with the Mdm2 mutant C464A (Mdm2 CA) lacking the E3 ligase activity 6, 41 to clarify whether NAT10 promoted Mdm2 ubiquitination through enhancing auto‐ubiquitination of Mdm2. Strikingly, Mdm2 CA was still ubiquitinated by purified NAT10 in cell‐free conditions (Fig 4D and E), demonstrating that NAT10 ubiquitinates Mdm2 rather than stimulating the auto‐ubiquitination of Mdm2. To exclude the existence of copurified E3s in the purified NAT10 from Sf9 insect cells, we purified NAT10 from bacteria, and it also ubiquitinated Mdm2 (Fig EV2E). In addition, NAT10 possesses auto‐ubiquitination capability (Fig EV2F) as most E3 ligases 42. Together, our data demonstrated that NAT10 is a novel E3 ligase for Mdm2.
NAT10 E3 ligase activity is located at residues 456–558
We thereafter searched for the conserved E3 ligase domains in NAT10 and found minor homology to any known E3 motifs such as HECT, RING finger, or U‐box domain (Fig EV2G). To map the active E3 domain of NAT10, we first established an assay to evaluate the activity of NAT10 in regulating p53 stability. In this assay, we silenced endogenous NAT10 by using a NAT10 siRNA (siRNA‐3) which specifically targets 3′‐UTR of NAT10 mRNA and ectopically expressed Flag‐NAT10 to rescue NAT10 knockdown‐induced p53 decrease (Fig EV3A). Then, a series of NAT10 deletion mutants (Fig 4F) were reintroduced into the NAT10‐depleted cells and p53 levels were evaluated. NAT10‐D5 failed to rescue p53 as the other NAT10 deletion mutants and full‐length NAT10 did (Fig 4G), suggesting that residues 456–558 may be its active fragment in regulating p53 level. The role of residues 456–558 was further confirmed by evaluation of NAT10‐D5 effect on p53 stability. Full‐length NAT10 abrogated the Mdm2‐mediated p53 degradation, while NAT10‐D5 failed to do so (Fig EV3B) though NAT10‐D5 still retains the ability to bind Mdm2 (Fig EV3C). To determine the capability of NAT10‐D5 in ubiquitinating Mdm2, in vitro ubiquitination assay was conducted with purified NAT10‐D5. As shown in Fig 4H, purified NAT10‐D5 lost the E3 ligase function to conjugate ubiquitin chain to GST‐Mdm2 CA. Similarly, purified bacterial NAT10‐D5 also lost the E3 activity for Mdm2 (Fig EV3D). We next examined the potential of NAT10 in promoting Mdm2 degradation in cells. Overexpression of either wild‐type NAT10 or the NAT10 GE mutant dramatically increased degradation of Mdm2, while NAT10 D5 lost this capability (Fig 4I). Importantly, the similar results were observed for the Mdm2 CA mutant that is more stable than the wild‐type Mdm2 because of lacking auto‐ubiquitination activity. Taken together, our data demonstrated that the E3 ligase activity lies in residues 456–558 of NAT10.
Figure EV3. NAT10 E3 ligase activity is located at residues 456–558.
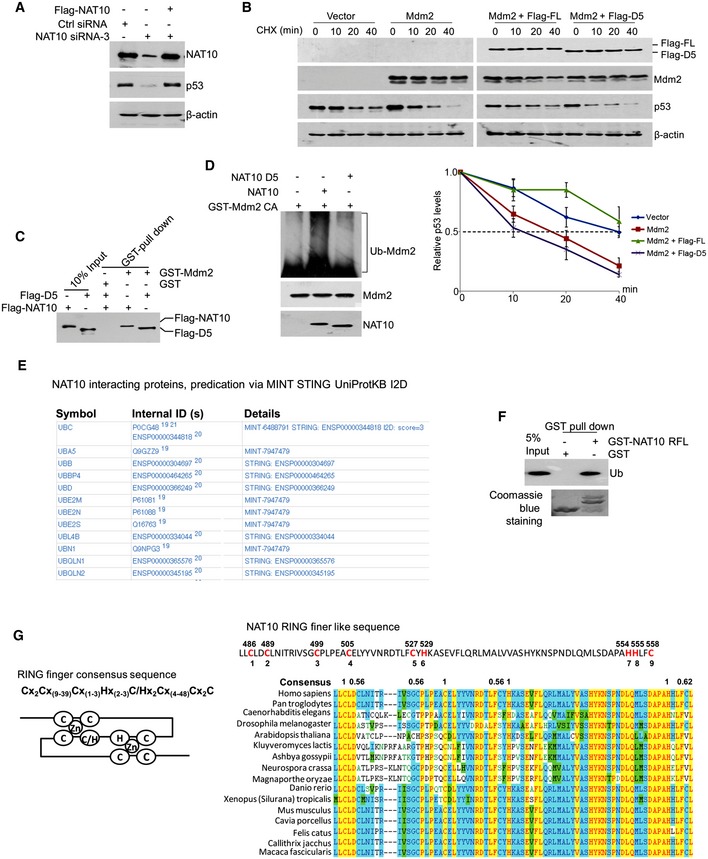
- U2OS cells were transfected with the indicated siRNAs or plasmids. Seventy‐two hours later, cells were harvested and Western blot was then performed for the indicated proteins.
- U2OS cells were transfected with the indicated constructs. Cells were harvested after CHX treatment for the indicated times and lysed. The p53 protein levels were analyzed by Western blot. The lower panel shows quantification of p53 protein levels relative to β‐actin. Error bars represent the SEM from three independent experiments in triplicates.
- GST pull‐down assay was performed with GST or GST‐Mdm2 fusion proteins and purified Flag‐NAT10 or its mutant (Flag‐D5).
- In vitro ubiquitination of Mdm2 by bacterial NAT10. Recombinant GST‐Mdm2 CA was incubated with bacterial NAT10 or D5 mutant in the presence of E1, E2, and Ub in vitro as described in Materials and Methods. The reaction products were resolved by SDS–PAGE, followed by Western blot for the indicated proteins.
- The potential NAT10 interacting proteins (UPS‐associated) were predicated via UniProtKB.
- GST pull‐down assay was performed with GST or GST‐NAT10 RFL fusion proteins and purified His‐Ub (RFL is the abbreviation of “RING finger‐like”, representing residues 456–558.)
- The sequence of NAT10 RING finger‐like motif. RING finger consensus sequence and schematic representation of RING finger (left). The sequence of NAT10 RING finger‐like motif and the potential zinc coordination residues are in red and numbered (top right). Alignment of NAT10 RING finger‐like domain showing the conservation of the potential zinc coordination residues (bottom right).
NAT10 regulates stress‐induced p53 activation
As a genome guardian, p53 is stabilized after DNA damage to maintain genome stability and determine cell fate 43. Since NAT10 regulates p53 in unstressed cells, we asked whether NAT10 regulates p53 stability and activation after DNA damage. Consistent with reported results 36, DNA damage agent actinomycin D induced increased p53 levels, acetylation at K120, and the expression of p53 target genes p21 and Mdm2 (Fig 5A). Importantly, NAT10 was also upregulated after DNA damage agent treatment including actinomycin D, etoposide, and doxorubicin in U2OS cells (Figs 5A and EV4A). This stress‐induced NAT10 upregulation was also observed in p53‐expressing cells A549 and SMMC‐7721 (Fig EV4B) but not in p53‐deficient cells H1299 and HCT116 (p53−/−) (Figs 5B and EV4C), suggesting that DNA damage‐induced increase of NAT10 is p53‐dependent. This hypothesis was confirmed in that NAT10 protein levels failed to change under stress conditions in U2OS cells when p53 was depleted (Fig EV4D). We further investigated if NAT10 regulates p53 activity in response to DNA damage. Depletion of NAT10 significantly decreased p53 levels after actinomycin D treatment in U2OS, A549, and SMMC‐7721 cells (Figs 5C and EV4E). Accordingly, acetylation of p53 at K120 as well as expression of p21, Puma, and Mdm2 was reduced upon knockdown of NAT10 in actinomycin D‐treated cells (Fig 5C). Moreover, knockdown of NAT10 also inhibited p53 activation induced by other DNA damage agents such as doxorubicin and UV irradiation (Fig EV4F). These results demonstrated that NAT10 regulates p53 stability and activation in response to DNA damage.
Figure 5. NAT10 regulates p53 activity in DNA damage response.
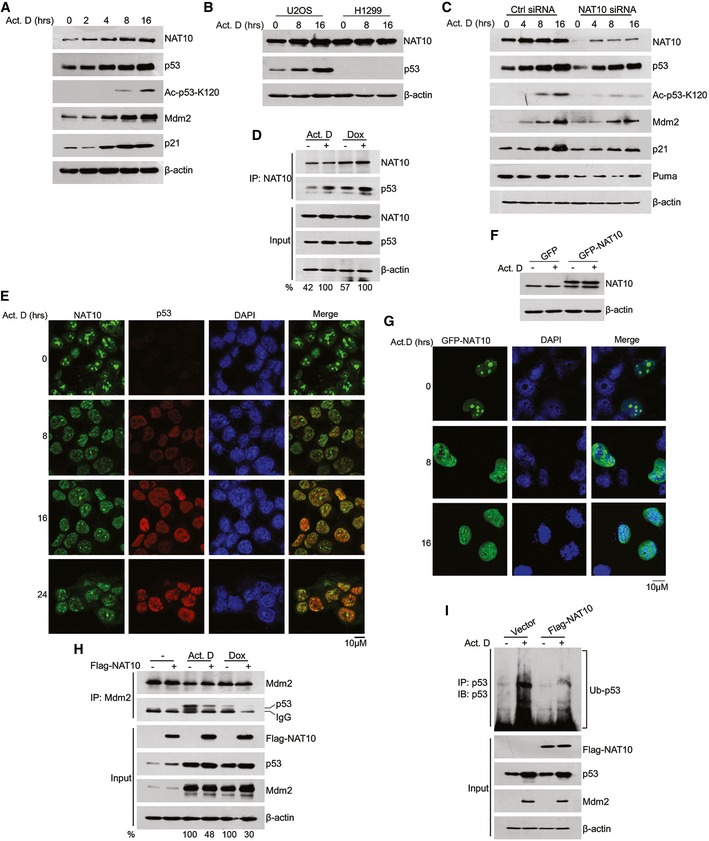
- U2OS cells were treated with 10 nM actinomycin D, and cell extracts were prepared at indicated time points. Proteins from extracts were analyzed by Western blot using the indicated antibodies.
- U2OS and H1299 cells were treated with actinomycin D for 12 h. Cells were harvested, and proteins from cell lysates were subjected to Western blot probed with the indicated antibodies.
- U2OS cells transfected with the indicated siRNAs were treated with actinomycin D and harvested at the indicated time points. Proteins from cell lysates were subjected to Western blot.
- HCT116 cells were treated with 10 nM of actinomycin D or 4 μM of doxorubicin (Dox) for 12 h. Cells were harvested, and total proteins were immunoprecipitated with anti‐NAT10 antibody. Immunoprecipitates were analyzed by Western blot for the indicated proteins.
- HCT116 cells were treated with actinomycin D for the indicated times. Cells were fixed and immunostained as indicated. Immunofluorescence images were taken by confocal microscopy. Scale bar represents 10 μm.
- H1299 cells were transfected with the indicated vectors. After treatment with actinomycin D, cells were harvested and the whole‐cell extracts were subjected to Western blot using the indicated antibodies.
- H1299 cells were transfected with GFP‐NAT10 vectors. After treatment with actinomycin D for 12 h, cells were fixed and nuclei were stained with DAPI. Scale bar represents 10 μm.
- HCT116 cells transfected with the indicated constructs were treated with actinomycin D or doxorubicin. Western blot was performed with total cell extracts and Mdm2‐specific immunoprecipitates for the indicated proteins.
- HCT116 cells were transfected with the indicated constructs. Cells were harvested after treatment with actinomycin D. p53 protein was immunoprecipitated with an anti‐p53 polyclonal antibody and immunoblotted with p53 monoclonal antibody (DO‐1).
Figure EV4. NAT10 regulates p53 activity in DNA damage response.
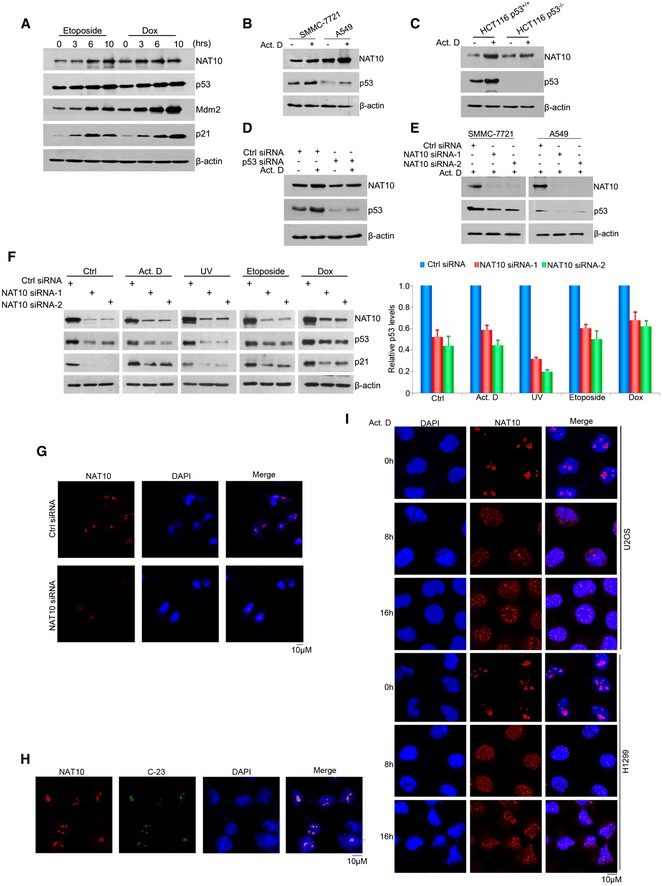
- U2OS cells were treated with etoposide (20 μM) or Dox (4 μM) for the indicated times. Cells were harvested and lysed. Western blot was then performed for the indicated proteins.
- A549 and SMMC‐7721 cells were treated with actinomycin D (10 nM). Twelve hours later, cells were harvested and the whole‐cell extracts were subjected to Western blot using the indicated antibodies.
- HCT116 p53+/+ and p53−/− cells were treated with actinomycin D (10 nM). Twelve hours later, cells were harvested and the whole‐cell extracts were subjected to Western blot using the indicated antibodies.
- U2OS cells were transfected with the indicated siRNAs and treated with actinomycin D (10 nM) for 12 h before harvest. The cells were then lysed, and the whole‐cell extracts were subjected to Western blot using the indicated antibodies.
- A549 and SMMC‐7721 cells were transfected with the indicated siRNAs. Seventy‐two hours later, cells were harvested after treatment with actinomycin D (10 nM) for 4 h. The total proteins were extracted, and Western blot was performed for the indicated proteins.
- U2OS cells were transfected with the indicated siRNAs. Seventy‐two hours later, cells were harvested after treatment with actinomycin D (10 nM), UV (50 J/m2), etoposide (20 μM), or Dox (4 μM) for 4 h. The cells were lysed, and Western blot was performed for the indicated proteins. Right panel: Quantification of p53 protein levels relative to β‐actin. Error bars represent the SEM of the experiments in triplicate.
- U2OS cells were transfected with the indicated siRNAs. Seventy‐two hours later, cells were fixed and stained with anti‐NAT10 antibodies, and nuclei were stained with DAPI. Scale bar represents 10 μm.
- U2OS cells were fixed and immunostained with the indicated antibodies and DAPI. Scale bar represents 10 μm.
- U2OS and H1299 cells were treated with actinomycin D (10 nM) for the indicated times. Then, cells were fixed and stained with anti‐NAT10 antibodies and DAPI. Scale bar represents 10 μm.
Next, we asked how NAT10 functions in stress‐induced p53 activation. To answer this question, we first performed immunoprecipitation assay to explore the interaction between NAT10 and p53 after DNA damage. As shown in Fig 5D, the interaction between NAT10 and p53 was enhanced by actinomycin D and doxorubicin treatment. Since NAT10 is a nucleolar protein 33, we thus wondered in which cellular compartment NAT10 interacts with p53 when DNA damage occurs. NAT10 is localized predominantly in the nucleolus, with much less expression in the nucleoplasm in both p53‐sufficient and p53‐deficient cells under normal conditions (Fig EV4G and H). However, in response to DNA damage, a portion of NAT10 translocated from the nucleolus to the nucleoplasm and colocalized with p53 in HCT116 p53+/+ cells (Fig 5E). To exclude the possibility that the nucleoplasmic NAT10 was due to an increase of NAT10 protein level instead of a translocation, we transfected H1299 cells with GFP‐NAT10. GFP‐NAT10 translocated into the nucleoplasm while its protein levels did not change upon actinomycin D treatment (Fig 5F and G). The DNA damage‐induced nucleoplasmic translocation of NAT10 was also observed in p53‐deficient H1299 cells as well as in p53‐sufficient U2OS cells (Fig EV4I). We speculated that NAT10 translocates to the nucleoplasm to interact with p53 and thus protects p53 from Mdm2‐mediated degradation. To validate this speculation, we evaluated the interaction between p53 and Mdm2 after DNA damage. As shown in Fig 5H, NAT10 inhibits the interaction between p53 and Mdm2 under stressed conditions. Further, ectopic expression of Flag‐NAT10 decreased p53 ubiquitination levels in cells sustaining DNA damage (Fig 5I). Taken together, these results indicated that NAT10 is required for p53 activation after DNA damage.
NAT10 regulates p53 function
Activation of p53 exerts its function in cell cycle arrest and apoptosis by transcriptionally regulating its target genes. As NAT10 increases p53 stability, we first analyzed if p21 promoter activity is regulated by NAT10. As shown in Figs 6A and EV5A, p21 promoter activity was reduced by knockdown of NAT10 in U2OS cells but was not significantly changed when p53 is depleted. In addition, knockdown of NAT10 significantly impaired transcriptional activation of p21 induced by actinomycin D treatment in U2OS cells only when p53 was present (Fig 6B). Further, depletion of NAT10 dramatically decreased transcriptional activation of p21, Puma, and Bax in U2OS cells, but failed to do so in H1299 cells (Fig EV5B and C). These results revealed that NAT10 regulates p53‐dependent transcriptional activity.
Figure 6. Effects of NAT10 on p53‐mediated transcriptional activity, cell cycle arrest, and apoptosis.
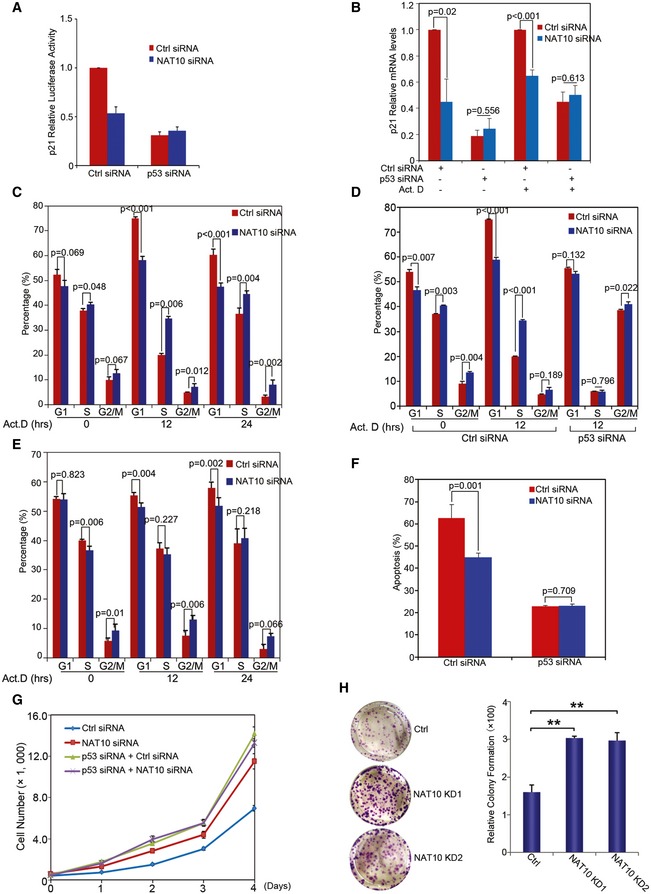
-
Ap21 promoter luciferase reporter plasmid was cotransfected with the indicated siRNAs into U2OS cells. Luciferase activity was determined as described in Materials and Methods.
-
BU2OS cells were transfected with siRNAs as indicated. Seventy‐two hours later, cells were harvested after treatment with actinomycin D for 12 h. Relative mRNA levels for the indicated genes were analyzed by RT–qPCR.
-
C–EU2OS cells (C and D) and H1299 cells (E) transfected with the indicated siRNAs were treated with actinomycin D and harvested at the indicated time points. Cell cycle profiles were determined by FACS. Results from three independent experiments are shown as mean ± SEM (two‐tailed Student's t‐test).
-
FU2OS cells were transfected with the indicated siRNAs. Cells were irradiated with 50 J/m2 UV. Forty‐eight hours later, apoptotic cells were determined. Quantification of apoptotic cells is presented (two‐tailed Student's t‐test).
-
GU2OS cells transfected with the indicated siRNAs were plated. Cell numbers were determined as described in Materials and Methods at the indicated time points.
-
HColony formation assay was performed with U2OS cells transfected with the indicated siRNAs. The right panel shows the quantification of formed colonies. **P < 0.01 (two‐tailed Student's t‐test).
Figure EV5. NAT10 is downregulated in CRC samples.
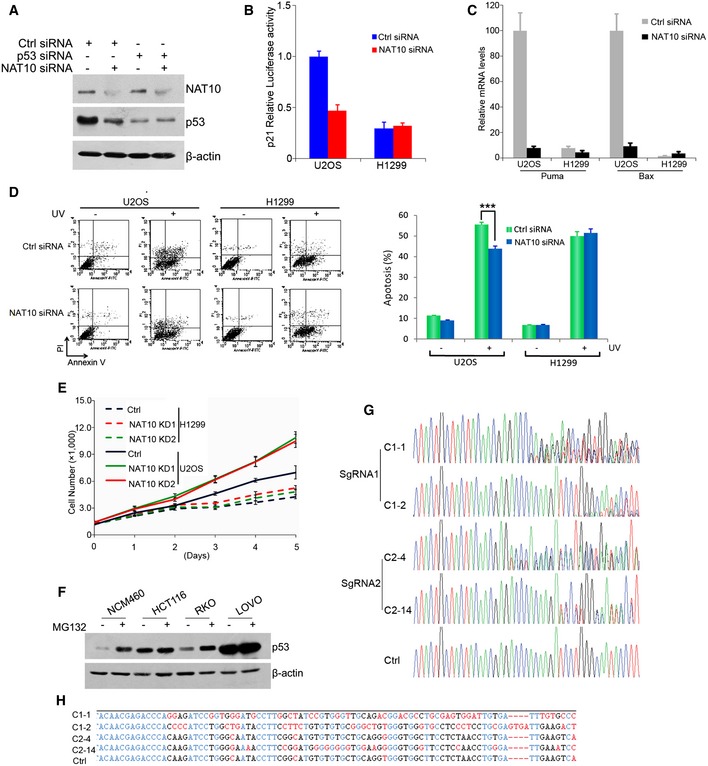
- U2OS cells were transfected with the indicated siRNAs. Seventy‐two hours later, cells were harvested and Western blot was performed for the indicated proteins.
- p21 promoter luciferase reporter plasmid was cotransfected with the indicated siRNAs into U2OS and H1299 cells. Luciferase activity was determined as described in Materials and Methods.
- U2OS cells or H1299 cells were transfected with siRNAs as indicated. Seventy‐two hours later, cells were irradiated with 50 J/m2 UV for 16 h. Relative mRNA levels for the indicated genes were analyzed by RT–qPCR.
- U2OS and H1299 cells were transfected with the indicated siRNAs. Cells were irradiated with 50 J/m2 UV. Forty‐eight hours later, apoptotic cells were determined. Quantification of apoptotic cells is shown in the right. ***P < 0.001 (two‐tailed Student's t‐test).
- U2OS and H1299 cells transfected with control or NAT10 siRNAs (NAT10 KD) were plated. Cell numbers were determined as described in Materials and Methods at the indicated time points.
- The CRC cell lines were harvested with or without MG132 treatment. Western blot was then performed for the indicated proteins.
- Verification of knockdown of NAT10 gene by Cas9 technology. HCT116 p53+/+ cells were cotransfected with the DNA expressing Cas9 and sgRNA. Single cell was plated into 96‐well plates, and monoclone was obtained. Genomic DNA was extracted from HCT116 NAT10 knockdown cell lines, and NAT10 gene was amplified by PCR. PCR products were subjected to DNA sequencing.
- Alignment of mutated NAT10 genes in the NAT10 knockdown cells as described in (G) and wild‐type NAT10 gene.
Next, we examined whether NAT10 affects p53‐mediated cell cycle arrest and apoptosis. Knockdown of NAT10 promoted G1/S transition and dramatically reduced actinomycin D‐induced G1 arrest in U2OS cells (Fig 6C). This is consistent with the changes of p53 and p21 levels. Furthermore, depletion of NAT10 had little effects on G1/S transition and actinomycin D‐induced G1 arrest in U2OS cells when p53 expression is ablated using RNAi approach (Fig 6D). In addition, NAT10 did not affect G1/S transition in H1299 cells (Fig 6E). These data indicated that NAT10 affects p53‐mediated cell cycle arrest. Unexpectedly, knockdown of NAT10 resulted in a significant increase in G2/M percentage in the presence or absence of actinomycin D treatment in H1299 cells (Fig 6E), suggesting that NAT10 might involve in cell cycle control of G2/M phase in a p53‐independent way. We further determined if depletion of NAT10 could affect p53‐dependent apoptosis. Knockdown of NAT10 significantly decreased UV‐mediated apoptosis in U2OS cells, but had little effect when p53 was depleted (Fig 6F). In addition, depletion of NAT10 had little effects on UV irradiation‐induced apoptosis in p53 null cells (Fig EV5D). These results demonstrated that NAT10 regulates p53‐mediated apoptosis. Finally, we investigated whether NAT10 affects cell proliferation. Knockdown of NAT10 significantly promoted cell proliferation in U2OS cells only when p53 was present, while it had no apparent effect on that in H1299 cells (Figs 6G and EV5E). In addition, knockdown of NAT10 significantly facilitated cell colony formation in U2OS cells (Fig 6H). Together, these data demonstrated that NAT10 regulates p53‐dependent transactivation, cell cycle control, and apoptosis.
NAT10 is downregulated in colorectal cancer
Given that p53 is an important tumor suppressor and NAT10 potentiates p53 functions, we examined whether NAT10 expression is altered in human cancers. We evaluated NAT10 expression in human colorectal cancers (CRCs) and corresponding adjacent non‐malignant tissues. As determined by quantitative PCR, the cancerous tissues had lower NAT10 mRNA levels, compared to their corresponding non‐malignant tissues (Fig 7A). To further confirm these results, NAT10 expression in CRC tissues was evaluated by immunohistochemistry. We scored the staining of NAT10 from 0 to 3 and arbitrarily designated staining score 0 as negative and 1–3 as positive. Representative staining and scores are shown in Fig 7B. These results suggested that NAT10 is downregulated in CRC tissues. We further examined the expression levels of NAT10 and p53 in CRC cell lines. Strikingly, we found a positive correlation between NAT10 and p53 expression (Fig 7C). This p53 expression could be increased by MG132 treatment (Fig EV5F), indicating that p53 levels are regulated by the proteasome pathway in these cells.
Figure 7. NAT10 is downregulated in CRC samples.
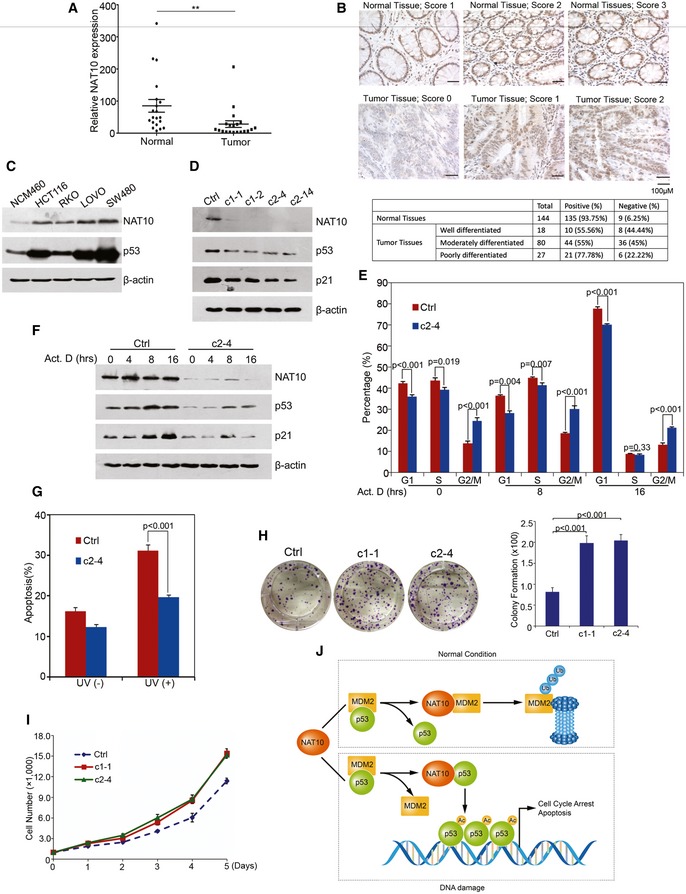
- Twenty pairs of fresh frozen CRC tissues and corresponding normal tissues were lysed, and total RNAs were extracted and subjected to RT–qPCR. **P < 0.01 (nonparametric Mann–Whitney t‐test).
- Immunohistochemical staining of NAT10 in CRC tissues and normal colon tissues. NAT10‐specific immunocomplex was detected with DAB as shown in brown color. Quantification of NAT10‐positive or NAT10‐negative CRC cases is shown in the lower table.
- Cell extracts were prepared from different human colon epithelial cell lines as indicated at the top. Proteins from the extracts were subjected to Western blot for the evaluation of NAT10 and p53.
- CRISPR‐Cas9 system‐mediated NAT10 gene editing. The genome targeting for knockdown NAT10 was performed as described in Materials and Methods. The HCT116 NAT10 knockdown cell lines were harvested, and Western blot was performed on the cell lysates for the indicated proteins.
- HCT116 Ctrl or c2‐4 cells with NAT10 disruption were treated with actinomycin D and harvested at the indicated time points. Cell cycle profiles were determined by FACS. Results from three independent experiments are shown as mean ± SEM (two‐tailed Student's t‐test).
- HCT116 Ctrl or c2‐4 cells with NAT10 disruption were treated with actinomycin D. Cells were harvested at the indicated time points and lysed. The total proteins were analyzed by Western blot using the indicated antibodies.
- HCT116 Ctrl or c2‐4 cells were irradiated with 80 J/m2 UV. Forty‐eight hours later, apoptotic cells were determined. Results from three independent experiments are shown as mean ± SEM (two‐tailed Student's t‐test).
- Colony formation assay were performed with HCT116 cells (Ctrl, c1‐1, and c2‐4). The right panel shows the quantification of formed colonies (two‐tailed Student's t‐test). Error bars represent the SEM from three independent experiments in triplicates.
- HCT116 c1‐1 and c2‐4 cell lines were plated and cell growth curves were determined as described in Fig 6F. Error bars represent the SEM of the triplicate experiments.
- The working model of p53 regulation by NAT10.
To further confirm the role of NAT10 in colorectal cell proliferation, we mutated NAT10 gene in HCT116 cells through CRISPR‐Cas9‐mediated genome editing. We selected cell lines carrying partially destroyed NAT10 gene with low NAT10 protein levels (Figs 7D and EV5G and H) and evaluated the effect of reduced expression of NAT10 on regulating p53 activity and cell proliferation. In addition to the lower expression levels of p53 and p21 as compared to the CRISPR‐Cas9 control cell lines (Fig 7D), Cas9 NAT10 KD cells displayed increased G1/S transition (Fig 7E), which is consistent with inactivation of p53 and p21 (Fig 7F), indicating that NAT10 depletion promotes cell cycle progression. Furthermore, disruption of NAT10 also resulted in a significant increase in G2/M percentage as knockdown of NAT10 did in U2OS and H1299 cells (Fig 7E). In addition, disruption of NAT10 dramatically decreased UV‐induced apoptosis (Fig 7G). Consequently, NAT10‐depleted cells showed enhanced ability of proliferation and colony formation (Fig 7H and I). These data further confirmed that NAT10 plays an important role in modulation of p53 stability and activity.
Discussion
Recently, new nucleolus–nucleoplasm networks of proteins which regulate p53 stability has been discussed by Zhang and Lu 15. The oncogenic stress‐induced ARF disrupts p53‐Mdm2 interaction and sequesters Mdm2 into the nucleoli, thus stabilizing and activating p53 44. Several ribosomal and nucleolar proteins, including RPL5, RPL11, RPL23, NPM/B23, and nucleostemin, interfere with p53‐Mdm2 interaction and contribute to p53 activation in response to ribosomal stress 18, 45, 46, 47, 48, 49. ARF/RPs are dispensable for DNA damage‐induced p53 activation, while some DNA damage signals activate p53 through the nucleolar disruption 50. These findings suggest that nucleolar stress‐induced p53 activation is far more complex than previously thought. This promoted us to consider the possibility that nucleolar disruption may trigger p53 activation through other mechanisms. In this report, we identified nucleolar protein NAT10 as a novel regulator for p53. Different from the ARF/RPs, NAT10 involvement in p53 regulation has dual mechanisms (Fig 7J). Under normal conditions, NAT10 acts as an E3 ligase for Mdm2 to promote Mdm2 degradation and stabilizes p53. While under DNA damage conditions, NAT10 prevents Mdm2–p53 interaction by binding to p53 and acetylates p53 at K120, thus contributing to p53 activation.
It has been widely accepted that auto‐ubiquitination regulates Mdm2 levels in cells, as Mdm2 RING domain is critical for the protein levels of Mdm2 itself 39. However, recent study demonstrated that the Mdm2 C462A mutant was still degraded rapidly in knock‐in mice though Mdm2 C462A abolishes its E3 activity, suggesting the Mdm2 auto‐ubiquitination is dispensable for Mdm2 stability and other E3 ligases for Mdm2 exist 51. PCAF has been reported to control Mdm2 stability with its intrinsic ubiquitin ligase activity 52. However, depletion of PCAF had little effect on p53‐dependent apoptosis induced by UV irradiation though PCAF is required for destabilization of Mdm2 upon DNA damage. Another reported E3 ligase for Mdm2 is SCF(β‐TrCP). SCF(β‐TrCP) promotes Mdm2 degradation after Mdm2 is phosphorylated by casein kinase I, indicating that SCF(β‐TrCP) might be the E3 ligase responsible for DNA damage‐induced Mdm2 degradation 53. Interestingly, a recently identified cytosolic E3 ligase for Mdm2, NEDD4‐1, mediates Mdm2 ubiquitination and promotes stabilization of Mdm2 54. Thus, the regulation of Mdm2 by its E3 ligases is far more complicated than we anticipated. In the present study, we identified NAT10 as a novel E3 ligase for Mdm2 through several lines of evidence: NAT10 physically interacts with Mdm2 (Fig 1), ubiquitinates both wild‐type and enzyme‐dead Mdm2 in vitro (Figs 4 and EV3), and enhances Mdm2 ubiquitination in cells. In addition, NAT10 overexpression shortened the half‐life of wild‐type and enzyme‐dead Mdm2 in cells. Therefore, our findings demonstrated that NAT10 controls Mdm2 stability independent of auto‐ubiquitination of Mdm2 in the absence of stress.
E3 ligases specifically recognize the substrates and bind an E2‐Ub thioester and transfer ubiquitins to substrates, facilitating the polyubiquitination of the substrates 55. In eukaryotes, E3s mainly fall into three classes, namely HECT, RING finger, and U‐box, according to the conserved domains for E3 function 56. Here, we identified NAT10 as a novel E3 ligase for Mdm2 without known E3 domain. We thus conducted a bioinformatics analysis which suggested that NAT10 might interact with E2 and ubiquitin‐associated proteins (Fig EV3E) and the central region of the residues 456–558, which is indispensable for the E3 ligase activity of NAT10, contains a “RING finger‐like” domain with high sequence consensus (Fig EV3G). Importantly, our in vitro experiment showed that NAT10 binds to ubiquitin (Fig EV3F). Thus, NAT10 might belong to another type of RING finger E3 ligase.
Under normal conditions, NAT10 promotes degradation of Mdm2 to stabilize p53, thus contributing to the maintenance of p53 protein levels. Upon DNA damage, NAT10 translocated from the nucleolus to the nucleoplasm as demonstrated in HCT116 p53+/+, U2OS, and H1299 cells (Figs 5E and G, and EV4I). In the nucleoplasm, it stabilizes p53 by protecting p53 from Mdm2‐induced degradation and also activates p53 by acetylating p53 at K120. It is notable that NAT10 was upregulated under stress conditions in a p53‐dependent way. Thus, NAT10 forms a positive regulation feedback with p53 in response to stress. It is known that nucleolar protein ARF stabilizes and activates p53 by sequestering Mdm2 into the nucleoli and disrupting p53‐Mdm2 interaction under oncogenic stress 57. In this report, we provide an alternative mechanism for a nucleolar protein to activate p53 in response to stress. Loss of this regulatory mechanism has evident effects on p53 response to DNA damage in terms of p53 accumulation and transactivation of its downstream genes as demonstrated in Figs 5 and 6.
Main outcomes of p53 activation are cell cycle arrest and apoptosis under DNA damage conditions, by which p53 functions in eliminating the damaged cells and maintains genome stability 4. p21 gene, the primarily identified as downstream gene of p53, encodes an inhibitor of cyclin‐dependent kinases and mediates the ability of p53 to inhibit cell proliferation 58. Depletion of NAT10 impairs p53‐dependent transcription activation of p21 but failed to do so in p53‐depleted cells after cellular stress (Figs 5C and 6B). Further, knockdown of NAT10 reduces p53‐mediated G1 arrest only in the presence of p53. We thus conclude that NAT10 regulates cell cycle in a p53‐dependent way. Unexpectedly, significant increases of G2/M percentage were observed when NAT10 was depleted either in p53‐sufficient cells or in p53‐deficient cells under both unstressed and stressed conditions (Figs 6C–E and 7E), suggesting that NAT10 is involved in G2/M progression in a p53‐independent way. Given that NAT10 acetylates microtubules and regulates cytokinesis as previously found 35, NAT10 might regulate G2/M progression by functioning on mitosis independent of p53. It has been found that acetylation at K120 is critical for p53‐mediated Puma activation and cellular apoptosis 28, 31. Our data showed that NAT10 enhances p53 acetylation at K120 as well as activation of Puma upon cellular stress (Figs 5C and EV5C). Moreover, knockdown of NAT10 desensitized cells to UV‐induced apoptosis in a p53‐dependent manner (Figs 6F, 7G and EV5D), suggesting that NAT10 participates in p53‐mediated apoptosis. Several MYST‐type HATs including Tip60, hMOF, and MOZ contribute to p53‐dependent apoptosis by promoting p53 K120 acetylation 28, 29, 30. NAT10 acetylates p53 at K120 in vitro and in cells with knockdown of hMOF (Figs 2D and EV1B). Thus, NAT10 may collaborate with other HATs in eliminating abnormal cells under diverse stress conditions and maintaining genome stability in p53 wild‐type cells. These data support the role of NAT10, via its effects on stability and acetylation of p53, as a novel regulatory component of the p53 pathway after DNA damage.
In summary, NAT10 activates p53 through acetylating p53 and counteracting Mdm2 action, which provides a novel mechanism for nucleolar protein as cellular stress sensor to activate p53 and keep cell in control. It is intriguing whether there exists a crosstalk between NAT10 and other nucleolar proteins in response to cellular stress. Recently, a NAT10 inhibitor, Remodelin, has provided a possibility for alleviating dystrophic and premature aging diseases by reducing senescence 34. Combining our observation that NAT10 is downregulated in colorectal carcinomas and is involved in the regulation of p53 function, NAT10 may have considerable implications as a target in cancer therapy.
Materials and Methods
Cell culture and transfections
U2OS, H1299, HCT116 p53+/+, HCT116 p53−/−, and HEK293T cell lines were maintained in DMEM medium. NCM460, RKO, LOVO, and SW480 cell lines were maintained in RPMI 1640 medium. All media were supplemented with 10% fetal bovine serum. Cells were transfected with plasmid DNA or siRNA RNA duplexes by Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. In transient transfection experiments, plasmid DNA was kept constant with empty vector.
Plasmids and antibodies
Flag‐ or GFP‐tagged NAT10 and NAT10 mutants (D1 to D9) were cloned into pCI‐neo or pEGFP‐C2. Similarly, Mdm2 was cloned into pCMV‐HA vector. Flag‐tagged p53 was cloned into pCI‐neo vector. For in vitro GST pull‐down assay, p53 or Mdm2 was cloned into pGEX‐4T‐1 vector. For protein purification, NAT10 or its mutants were cloned into pFast‐Bac1 vector. p53 was cloned into pET28a vector. pGL3‐p21‐Luc is a kind gift from Dr. Y. Shang. All PCR products were confirmed by DNA sequencing. Mutant plasmids including pCI‐neo‐Flag NAT10 G641E, pGEX‐4T‐1 Mdm2 C464A, pET28a‐His p53 K120R, pCI‐neo‐Flag‐p53 K120R were obtained by mutagenesis using the QuikChange Site‐Directed Mutagenesis Kit (Stratagene) according to the manufacturer's protocol. The presence of the mutations in the constructed plasmids was confirmed by DNA sequencing. For genome editing, pc3‐U6‐NAT10‐g1‐CMV‐Red, pc3‐U6‐NAT10‐g2‐CMV‐Red, pc3‐U6‐guide‐CMV‐Red, Topo‐Cas9‐IRES‐EGFP were purchased from Shanghai Biomodel Organism Co. Ltd.
Anti‐p53 (DO‐1), anti‐p53 (FL‐393), anti‐p21 (817), anti‐Bax (P‐19), anti‐actin (C‐11), anti‐Mdm2 (SMP14), and anti‐Mdm2 (C‐18) were purchased from Santa Cruz Biotechnology. Anti‐p53 (9282), anti‐Puma (4976), and anti‐Ub (P4D1) were purchased from Cell Signaling Technology. Anti‐Flag (F1804), anti‐Flag (F3165), and anti‐HA (H6908) were purchased from Sigma. Anti‐acetylated lysine (Clone 4G12) was purchased from Millipore. Anti‐acetylated‐p53 (AcK120) was purchased from Abcam. Anti‐NAT10 was a gift from Dr. B. Zhang.
RNA interference
For silencing NAT10, siRNA‐1 5′‐CAGCACCACUGCUGAGAAUAAGA‐3′, siRNA‐2 5′‐CCGAAUCCGGAUUCUCAUU‐3′, and siRNA‐3 5′‐ UUGCCACGAGUCUCUCUCUUC‐3′ were used. For silencing p53, 5′‐GACUCCAGUGGUAAUCUAC‐3′ was used.
GST pull‐down assay
GST fusion proteins were prepared following standard protocol. For in vitro binding assays, GST fusion proteins bound to the Glutathione Sepharose 4B (GE Healthcare) were incubated with purified proteins or cell lysate. After washing, the bound proteins were separated by SDS–PAGE and immunoblotted with indicated antibodies.
Coimmunoprecipitation assay
Cell lysates were prepared in Buffer A (25 mM Tris–Cl pH 7.5, 150 mM KCl, 1 mM DTT, 2 mM EDTA, 0.5 mM PMSF, and 0.2% Nonidet P‐40) and used directly for immunoprecipitation. Antibodies were coupled with a 50% suspension of protein A‐Sepharose beads (GE Healthcare) in IPP500 (500 mM NaCl, 10 mM Tris–Cl pH 8.0, 0.2% Nonidet P‐40). Coupled beads were incubated with cell lysates for 2 h at 4°C. After washing, precipitates were analyzed by Western blot using the indicated antibodies.
In vitro ubiquitination assay
Purified GST or GST‐Mdm2 (500 ng) was incubated with the indicated E3 (100 ng) in the presence of 50 ng UBE1 (Sigma), 50 ng UBCH5b (Sigma), and 10 μg ubiquitin (Sigma) in final volume of 20 μl of ubiquitination reaction buffer (250 mM Tris–Cl pH 7.5, 50 mM NaCl, 1 mM DTT, 2 mM ATP, and 2 mM MgCl2). After incubation at 30°C for 2 h, reaction mixtures were separated by SDS–PAGE and analyzed by Western blot using the indicated antibodies.
In vitro acetylation assay
This assay was carried out as described previously with some modifications 28. Briefly, His‐p53 or its mutants were purified from E. coli. Flag‐NAT10 or its mutants were purified from Sf9 cells. Twenty microliters reaction mixture contained 50 mM Tris–Cl pH 7.9, 10% glycerol, 0.1 mM EDTA, 1 mM PMSF, 10 mM sodium butyrate, 10 μM acetyl‐CoA, 200 ng of purified His‐p53 or His‐p53‐K120R, and 20 ng of Flag‐NAT10. The reaction mixtures were incubated at 30°C for 1 h and separated by SDS–PAGE followed by immunoblot.
Luciferase assay
U2OS and H1299 cells were seeded at a density of 80,000 cells per well in 24‐well plates. The next day, cells were cotransfected with p21 promoter luciferase reporter constructs or pGL3‐basic reporter control plasmid and a Renilla luciferase control reporter vector (Promega) used for normalizing transfection efficiency. Twenty‐four hours later, the luciferase assays were carried out according to the manufacturer's instructions (Dual‐Luciferase Reporter Assay System; Promega). Data were presented as relative luciferase activity normalized for expression of Renilla luciferase vector as measured by Renilla luciferase activity.
Flow cytometry and apoptosis assay
For DNA content analysis, cells were trypsinized, washed with PBS, and fixed in 75% ice‐cold ethanol at 4°C overnight. Cells were then rehydrated in PBS. Following RNase digestion, cells were stained with propidium iodide. Flow cytometry analysis was performed using red (propidium iodide) emission (at 630 nm). Data from 10,000 cells were collected and analyzed by using CellQuest software (Becton Dickinson). For apoptosis analysis, cells were double‐stained with annexin V and propidium iodide (KeyGen) and subjected to flow cytometric analysis.
Cell growth assay
Cell growth was analyzed using MTS reagent (Promega) according to the manufacturer's directions. U2OS, H1299, or HCT116 cells transfected with the indicated siRNAs (1,000 cells per well) were plated on 96‐well plates and grown on 10% serum containing media. MTS was added to cells, and absorbance at 490 nm was measured at day 0, 1, 2, 3, 4, and 5. Cell numbers were estimated as previously described 59.
Colony formation assay
Cells were split and counted. Five hundred cells were seeded into 6‐well plates and cultured with 10% serum containing media for about 12 days. Cells were fixed and stained with crystal violet, and visible colonies were counted.
CRISPR‐Cas9‐mediated genome editing
HCT116 cells were cotransfected with Topo‐Cas9‐IRES‐EGFP and pc3‐U6‐NAT10‐g1‐CMV‐Red or pc3‐U6‐NAT10‐g1‐CMV‐Red. Twenty‐four hours later, the double‐positive cells (green & red) were sorted by flow cytometry and cultured for 3 days. Then, single cells were seeded into 96‐well plates. One month later, independent monoclones were selected, and mutation of NAT10 gene was verified by DNA sequencing and knockdown of NAT10 protein was evaluated by Western blot. The NAT10 knockdown cell lines were maintained in DMEM medium, supplemented with 10% serum. For editing NAT10 gene, the sgRNAs, #1 ACATGCCGAAGGTATTGCCC and #2 CAACGAGACCCACAAGATCC, were used.
Immunofluorescence assay
Cells were plated on coverslips in 6‐well plates and treated as indicated. Then, the cells were fixed with 4% paraformaldehyde for 15 min at room temperature and permeabilized using 0.2% Triton X‐100 for 10 min at room temperature. Cells were then incubated with 10% goat serum for 30 min at 37°C. Primary antibodies were incubated with the cells overnight at 4°C and then washed with PBS for 20 min. FITC‐conjugated anti‐rabbit antibody and TRITC‐conjugated anti‐mouse antibody were added and incubated for 1 h at room temperature. Finally, after a 20‐min PBS washing, cells were stained with DAPI for 15 min at room temperature to visualize the nuclei.
Real‐time quantitative PCR
Real‐time qPCR was performed using the ABI 7500/7500 Fast Real‐Time PCR systems (Applied Biosystems). The human β‐actin and GAPDH were used as internal controls. All real‐time data were analyzed by comparative C t method and normalized to β‐actin or GAPDH.
Protein purification
Flag‐NAT10‐His or its mutants were cloned into pFast‐Bac1. The recombinant baculoviruses were generated with the Bac‐to‐Bac Baculovirus Expression System (Invitrogen). Recombinant proteins were purified from baculovirus‐infected Sf9 cells using Ni‐NTA agarose (Qiagen) according to the manufacturer's instructions. His‐p53, His‐p53‐K120R, and all GST recombinant proteins were expressed in E. coli strain BL21 (DE3; Tiangen), treated with isopropyl‐β‐D‐thiogalactoside to induce fusion protein expression, lysed by sonication, and purified using the Glutathione Sepharose 4B (GE Healthcare) for GST proteins or using Ni‐NTA agarose (Qiagen) for His‐fusion proteins, respectively. Purified proteins were separated on SDS–PAGE followed by Coomassie blue staining and Western blotting.
Sequential immunoprecipitation and LC‐MS/MS
U2OS cells were transfected with plasmids expressing Flag or Flag‐NAT10. Forty‐eight hours later, the cells were collected and lysed in Flag lysis buffer (50 mM Tris–Cl pH 7.4, 150 mM NaCl, 10 mM NaF, 1 mM EDTA, 0.5% Triton X‐100, 10% glycerol, and fresh protease inhibitor cocktail). Cell extracts were immunoprecipitated using anti‐Flag monoclonal antibody M2‐conjugated affinity gel (Sigma) followed by elution with Flag peptide. The eluted material was resolved by SDS–PAGE and visualized by silver staining. The bands were cut from SDS–PAGE gel, fully trypsinized, and analyzed by Q‐Extractive liquid chromatography–tandem mass spectrometry (LC‐MS/MS) using mass spectrometer (Thermo). Mass spectrometry was carried out at Protein Chemistry Facility at the Center for Biomedical Analysis of Tsinghua University, and data were processed using the Proteome Discoverer software (Version 1.4).
Statistical analysis
Student's t‐test was used in all cellular experiments, and the results from three independent experiments are presented as mean ± SEM. The nonparametric Mann–Whitney t‐test was used for human sample analysis. Data collection and analysis were not performed with blinding to the conditions of the experiments. Randomization was followed in all experiments.
Author contributions
XL and XD designed the project; XL, YT, CZ, YZ, LZ and PR performed the experiments; XL, JL, HD, YK and XD prepared the manuscript. All authors contributed to and commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Source Data for Figure 6C
Acknowledgements
We thank Dr. Qihua He for assistance with confocal microscopy, Dr. Hounan Wu for flow cytometry, and Dr. Yan Gao for assisting in mass spectrometric analysis. We also thank Prof. Bo Zhang for providing us the anti‐NAT10 antibody. This work was supported by grants from the National Natural Science Foundation of China (Grant No. 81171877, 81371868, and 81321003), Innovation Team of Ministry of Education (IRT13001), and grant from the 973 Program (Grant No. 2010CB529303).
EMBO Reports (2016) 17: 349–366
References
- 1. Hussain SP, Harris CC (1999) p53 mutation spectrum and load: the generation of hypotheses linking the exposure of endogenous or exogenous carcinogens to human cancer. Mutat Res 428: 23–32 [DOI] [PubMed] [Google Scholar]
- 2. Vogelstein B, Lane D, Levine AJ (2000) Surfing the p53 network. Nature 408: 307–310 [DOI] [PubMed] [Google Scholar]
- 3. Vousden KH, Lu X (2002) Live or let die: the cell's response to p53. Nat Rev Cancer 2: 594–604 [DOI] [PubMed] [Google Scholar]
- 4. Riley T, Sontag E, Chen P, Levine A (2008) Transcriptional control of human p53‐regulated genes. Nat Rev Mol Cell Biol 9: 402–412 [DOI] [PubMed] [Google Scholar]
- 5. Brooks CL, Gu W (2003) Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr Opin Cell Biol 15: 164–171 [DOI] [PubMed] [Google Scholar]
- 6. Honda R, Tanaka H, Yasuda H (1997) Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett 420: 25–27 [DOI] [PubMed] [Google Scholar]
- 7. Montes de Oca Luna R, Wagner DS, Lozano G (1995) Rescue of early embryonic lethality in mdm2‐deficient mice by deletion of p53. Nature 378: 203–206 [DOI] [PubMed] [Google Scholar]
- 8. Jones SN, Roe AE, Donehower LA, Bradley A (1995) Rescue of embryonic lethality in Mdm2‐deficient mice by absence of p53. Nature 378: 206–208 [DOI] [PubMed] [Google Scholar]
- 9. Thut CJ, Goodrich JA, Tjian R (1997) Repression of p53‐mediated transcription by MDM2: a dual mechanism. Genes Dev 11: 1974–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kubbutat MH, Jones SN, Vousden KH (1997) Regulation of p53 stability by Mdm2. Nature 387: 299–303 [DOI] [PubMed] [Google Scholar]
- 11. Momand J, Jung D, Wilczynski S, Niland J (1998) The MDM2 gene amplification database. Nucleic Acids Res 26: 3453–3459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Forslund A, Zeng Z, Qin LX, Rosenberg S, Ndubuisi M, Pincas H, Gerald W, Notterman DA, Barany F, Paty PB (2008) MDM2 gene amplification is correlated to tumor progression but not to the presence of SNP309 or TP53 mutational status in primary colorectal cancers. Mol Cancer Res 6: 205–211 [DOI] [PubMed] [Google Scholar]
- 13. Shibagaki I, Tanaka H, Shimada Y, Wagata T, Ikenaga M, Imamura M, Ishizaki K (1995) p53 mutation, murine double minute 2 amplification, and human papillomavirus infection are frequently involved but not associated with each other in esophageal squamous cell carcinoma. Clin Cancer Res 1: 769–773 [PubMed] [Google Scholar]
- 14. Brooks CL, Gu W (2006) p53 ubiquitination: Mdm2 and beyond. Mol Cell 21: 307–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang Y, Lu H (2009) Signaling to p53: ribosomal proteins find their way. Cancer Cell 16: 369–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Honda R, Yasuda H (1999) Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J 18: 22–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang Y, Xiong Y, Yarbrough WG (1998) ARF promotes MDM2 degradation and stabilizes p53: ARF‐INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 92: 725–734 [DOI] [PubMed] [Google Scholar]
- 18. Dai MS, Lu H (2004) Inhibition of MDM2‐mediated p53 ubiquitination and degradation by ribosomal protein L5. J Biol Chem 279: 44475–44482 [DOI] [PubMed] [Google Scholar]
- 19. Zhang Y, Wolf GW, Bhat K, Jin A, Allio T, Burkhart WA, Xiong Y (2003) Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53‐dependent ribosomal‐stress checkpoint pathway. Mol Cell Biol 23: 8902–8912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dai MS, Zeng SX, Jin Y, Sun XX, David L, Lu H (2004) Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol Cell Biol 24: 7654–7668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brooks CL, Gu W (2010) New insights into p53 activation. Cell Res 20: 614–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sherr CJ (2006) Divorcing ARF and p53: an unsettled case. Nat Rev Cancer 6: 663–673 [DOI] [PubMed] [Google Scholar]
- 23. Gu W, Roeder RG (1997) Activation of p53 sequence‐specific DNA binding by acetylation of the p53 C‐terminal domain. Cell 90: 595–606 [DOI] [PubMed] [Google Scholar]
- 24. Luo J, Su F, Chen D, Shiloh A, Gu W (2000) Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 408: 377–381 [DOI] [PubMed] [Google Scholar]
- 25. Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W (2001) Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 107: 137–148 [DOI] [PubMed] [Google Scholar]
- 26. Luo J, Li M, Tang Y, Laszkowska M, Roeder RG, Gu W (2004) Acetylation of p53 augments its site‐specific DNA binding both in vitro and in vivo . Proc Natl Acad Sci USA 101: 2259–2264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krummel KA, Lee CJ, Toledo F, Wahl GM (2005) The C‐terminal lysines fine‐tune P53 stress responses in a mouse model but are not required for stability control or transactivation. Proc Natl Acad Sci USA 102: 10188–10193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tang Y, Luo J, Zhang W, Gu W (2006) Tip60‐dependent acetylation of p53 modulates the decision between cell‐cycle arrest and apoptosis. Mol Cell 24: 827–839 [DOI] [PubMed] [Google Scholar]
- 29. Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB (2006) Acetylation of the p53 DNA‐binding domain regulates apoptosis induction. Mol Cell 24: 841–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rokudai S, Laptenko O, Arnal SM, Taya Y, Kitabayashi I, Prives C (2013) MOZ increases p53 acetylation and premature senescence through its complex formation with PML. Proc Natl Acad Sci USA 110: 3895–3900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W (2012) Tumor suppression in the absence of p53‐mediated cell‐cycle arrest, apoptosis, and senescence. Cell 149: 1269–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lv J, Liu H, Wang Q, Tang Z, Hou L, Zhang B (2003) Molecular cloning of a novel human gene encoding histone acetyltransferase‐like protein involved in transcriptional activation of hTERT. Biochem Biophys Res Commun 311: 506–513 [DOI] [PubMed] [Google Scholar]
- 33. Kong R, Zhang L, Hu L, Peng Q, Han W, Du X, Ke Y (2011) hALP, a novel transcriptional U three protein (t‐UTP), activates RNA polymerase I transcription by binding and acetylating the upstream binding factor (UBF). J Biol Chem 286: 7139–7148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Larrieu D, Britton S, Demir M, Rodriguez R, Jackson SP (2014) Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science 344: 527–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shen Q, Zheng X, McNutt MA, Guang L, Sun Y, Wang J, Gong Y, Hou L, Zhang B (2009) NAT10, a nucleolar protein, localizes to the midbody and regulates cytokinesis and acetylation of microtubules. Exp Cell Res 315: 1653–1667 [DOI] [PubMed] [Google Scholar]
- 36. Tang Y, Zhao W, Chen Y, Zhao Y, Gu W (2008) Acetylation is indispensable for p53 activation. Cell 133: 612–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lu L, Berkey KA, Casero RA Jr (1996) RGFGIGS is an amino acid sequence required for acetyl coenzyme A binding and activity of human spermidine/spermine N1acetyltransferase. J Biol Chem 271: 18920–18924 [DOI] [PubMed] [Google Scholar]
- 38. Li M, Luo J, Brooks CL, Gu W (2002) Acetylation of p53 inhibits its ubiquitination by Mdm2. J Biol Chem 277: 50607–50611 [DOI] [PubMed] [Google Scholar]
- 39. Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM (2000) Mdm2 is a RING finger‐dependent ubiquitin protein ligase for itself and p53. J Biol Chem 275: 8945–8951 [DOI] [PubMed] [Google Scholar]
- 40. Honda R, Yasuda H (2000) Activity of MDM2, a ubiquitin ligase, toward p53 or itself is dependent on the RING finger domain of the ligase. Oncogene 19: 1473–1476 [DOI] [PubMed] [Google Scholar]
- 41. Kubbutat MH, Ludwig RL, Levine AJ, Vousden KH (1999) Analysis of the degradation function of Mdm2. Cell Growth Differ 10: 87–92 [PubMed] [Google Scholar]
- 42. Lorick KL, Jensen JP, Fang S, Ong AM, Hatakeyama S, Weissman AM (1999) RING fingers mediate ubiquitin‐conjugating enzyme (E2)‐dependent ubiquitination. Proc Natl Acad Sci USA 96: 11364–11369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vousden KH, Prives C (2009) Blinded by the light: the growing complexity of p53. Cell 137: 413–431 [DOI] [PubMed] [Google Scholar]
- 44. Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar‐Sagi D (1999) Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol 1: 20–26 [DOI] [PubMed] [Google Scholar]
- 45. Lohrum MA, Ludwig RL, Kubbutat MH, Hanlon M, Vousden KH (2003) Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell 3: 577–587 [DOI] [PubMed] [Google Scholar]
- 46. Jin A, Itahana K, O'Keefe K, Zhang Y (2004) Inhibition of HDM2 and activation of p53 by ribosomal protein L23. Mol Cell Biol 24: 7669–7680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kurki S, Peltonen K, Latonen L, Kiviharju TM, Ojala PM, Meek D, Laiho M (2004) Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2‐mediated degradation. Cancer Cell 5: 465–475 [DOI] [PubMed] [Google Scholar]
- 48. Meng L, Lin T, Tsai RY (2008) Nucleoplasmic mobilization of nucleostemin stabilizes MDM2 and promotes G2‐M progression and cell survival. J Cell Sci 121: 4037–4046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lee S, Kim JY, Kim YJ, Seok KO, Kim JH, Chang YJ, Kang HY, Park JH (2012) Nucleolar protein GLTSCR2 stabilizes p53 in response to ribosomal stresses. Cell Death Differ 19: 1613–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rubbi CP, Milner J (2003) Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J 22: 6068–6077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Itahana K, Mao H, Jin A, Itahana Y, Clegg HV, Lindstrom MS, Bhat KP, Godfrey VL, Evan GI, Zhang Y (2007) Targeted inactivation of Mdm2 RING finger E3 ubiquitin ligase activity in the mouse reveals mechanistic insights into p53 regulation. Cancer Cell 12: 355–366 [DOI] [PubMed] [Google Scholar]
- 52. Linares LK, Kiernan R, Triboulet R, Chable‐Bessia C, Latreille D, Cuvier O, Lacroix M, Le Cam L, Coux O, Benkirane M (2007) Intrinsic ubiquitination activity of PCAF controls the stability of the oncoprotein Hdm2. Nat Cell Biol 9: 331–338 [DOI] [PubMed] [Google Scholar]
- 53. Inuzuka H, Tseng A, Gao D, Zhai B, Zhang Q, Shaik S, Wan L, Ang XL, Mock C, Yin H et al (2010) Phosphorylation by casein kinase I promotes the turnover of the Mdm2 oncoprotein via the SCF(beta‐TRCP) ubiquitin ligase. Cancer Cell 18: 147–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xu C, Fan CD, Wang X (2015) Regulation of Mdm2 protein stability and the p53 response by NEDD4‐1 E3 ligase. Oncogene 34: 281–289 [DOI] [PubMed] [Google Scholar]
- 55. Metzger MB, Hristova VA, Weissman AM (2012) HECT and RING finger families of E3 ubiquitin ligases at a glance. J Cell Sci 125: 531–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Berndsen CE, Wolberger C (2014) New insights into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol 21: 301–307 [DOI] [PubMed] [Google Scholar]
- 57. Tao W, Levine AJ (1999) P19(ARF) stabilizes p53 by blocking nucleo‐cytoplasmic shuttling of Mdm2. Proc Natl Acad Sci USA 96: 6937–6941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. el‐Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75: 817–825 [DOI] [PubMed] [Google Scholar]
- 59. Yuan J, Luo K, Zhang L, Cheville JC, Lou Z (2010) USP10 regulates p53 localization and stability by deubiquitinating p53. Cell 140: 384–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File
Source Data for Figure 6C