

Abstract
The quality of mitochondria, essential organelles that produce ATP and regulate numerous metabolic pathways, must be strictly monitored to maintain cell homeostasis. The loss of mitochondrial quality control systems is acknowledged as a determinant for many types of neurodegenerative diseases including Parkinson's disease (PD). The two gene products mutated in the autosomal recessive forms of familial early‐onset PD, Parkin and PINK1, have been identified as essential proteins in the clearance of damaged mitochondria via an autophagic pathway termed mitophagy. Recently, significant progress has been made in understanding how the mitochondrial serine/threonine kinase PINK1 and the E3 ligase Parkin work together through a novel stepwise cascade to identify and eliminate damaged mitochondria, a process that relies on the orchestrated crosstalk between ubiquitin/phosphorylation signaling and autophagy. In this review, we highlight our current understanding of the detailed molecular mechanisms governing Parkin‐/PINK1‐mediated mitophagy and the evidences connecting Parkin/PINK1 function and mitochondrial clearance in neurons.
Keywords: mitochondria, mitophagy, Parkin, Parkinson's disease, PINK1
Subject Categories: Autophagy & Cell Death; Neuroscience; Post-translational Modifications, Proteolysis & Proteomics
Glossary
- AAA
ATPases associated with various cellular activities
- APC/C
anaphase‐promoting complex/cyclosome
- ATG
autophagy related
- ATP
adenosine triphosphate
- CCCP
carbonyl cyanide m‐chlorophenyl hydrazine
- CHCHD2
coiled‐coil‐helix‐coiled‐coil‐helix domain‐containing 2
- CRISPR
clustered regularly interspaced short palindromic repeat
- DFCP1
double FYVE domain‐containing protein 1
- DUB
deubiquitinating enzyme
- ER
endoplasmic reticulum
- FACS
fluorescence‐activated cell sorting
- GABARAP
GABA(A) receptor‐associated protein
- GAP
GTPase‐activating protein
- GTP
guanosine triphosphate
- HECT
homologous to the E6AP carboxyl terminus
- IBR
in‐between‐RING
- ITC
isothermal titration calorimetry
- KO
knockout
- LIR
LC3‐interacting region
- LRRK2
leucine‐rich repeat kinase 2
- MDV
mitochondrial‐derived vesicle
- Mfn1/2
mitofusin‐1/2
- MIA
mitochondrial intermembrane space assembly
- Miro1
mitochondrial Rho 1
- MiT/TFE
microphthalmia/transcription factor E
- MPP
mitochondrial processing peptidase
- MPTP
1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine
- MS
mass spectrometry
- mTOR
mammalian target of rapamycin
- NBR1
neighbor of BRCA1 gene 1
- NDP52
nuclear dot protein 52
- NDUFA10
NADH:ubiquinone oxidoreductase subunit A10
- NMR
nuclear magnetic resonance
- PAGE
polyacrylamide gel electrophoresis
- PARL
Presenillin‐associated rhomboid‐like
- PD
Parkinson's disease
- PE
phosphatidylethanolamine
- PI3K
phosphatidylinositol 3‐kinase
- PI3P
phosphatidylinositol 3‐phosphate
- PINK1
PTEN‐induced putative kinase 1
- RBR
RING‐in‐between‐RING
- REP
repressor element of Parkin
- RING
really interesting new gene
- ROS
reactive oxygen species
- shRNA
small hairpin RNA
- TAX1BP1
Tax1 (human T‐cell leukemia virus type I) binding protein 1
- TBK1
TANK1‐binding kinase1
- TFEB
transcription factor EB
- TIM
translocase of the inner mitochondrial membrane
- TOM (TOMM)
translocase of outer mitochondrial membrane
- UBD
ubiquitin binding
- Ubl
ubiquitin‐like
- UBR
ubiquitin protein ligase E3 component n‐recognin
- ULK1
unc‐51 like autophagy activating kinase 1
- UPRmt
mitochondrial unfolded protein response
- USP
ubiquitin‐specific protease
Mitochondria and Parkinson's disease
After symbiosis of α‐proteobacteria in pre‐eukaryotic cells, mitochondria became essential organelles in eukaryotic cells. They not only generate ATP through an electron transport chain system, but also function as scaffolds for many cellular metabolic pathways such as iron‐sulfur cluster biogenesis, amino acid synthesis and lipid metabolism, and regulation of apoptosis. However, in compensation for the cellular energy production and the control of cell homeostasis, mitochondria are presented with a number of obstacles that must be overcome. One of the obstacles is the generation of ROS as a byproduct of the oxidative phosphorylation process, which damages proteins, lipids, and mitochondrial DNAs. Although minor amounts of damage to the mitochondria can be nullified by the redistribution of recycled contents via fusion/fission cycles 1 and/or by intraorganellar quality control such as proteolysis 2, excessive damage will disrupt the membrane potential across the inner membrane, eventually leading to cell death. For these reasons, dysfunctional mitochondria (which sporadically appear with some frequency) with significantly impaired membrane potential must be properly eliminated; otherwise, the condition can lead to a deterioration in cell homeostasis and potentially to the development of neurodegenerative disorders. Of note, post‐mitotic neuronal cells in particular require robust surveillance systems for assessing mitochondrial quality due to their high energy demand.
PD is a highly prevalent neurodegenerative disorder (affects ~2% of those 65 years of age and older) that is clinically characterized by movement‐related symptoms including rigidity, tremor, postural instability, and gait disturbance. The PD motor symptoms result from the massive degeneration of dopaminergic neurons in the substantia nigra, causing a 70–80% depletion in dopamine levels 3. In the remaining neuronal cells, cytosolic protein aggregates called Lewy bodies, the primary structural component of which is α‐synuclein, can be observed 4. Since the majority of PD is sporadic, it is quite difficult to ascertain a clear pathogenic mechanism. Despite occurring with much less frequency, however, autosomal dominant and recessive genetic forms of PD have been identified among early‐onset parkinsonism patients. Over the past 15 years, genetic researchers have identified a diversity of genes that either contribute to monogenic forms of PD or contribute as risk factors to the development of the disorder 5. For example, the genes SNCA (α‐synuclein) and LRRK2 function in autosomal dominant PD, while the genes PARKIN, PINK1, and DJ‐1 are causal for recessive PD. To date, nearly 30 distinct chromosomal regions implicated in the complex etiology mechanism have been identified even in the familial cases of PD; however, recent molecular studies have unequivocally shown the critical roles played by Parkin and PINK1 6.
Parkin was first identified in 1998 as a gene product mutated in recessive forms of familial parkinsonism in Japanese patients 7. PARKIN encodes a 465‐aa protein that belongs to the E3 ubiquitin ligase enzyme family 8, 9, 10. To date, more than 100 loss‐of‐function mutations have been identified in the PARKIN gene with many mutations prevalent among familial cases in which onset of the disorder occurs in those younger than 30 years old.
The second most common gene product associated with autosomal recessive juvenile parkinsonism is PINK1, which was initially identified in 2001 from an Italian family 11 and then later described in several European families 12. PINK1 is a 581‐aa protein expressed ubiquitously 13. According to the amino acid sequence, PINK1 harbors a mitochondrial targeting sequence followed by a hydrophobic segment at the N‐terminus and a large kinase domain at the C‐terminus.
Potential linkage between mitochondrial dysfunction and PD emerged from multiple lines of evidence 14. For example, uptake of MPTP (a byproduct of “synthetic heroin”) 15, paraquat 16, and rotenone 17 (chemical herbicide and pesticide), all of which cause a deficiency in mitochondrial respiratory chain function, led to parkinsonian symptoms in humans and animal models. Recently, another causative gene of the autosomal dominant form of familial PD, CHCHD2, was identified from a genome‐wide linkage analysis 18. The gene encodes a 151‐aa protein with twin CX9C motifs that localize the protein in the mitochondrial intermembrane space. Interestingly, the mitochondrial intermembrane space assembly (MIA) pathway involved in import of the precursor and subsequent maturation of Mix17 (the yeast homologue of CHCHD2) is related to electron transfer to the respiratory chain complex 19, 20. Although further functional studies are required, CHCHD2 is the first mitochondrial causal gene product, mutation of which can affect mitochondrial integrity through functional depression of the respiratory complexes.
In addition, Drosophila studies have shown that the loss of either parkin or pink1 function results in phenotypes similar to mitochondrial impairments such as muscle degeneration and male sterility 21, 22, 23, 24. Overexpressed Parkin can partially compensate for some pink1 loss‐of‐function (i.e. mitochondrial abnormality). This suppression is not derived from the simple protection of apoptotic cell death. Mutant flies that have lost both parkin and pink1 do not exhibit stronger phenotypes than those seen with either mutant alone. These in vivo studies strongly suggest that Parkin and PINK1 function in a common pathway that maintains mitochondrial integrity.
Cytosolic E3 ligase Parkin is recruited to damaged mitochondria for autophagic degradation
Ubiquitin plays pivotal roles in many different cellular functions including protein degradation, signaling, endocytosis, and the immune system. While E1s (ubiquitin‐activating enzymes) and E2s (ubiquitin‐conjugating enzymes) activate ubiquitin via thioester intermediates, E3 ubiquitin ligases, the final enzymes in the ubiquitination cascade, transfer the ubiquitin moiety from the E2 to a lysine residue on protein substrates 25, 26, 27. While at least 1 E1 and about 40 E2s are encoded in the human genome, the abundance and diversity of E3 ligases (roughly 500–1,000, but we cannot determine the exact number because of subset diversity) is striking with an even greater number of proteins thought to undergo ubiquitination. This abundance indicates that E3 ligases are the key factors in providing the substrate specificity essential to the ubiquitin network. Because ubiquitin itself can also serve as a ubiquitination site to form polymeric ubiquitin chains, different chain linkages can be formed. MS‐based proteomic and biochemical approaches showed that all lysine residues (K6, K11, K27, K29, K33, K48, and K63) 28 and an N‐terminal methionine residue 29 can serve as ubiquitination sites. In fact, the K48‐linked chain, the most common and abundant ubiquitin chain, has been identified as a signal for proteasome‐mediated degradation, while the K63‐linked chain induces clearance of the substrate protein via the autophagy‐lysosome pathway 30, 31, 32 and activates the DNA damage response 33. The E3 ligases are classified into at least four types: HECT (homologous to the E6AP carboxyl terminus), RING (really interesting new gene), U‐box, and RBR (RING‐in‐between‐RING). Although RBR‐type ligases contain two RING domains (RING1 and RING2), they receive ubiquitin on a cysteine in the RING2 domain via a thioester intermediate like HECT, functioning as HECT/RING hybrids 34. Another feature of RBR ligases is that their ligase activities are normally autoinhibited.
Parkin is an RBR‐type E3 ligase that normally localizes in the cytosol as an autoinhibited form. Parkin was first proposed to ubiquitinate misfolded proteins for proteasome‐dependent degradation. While this hypothesis may have explained the accumulation of protein aggregates in neuronal cells of PD patients, it could not explain the relationship between Parkin and mitochondrial integrity observed in the Drosophila studies.
In 2008, Richard Youle's group reported that cytosolic Parkin is recruited to damaged mitochondria for its degradation through an autophagy pathway, which undoubtedly opened a new research field termed Parkin‐mediated mitophagy 35. When mitochondria lose their membrane potential following the addition of a chemical compound like CCCP (see Box Box 1), cytosolic Parkin is recruited to the damaged mitochondria. Once Parkin reaches the mitochondrial outer membrane, its E3 activity is fully activated 36 and various mitochondrial outer membrane proteins are ubiquitinated 37 (the detailed mechanism is discussed later). Because of the robust ubiquitination, p97 and proteasomes are also recruited to mitochondria and a portion of the outer membrane proteins, such as Mfn1/Mfn2, is thought to be degraded via the proteasome 38. Mfn1/Mfn2 are integrated in the outer membrane exposing the large GTPase domain to the cytosol for involvement in the mitochondrial fusion 39. While the necessity of proteasomal degradation for downstream autophagy activity and clearance of damaged mitochondria remains a matter of debate 40, 41, rapid poly‐ubiquitination of Mfn1/Mfn2, which occurs within 1 h of CCCP treatment, is important to segregate damaged mitochondria from the healthy mitochondrial network 42. Hexameric AAA+ ATPase p97 (also known as VCP) is a multifunctional protein primarily involved in ubiquitin‐dependent proteolysis 43. While different cofactors that bind to p97 specify the distinct localization and function 44, the Npl4/Ufd1 heterodimer contributes to some degree to the degradation of outer membrane proteins during mitophagy 45, 46. In yeast, another cofactor termed Vms1 was reported to recruit Cdc48 (yeast homologue of p97) to mitochondria following mitochondrial oxidative stress 47. Vms1 is highly conserved from yeast to humans, but mammalian Vms1 function especially in Parkin‐mediated mitophagy remains unclear. In addition to Mfn1/Mfn2, Miro1 (involved in mitochondrial transport along microtubules), MitoNEET/CISD1 (a 2Fe‐2S containing protein), and TOMM70 (an import receptor of mitochondrial precursor proteins), all of which are integral mitochondrial proteins, were identified in quantitative proteomic approaches as proteins that are significantly reduced in response to Parkin recruitment 41, 48. Miro1 contains GTPase and EF‐hand domains that are crucial for connecting mitochondria to the microtubule network through associations with its binding partners, Milton and kinesin heavy chain. Studies using primary neurons revealed that rapid Miro1 degradation arrests microtubule‐dependent mitochondrial trafficking, thereby preventing damaged mitochondria from moving, especially to the axon terminal 49. Although Mfn1/Mfn2 and Miro1 were reported to be poly‐ubiquitinated by endogenous Parkin 38, 49, 50, 51, 52, the physiological importance underlying degradation of the other Parkin substrates is unknown. Because a broad group of outer membrane proteins are ubiquitinated by both endogenous and exogenous Parkin following CCCP treatment 37, it is clear that Parkin does not possess rigorous substrate specificity. This characteristic is compatible with a positive feedback ubiquitination amplification model (discussed later). Prolonged CCCP treatment (for 24–48 h) selectively degrades the mitochondria in an autophagy‐dependent manner. Since the first report by Richard Youle's group, many other groups have confirmed Parkin recruitment to damaged mitochondria and have elucidated the molecular mechanism leading to Parkin recruitment and the subsequent clearance of mitochondria.
Box 1.
We would like to highlight the various tools that have been utilized so far in elucidating the mechanism of Parkin‐/PINK1‐mediated mitophagy.
- Chemical compounds triggering Parkin translocation:
-
CCCPCarbonyl cyanide m‐chlorophenyl hydrazine (CCCP) is an ionophore that disrupts the mitochondrial proton gradient by allowing protons to cross lipid bilayers. CCCP is the chemical compound most frequently used to trigger PINK1 accumulation following Parkin translocation. However, CCCP affects lysosomal and Golgi pH and LC3 lipidation in a Parkin‐independent manner 158, 159, 160, 161. Therefore, interpretation of data on autophagy function during CCCP‐induced mitophagy should be carefully considered.
-
ValinomycinValinomycin is a potassium‐selective ionophore that accelerates the transport of potassium ion across the membrane. In the presence of valinomycin, mitochondria take up potassium at the expense of the proton gradient, resulting in dissipation of the membrane potential. Similar to CCCP, valinomycin induces extensive PINK1 accumulation and Parkin translocation.
-
Antimycin AAntimycin A binds to the Qi site of cytochrome c reductase in complex III and inhibits electron transfer from cytochrome b to cytochrome c, which leads to a collapse in the membrane potential 162. Antimycin A alone or in combination with oligomycin (an inhibitor of the FoF1 ATP synthase) also induces Parkin translocation.
-
Paraquat
-
Rotenone
-
-
Mito‐KillerRed
A genetically encoded photo‐sensitizer named KillerRed is a dimeric red fluorescent protein developed from the hydrozoan chromoprotein anm2CP 165. Photo‐activation of KillerRed with light at 540–580 nm can generate ROS. Therefore, mitochondria‐targeted KillerRed (mito‐KillerRed) can induce ROS‐mediated damage in the matrix in a spatiotemporally controlled manner that more closely mimics physiological conditions than CCCP treatment. Photo‐activating mito‐KillerRed in a specific area was reported to induce local Parkin recruitment 156, 166, 167, 168.
-
ΔOTC
ΔOTC is an ornithine transcarbamylase mutant lacking 85 aa (residues 30–114) from the N‐terminus and is a model substrate for activating the mitochondrial unfolded protein response (UPRmt) in mammalian cells 169 and in flies 170. It can be targeted to the mitochondria as well as wild‐type OTC, but forms insoluble aggregates in the matrix. When expressing ΔOTC in cultured cells, PINK1 accumulation and subsequent Parkin translocation were observed without the loss of membrane potential, probably due to clogging of the TIM23 translocation channel 171.
-
Mito‐Keima
Keima is a novel fluorescent protein probe used to detect autolysosome formation. While Keima has an emission peak at 620 nm, the excitation spectrum varies under different pH conditions (Ex 440 nm in a neutral state and Ex 586 nm in an acidic state). Therefore, Keima fused with a mitochondrial matrix targeting signal (mito‐Keima) in conjunction with fluorescent microscopy can function as a reporter for delivery of damaged mitochondria to the lysosome 172. Furthermore, when combined with FACS, a small amount of mitophagy can be monitored quantitatively in an unbiased way 131, 147.
-
Phos‐tag PAGE
Phos‐tag is a molecule that selectively binds to phosphorylated ions 173. SDS–PAGE containing an acrylamide‐pendant Phos‐tag ligand (Phos‐tag PAGE) can separate phosphorylated and non‐phosphorylated forms of proteins such as PINK1, Parkin, and ubiquitin 62, 68.
-
Recombinant Ser65‐phosphorylated ubiquitin
Using the amber suppression technique, it is possible to incorporate phosphoserine (as well as its non‐hydrolyzable analog) at a desired position in the target protein in bacterial cells, for example, Ser65‐phosphorylated ubiquitin 174, 175, 176. Thus, milligram quantities of the recombinant protein free from contamination of the non‐phosphorylated form can be prepared, as previously used to demonstrate Parkin activation 79.
-
Phosphorylated ubiquitin antibody
Novel antibodies recognizing Ser65‐phosphorylated ubiquitin, but not non‐phosphorylated ubiquitin, confirm that the phosphorylated ubiquitin signal increases with mitochondrial stress in a PINK1‐dependent manner under endogenous conditions. Importantly, the signal is detected in human postmortem brain sections of the substantia nigra and increases with age and disease. Therefore, this can be used as a potential biomarker for PD 177.
PINK1 as a mitochondrial stress sensor
PINK1 functions upstream of Parkin recruitment 36, 53, 54, 55, 56. Following disruption of the mitochondrial membrane potential, the serine/threonine kinase PINK1 switches the import pathway from the inner membrane to the outer membrane where associations with the TOM complex stabilize PINK1 on the outer membrane 57 (Fig 1). The TOM complex is a major protein translocator complex consisting of multi‐subunits 58, 59. Knockout (KO) studies revealed that one of the subunits of the TOM complex, TOMM7, is essential for inserting PINK1 into the outer membrane via the TOM complex (Fig 1). TOMM7‐KO cells though retain normal protein import efficiencies into the matrix or inner membrane from the cytosol 60. When PINK1 is stabilized on the outer membrane, it forms a large complex (~700 kDa on blue‐native PAGE) comprising the TOM machinery and at least two PINK1 molecules 57, 61. PINK1 dimers contribute to the intermolecular autophosphorylation of residues S228 and S402 that will be conformationally close to one another 62, 63 (Fig 1). Inhibiting autophosphorylation prevents Parkin translocation despite accumulation on the outer membrane, suggesting that PINK1 is not only quantitatively but also qualitatively regulated. Unexpectedly, residues 34–90, rather than the PINK1 transmembrane segment, are required for outer membrane localization in damaged mitochondria. This unique signal presumably interacts with the TOM complex when the primary targeting signal is blocked 64. The “association” of PINK1 with the TOM complex though is dispensable for Parkin recruitment to the mitochondria since ectopically targeted PINK1 can recruit cytosolic Parkin to other organelle membranes, such as the peroxisome or lysosome, that lack the TOM machinery 57. Furthermore, this also suggests that mitochondrial proteins (if they are specifically on mitochondria) are dispensable; the exceptions are PINK1 and the import machinery that is essential for PINK1 outer membrane localization for Parkin translocation.
Figure 1. PINK1 accumulation in the outer membrane of the damaged mitochondria.
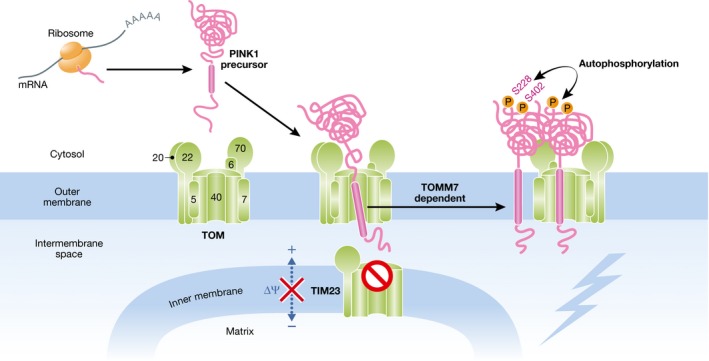
The newly synthesized PINK1 precursor is targeted to the damaged mitochondria. Because of the loss of membrane potential, the PINK1 precursor is not allowed to enter the inner membrane via the TIM23 complex. Instead, the PINK1 precursor is inserted into the outer membrane through the TOM complex in a TOMM7‐dependent manner. PINK1 stabilized on the outer membrane then forms a large complex with the TOM complex and undergoes intermolecular autophosphorylation at residues S228 and S402. 5, 6, 7, 20, 22, 40, and 70 denote TOMM5, TOMM6, TOMM7, TOMM20, TOMM22, TOMM40, and TOMM70, respectively.
Molecular mechanism of Parkin recruitment
What molecular mechanism drives Parkin recruitment to the damaged mitochondria? The answer of this key question has been solved incrementally. The first clue was that PINK1 kinase activity is essential for Parkin recruitment. Kinase‐inactivated PINK1 cannot recruit Parkin even when it accumulates on the mitochondria. Therefore, Parkin stable recruitment is mediated by the enzymatic (phosphorylation) activity of PINK1. The second clue came from the fact that Parkin E3 ligase activity itself is also essential for Parkin stable recruitment. As Parkin is a member of the RBR‐type E3 ligases, a ubiquitin molecule is transferred from the E2 to the conserved and catalytic Cys431 of Parkin via thioester formation before loading ubiquitin onto the substrate. A Parkin C431A mutation inhibits stable translocation to the damaged mitochondria 65, 66, 67. Furthermore, monitoring ubiquitination of N‐terminally fused GFP as a pseudosubstrate revealed that pathogenic mutations such as K161N, K211N, and T240R that impede Parkin translocation to mitochondria also disable the E3 ligase activity 36, 67.
In 2012, the Hattori and Muqit groups independently reported that PINK1 phosphorylates Ser65 in the Ubl (ubiquitin‐like) domain of Parkin. Shiba‐Fukushima et al 68 monitored the phosphorylation status of Parkin by phos‐tag PAGE (see Box Box 1) following disruption of the membrane potential and found that the Parkin Ser65 residue was phosphorylated in a PINK1‐dependent manner. Kondapalli et al tested whether catalytically active PINK1 69 directly phosphorylates various PD‐associated proteins in vitro and identified Parkin Ser65 as a PINK1 phosphorylation site. Muqit and our groups also observed that phosphorylated Parkin enhances E3 ligase activity, consistent with the idea that Parkin ligase activation and recruitment are coupled 70, 71. However, observations that Parkin translocation was not completely inhibited by a phosphorylation‐deficient S65A mutation raised the possibility that another PINK1 substrate was needed for Parkin translocation.
Early in 2014, three groups independently reported that PINK1‐mediated phosphorylation of ubiquitin at Ser65 plays an important role in Parkin activation 72, 73, 74. Similar findings were reported later 67, 75. Phosphorylation and ubiquitination are two major post‐translational modifications, and this finding represented the first example in which ubiquitin, a post‐translational modifier itself, also underwent phosphorylation 76. Kane et al first found that when overexpressed, all Ser/Thr residues conserved in human and Drosophila Parkin and even the whole Ubl domain where the Ser65 residue resides are not essential for the recruitment, and then identified a PINK1‐dependent phosphorylated ubiquitin peptide by MS. Kazlauskaite et al and Koyano et al also utilized MS approaches. The latter group built on structural and sequence similarities between ubiquitin and the Parkin Ubl domain to facilitate MS‐based confirmation of ubiquitin Ser65 phosphorylation. These groups reached the following conclusions: Endogenous PINK1 phosphorylates ubiquitin at Ser65 and phosphorylated ubiquitin activates Parkin E3 ligase activity 67, 75, 77. Both overexpression of a phosphorylation‐deficient S65A ubiquitin mutant 74 and replacement of endogenous ubiquitin with a S65A mutant 78, 79 inhibited Parkin translocation, indicating that ubiquitin phosphorylation by PINK1 is essential for Parkin stable recruitment. Furthermore, since PINK1 phosphorylates poly‐ubiquitin chains 67, 75, 80 and phosphorylated Parkin tightly binds phosphorylated poly‐ubiquitin chains 67, 78, 79, a positive feedback ubiquitination cycle that accelerates both Parkin translocation and poly‐ubiquitin chain formation on the surface of the damaged mitochondria was proposed 67, 79 (Fig 2). A few hours of depletion of the mitochondrial membrane potential can recruit most of the cytosolic Parkin onto the mitochondria even when overexpressed. Only a positive feedback amplification process can explain this robust translocation mechanism.
Figure 2. Positive feedback ubiquitination cycles induced by Parkin and ubiquitin chain formation on damaged mitochondria.
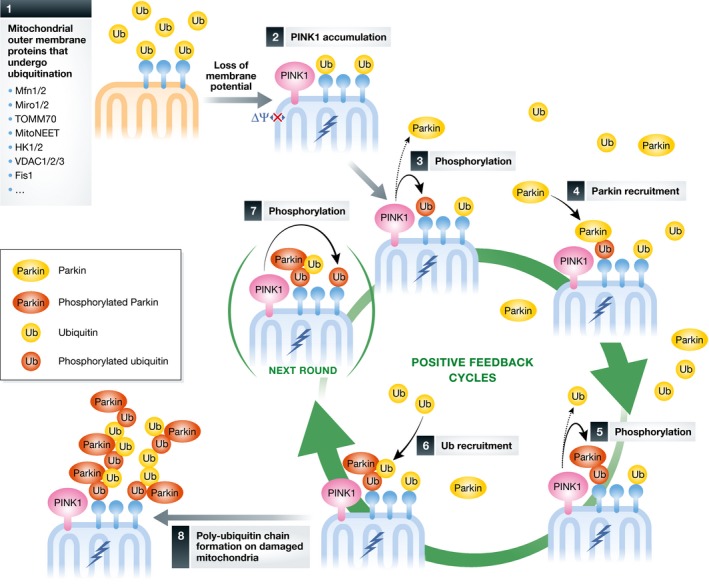
Although most of the ubiquitin diffuses in the cytosol, a fraction should reside on the outer membrane of healthy mitochondria since the ubiquitin system also contributes to the turnover of mitochondrial proteins under normal conditions 178 (Step 1). Following dissipation of the membrane potential, PINK1 is stabilized on the damaged mitochondria (Step 2). PINK1 can then phosphorylate the ubiquitin that is conjugated to the mitochondrial proteins, or PINK1 may also phosphory‐late Ser65 of cytosolic Parkin (Step 3). Of note, phosphorylated ubiquitin stably stays on the mitochondria because hydrolysis of phosphorylated ubiquitin chain by DUBs is impaired 75. Through higher affinity with phosphorylated ubiquitin, cytosolic Parkin is recruited to and retained on the mitochondria (Step 4). PINK1 further phosphorylates Parkin on the mitochondria. PINK1 may also phosphorylate Ser65 of cytosolic ubiquitin (Step 5). The fully activated, phosphorylated Parkin can then elongate ubiquitin chains or generate a new ubiquitinated substrate from cytosolic‐free ubiquitin. In other words, cytosolic ubiquitin is recruited to the mitochon‐dria through a ubiquitination reaction by activated Parkin (Step 6). The ubiquitin on the mitochondrial substrate is phosphorylated by PINK1 (Step 7 is the next round of the Step 3). Positive feedback amplification cycles (Steps 3–7) result in both Parkin and ubiquitin recruitment to and poly‐ubiquitin chain formation on the damaged mitochondria (Step 8).
Several deubiquitinating enzymes (DUBs) that counteract Parkin E3 ubiquitin ligases by catalyzing the removal of ubiquitin from substrates have been reported to regulate mitophagy. USP30 is a mitochondria‐anchored DUB that specifically cleaves Parkin‐generated K6‐ and K11‐ and K63‐linked ubiquitin chains on mitochondria 67, 81 and in vitro 75. USP30 overexpression inhibits mitophagy, whereas USP30 shRNA or overexpression of enzymatically inactive USP30 enhances mitophagy, indicating that USP30 directly opposes Parkin function. Moreover, knockdown of Drosophila USP30 largely rescued mitochondrial morphology defects in the flight muscles caused by parkin or pink1 mutants and reversed defects in dopamine levels against paraquat treatment in vivo 82. USP8, which normally regulates endosomal trafficking 83, has also been proposed as a regulator of Parkin recruitment to mitochondria 84. siRNA knockdown of USP8 impaired the recruitment of Parkin to damaged mitochondria and up‐regulated Parkin levels 84. While USP8 deubiquitinates the K6‐linked ubiquitin chain on Parkin, it does not hydrolyze ubiquitinated chains on mitochondrial substrates. Overexpression of USP15 was reported to reduce K48‐ and K63‐linked ubiquitin chains on damaged mitochondria in response to mitophagy 85. The antagonistic relationship between Parkin and USP15 was further investigated in a fly model in vivo. The physiological roles of DUB, in particular how Parkin and DUBs coordinately regulate mitochondrial fidelity, however, remain to be fully investigated.
Structural insights into Parkin E3 activity
Parkin structurally consists of Ubl, RING0 (also referred to as unique Parkin domain/UPD), RING1, IBR (in‐between‐RING), and RING2 domains. Crystal structures including full‐length rat Parkin 86 and Ubl‐deleted human Parkin 87, 88 revealed an autoinhibited state. The Ubl domain has a similar structural fold to ubiquitin and the Ser65 residue phosphorylated by PINK1 is also conserved. RING0, the zinc finger fold unique to Parkin, is connected to the Ubl domain through a structurally disordered linker, the length and sequence of which diverge across multiple species. Three additional zinc finger fold domains RING1, IBR, and RING2 form a minimal functional unit for RBR‐type ligases. A conserved two‐turn helix linker termed REP (repressor element of Parkin) is found between the IBR and RING2 domains. Based on the crystal structure, the E2 binding site in the RING1 domain is occluded by REP and the catalytic Cys431 in the RING2 domain where ubiquitin is transferred from E2 enzyme is buried in the RING0 domain. Parkin E3 ubiquitin ligase activity was likewise reported to be normally autoinhibited by the N‐terminal Ubl domain 89. This finding was consistent with the improved human Parkin structure following deletion of the linker between Ubl and RING0 90, 91. The Ubl domain mainly interacts with the α‐helix (261–274 aa) of the RING1 domain, which spatially blocks E2 enzyme accessibility.
Because complete activation of the Parkin E3 ligase necessitates binding of phosphorylated ubiquitin, it is likely that this binding triggers a conformational change in Parkin. However, the interaction between Parkin and phosphorylated ubiquitin does not utilize the catalytic Cys431 of Parkin or the C‐terminal glycine residue in ubiquitin, indicating that phosphorylated ubiquitin is not loaded onto Parkin Cys431 for activation 67, 72, 75, 79. How does phosphorylated ubiquitin bind to Parkin? Recently, five independent groups concurrently identified Parkin amino acid residues that are crucial for phosphorylated ubiquitin binding 90, 91, 92, 93, 94. A crystal structure of the Pediculus humanus (a species of louse that infects humans) Parkin complexed with phosphorylated ubiquitin via disulfide covalent linkages showed that A152, H304, A307, and Y314 residues of P. humanus Parkin (corresponding to K151, H302, R305, and Y312 residues in human Parkin, respectively) form a pocket that interacts with the phosphorylated serine in ubiquitin 93. Site‐specific photo‐crosslinking methods combined with MS and computational modeling also identified the same binding surface between phosphorylated ubiquitin and full‐length rat Parkin 94. When the phosphate‐binding pocket is mutated, the affinity for phosphorylated ubiquitin and Parkin translocation onto the damaged mitochondria were impeded. Moreover, the L283P, G284R, and C352G pathogenic Parkin mutations in the IBR and RING1 domains inhibited interactions with phosphorylated ubiquitin 94. One of the dynamic conformational changes is that a kinked helix at Gly319 in inactivated Parkin rearranges to form a straight long α‐helix when binding to phosphorylated ubiquitin. This affects the position of the IBR domain, which will in turn stretch the IBR‐REP linker and unlock the inhibitory interactions between the E2 binding region and the REP, and the interactions between Cys431 and RING0 93. Furthermore, the Parkin conformational change upon phosphorylated ubiquitin binding also enhances Parkin Ser65 phosphorylation by PINK1 91, 92, 93. Mutational analyses, NMR, and ITC‐based experiments showed that binding phosphorylated ubiquitin promotes dissociation of the Ubl domain from the α‐helix (amino acids 261–274, the opposite side of the phosphorylated ubiquitin binding region) in the RING1 domain. This further contributes to enhanced affinity for the E2 enzyme 79, 90, 91, 92, 93. In cells, phosphorylation‐dependent dissociation of the Ubl domain may have additional roles in mitophagy (e.g. association of the proteasome through Rpn13) 95.
In summary, despite structural and sequence similarities between ubiquitin and the Ubl domain of Parkin, they function antagonistically. Binding phosphorylated ubiquitin to Parkin and dissociation of phosphorylated Ubl from the Parkin core allosterically induce a Parkin conformational change from the intramolecular inhibited state to the maximal E3 active state, which was established and quantitatively measured by proteomics 79.
In sharp contrast to Parkin, there is limited information on the PINK1 structure. PINK1 possesses several motifs conserved among protein kinases. Homology modeling has been used to aid PINK1 structural prediction 96, 97, but no NMR or crystal structures of PINK1 from any species have been solved. Because PINK1 efficiently phosphorylates ubiquitin and the Parkin Ubl domain, it should recognize the ubiquitin‐fold as a substrate. Furthermore, Ubl or ubiquitin I44A mutations, or replacement of amino acid residues around the I44 patch of ubiquitin with an unnatural amino acid harboring a bulky side chain, inhibited Ser65 phosphorylation, suggesting that this region is functionally important 93, 94.
Ubiquitin system for PINK1 degradation
As mentioned above, PINK1 is the key factor in activating the ubiquitin system by phosphorylating both Parkin and ubiquitin in mitophagy. In addition to this, the ubiquitin system is also essential for rapid degradation of PINK1 in healthy mitochondria. PINK1 has a classical N‐terminal mitochondrial targeting sequence followed by a hydrophobic transmembrane segment. Once PINK1 is synthesized on cytosolic ribosomes, it is targeted to the mitochondria. The TOM complex recognizes the N‐terminal mitochondrial targeting sequence and allows the substrate to cross the outer membrane. N‐terminal presequence‐containing substrates are then transferred to the TIM23 complex (protein translocator in the inner membrane) for crossing or entering the inner membrane 98. In the case of the PINK1 precursor, the N‐terminal presequence is cleaved by MPP in the matrix, and the following hydrophobic segment is captured by the TIM23 complex, inserted into the inner membrane, and subjected to the second cleavage by PARL between A103 and F104 99, 100, 101, 102, 103. Interestingly, PARL cleavage releases the cleaved PINK1 back to the cytosol where the newly exposed N‐terminal phenylalanine residue is recognized by UBR E3 ligases 104 and rapidly degraded by the proteasome via the N‐end rule pathway 105 (Fig 3A). Protein translocation via the TOM/TIM23 pathway is believed to occur at the contact site where the distance from the outer membrane to the matrix is ~50–60 amino acid residues long 106, 107. Consequently, the large C‐terminal kinase domain remains outside the mitochondria if the transmembrane segment is arrested for an extended period of time in the TIM23 complex. Rhomboid proteases including PARL preferentially recognize a helix‐breaking glycine‐/proline‐rich segment, which is found in human PINK1 108. On the other hand, a proline residue in the transmembrane segment disfavors the lateral release from the TIM23 complex 109. These features may determine the unique PINK1 retrotranslocation pathway. Alternatively, other proteases in the mitochondria such as ClpXP and m‐AAA may also function in rapid PINK1 turnover 103 (Fig 3A).
Figure 3. Ephemeral life of PINK1 in the healthy mitochondria.
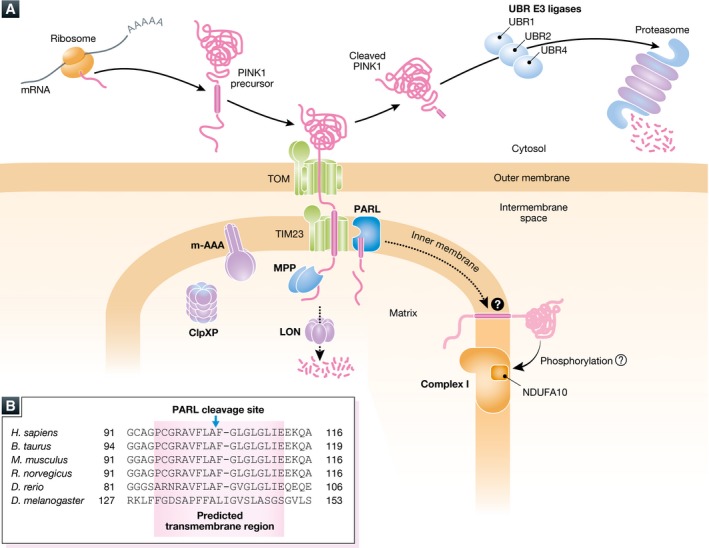
(A) The newly synthesized PINK1 precursor on the cytosolic ribosomes is targeted to the mitochondria. After crossing the outer membrane through the TOM complex, the N‐terminal mitochondrial targeting sequence is cleaved by MPP in the matrix. The following transmembrane segment is recognized by the TIM23 complex and received second cleavage by PARL between A103 and F104. Most of the cleaved PINK1 is released to the cytosol where the newly N‐terminal phenylalanine residue of the cleaved PINK1 is recognized by the N‐end rule UBR ligases (UBR1, UBR2, and UBR4 that preferentially recognize type‐2 N‐degrons) for proteasomal degradation. A matrix ATPase associated with diverse cellular activities (m‐AAA) protease is composed of AFG3L2 and paraplegin and has the active site oriented toward the matrix. ClpXP is a matrix ATP‐dependent protease composed of hetero‐oligomeric ATP‐binding subunits and proteolytic subunits. m‐AAA and ClpXP also participate in PINK1 degradation. Another ATP‐dependent LON protease also contributes, particularly in Drosophila, to PINK1 degradation in the matrix. As phosphorylation of NDUFA10 in the respiratory chain complex I is reduced in Pink1‐KO mice, a small amount of PINK1 retained in the inner membrane might be involved in the maintenance of complex I through phosphorylation. (B) Amino acid sequence alignment of the transmembrane region of PINK1. Amino acid sequences of the predicted transmembrane segments (pink‐colored box) from the indicated species are shown 99. The transmembrane regions are well conserved from zebrafish (Danio rerio) and humans (Homo sapiens), while the transmembrane segment in fly (Drosophila melanogaster) is less conserved with fewer glycine/proline residues. The PARL cleavage site of human PINK1 between A103 and F104 is also shown.
While most of the PINK1 is degraded by the proteasome, the possibility that a small fraction of the PINK1 stays in the healthy mitochondria cannot be excluded. Indeed, phosphorylation of Ser250 in the complex I subunit, NDUFA10, is reduced in the liver and brains of Pink1‐KO mice 110. Although whether PINK1 directly phosphorylates NDUFA10 remains unclear, deficiencies in complex I have been found in PD patients 111, 112. Furthermore, the LON protease in the mitochondrial matrix is reported to degrade PINK1 in Drosophila 113. The transmembrane segment and PARL cleavage site of PINK1 are well conserved from zebrafish to humans, but less so in fly, suggesting that the PINK1 degradation pathway varies depending on the species (Fig 3B).
In summary, a continuous process of mitochondrial import and degradation maintain PINK1 at extremely low levels on healthy mitochondria. In other words, this system functions as a sensitive sensor for the rapid detection of mitochondrial damage.
Autophagy
Autophagy is essential for Parkin‐mediated mitochondrial degradation because ATG5‐KO cells impede the elimination of the damaged mitochondria even when Parkin is recruited to them 35. Autophagy is a major intracellular degradation system that sometimes functions in consort with the ubiquitin‐proteasome system 114, 115. Both autophagy and proteasomes are well conserved in eukaryotes, but autophagy can encapsulate cytoplasmic materials including bigger protein aggregates and/or unwanted organelles for bulk degradation through the fusion with the lysosome, while proteasomes degrade ubiquitinated protein substrates singly. Although autophagy was traditionally regarded as a non‐selective process to keep up with the demand for energy under starvation conditions, evidence is accumulating that indicates many different types of selective autophagy for eliminating specific unwanted organelles, such as peroxisomes (pexophagy) and damaged mitochondria (mitophagy), and infecting pathogens (xenophagy) 116, 117, 118. An astonishing number of in vivo studies indicate that autophagy deficiency is associated with many diseases such as neurodegenerative disorders, cancer, microbial infection, and aging 119.
So far, numerous proteins that are essential for autophagy have been identified, many of which are evolutionarily conserved from yeast to humans 120, 121. These Atg proteins form several functional units. The most upstream autophagy regulator is the ULK1 complex, which consists of ULK1, Atg13, FIP200, and Atg101 in mammals. In starvation‐induced autophagy, mTOR negatively regulates the ULK1 complex through phosphorylation of ULK1 and Atg13. Following Parkin translocation, the ULK1 complex transiently forms foci on the mitochondria (or contact site between mitochondria and ER) 122 (Fig 4). mTOR1 inhibition was reported to induce mitophagy in consort with the loss of membrane potential 123. Another autophagy regulator, the PI3K complex, is also transiently recruited to mitochondria in an early stage of mitophagy. The PI3K complex, which generates PI3P, consists of Beclin1, Atg14L, Vps15, and Vps34 in mammals, although Beclin1‐Vps15‐Vps34 also forms other stable complexes with UVRAG and/or Rubicon for endosomal trafficking 124, 125. DFCP1 diffusely localizes on the ER and Golgi membranes under normal conditions, but upon mitophagy stimulation, it extensively accumulates at a spot where the ER and mitochondria are contacted (Fig 4). Since the FYVE domain in DFCP1 binds to PI3P, DFCP1 may serve as an appropriate reporter of PI3P‐enriched isolation membranes at downstream of the ULK1 and PI3K complexes 126, 127. Atg9A is the only known multi‐spanning membrane protein among the essential Atg core proteins and behaves uniquely. Atg9A resides on a small vesicular structure that shuttles between the cytosol, trans‐Golgi network, and endosomes under normal conditions. The two ubiquitin‐like conjugation systems, Atg5‐12 and PE (phosphatidylethanolamine)‐LC3, are important for elongation and/or complete encapsulation of the isolation membrane. The C‐terminal glycine in Atg12 is activated by Atg7 (E1‐like) and Atg10 (E2‐like) and is finally covalently conjugated to Atg5. In contrast, the C‐terminal arginine in LC3 (Atg8 homologue) is first cleaved by Atg4 to expose a glycine residue, and then, LC3 is conjugated to PE through Atg7 (E1‐like) and Atg3 (E2‐like) activation to become lipidated LC3. Mammals encode at least six Atg8 homologues (LC3A, LC3B, LC3C, GABARAP, GABARAPL1, and GABARAPL2) and both the LC3 subfamily and GABARAP subfamily undergo PE conjugation. Although distinct roles in starvation‐induced autophagy have been proposed 128, their roles in mitophagy remain unknown.
Figure 4. Activation and recruitment of autophagy machineries during Parkin‐/PINK1‐mediated mitophagy.
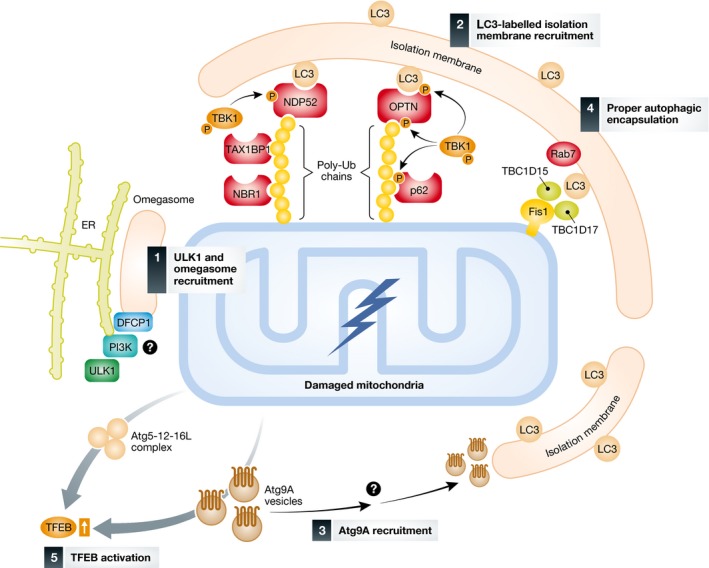
Following the generation of poly‐ubiquitin chains on damaged mitochondria, the indicated autophagy proteins including adaptors and regulators are recruited to the mitochondria in a multi‐independent process. (i) While autophagosomes form at ER–mitochondria contact sites under starvation‐induced autophagy 179, how the ULK1 and PI3K complexes and the omegasome marked by DFCP1 recognize mitochondrial damage remains unknown. (ii) Poly‐ubiquitin chains on the mitochondria are directly recognized by the autophagy adaptors, in particular NDP52 and optineurin (OPTN), which are phosphorylated by TBK1, and promote the recruitment of the LC3‐labeled isolation membrane. (iii) Atg9A vesicles are independently recruited to the mitochondria through an unknown mechanism. (iv) Mitochondria‐localized Rab‐GAPs, TBC1D15 and TBC1D17, via interaction of Fis1 regulate proper autophagosomal formation by modulating Rab7 activity. (v) Upon mitophagy stimulation, a regulator of autophagy‐lysosome biogenesis, TFEB (as well as MITF and TFE3), is activated in an Atg5‐ and Atg9A‐dependent manner.
Later steps of mitophagy link to autophagy
How are autophagy regulators recruited to the damaged mitochondria? The simple model proposed in 2010 was that p62, which has an ability to bind both LC3 and ubiquitin, serves as the adaptor in recruiting Atg proteins (at least lipidated LC3) to the poly‐ubiquitinated surface of the mitochondrial outer membrane 53. However, p62‐KO cells can still degrade the damaged mitochondria through the autophagy machinery, raising the possibility that binding between p62 and LC3 is not sufficient for mitophagy 129, 130. In addition to p62, mammals also express at least four additional autophagy adaptors, NDP52, NBR1, optineurin, and TAX1BP1. All possess an LC3‐interacting region (LIR), an ubiquitin‐associated domain (UBD), and a dimerization (or oligomerization) domain. Therefore, these autophagy adaptors are believed to bridge the LC3‐labeled isolation membrane and ubiquitinated cargo. To investigate the individual role of autophagy adaptors in mitophagy, two groups recently used CRISPR/Cas9 genome editing to knockout individually and different combinations of the five autophagy adaptors. Using a detailed mitophagy assay, these two studies found that NDP52 and optineurin primarily (and TAX1BP1 to some extent) function as autophagy adaptors during Parkin‐/PINK1‐mediated mitophagy, while p62 and NBR1 are not essential 131, 132. Ectopic targeting of the PINK1 kinase domain to the outer mitochondrial membrane in the absence of Parkin and mitochondrial stress recruits optineurin and NDP52, but not p62 131. This recruitment may suggest that phosphorylated ubiquitin chains generated locally on the mitochondria by PINK1 serve as a receptor for the selective autophagy machinery. However, Heo et al showed that a K63‐linked phosphorylated ubiquitin chain binds poorly to recombinant optineurin. This discrepancy can be explained if Parkin‐dependent optineurin phosphorylation changes the binding affinity of optineurin to ubiquitin chains 132. TBK1 participates in this step by phosphorylating optineurin (S177) 133 and p62 (S403) 134, which enhances the interactions with LC3 and ubiquitin, respectively 131, 132. Moreover, TBK1 also phosphorylates other sites of optineurin at S473 and S513 to enhance binding ability to poly‐ubiquitinated chains 132, 135 (Fig 4). TBK1 itself is activated by Ser172 phosphorylation (PINK1‐dependent but not autocatalytic) triggered by binding of optineurin to the ubiquitin chain on damaged mitochondria 132. Interestingly, these studies imply many functional similarities between mitophagy and xenophagy such as the involvement of NDP52/optineurin and TBK1 phosphorylation. Since the mitochondrion is evolutionary derived from α‐proteobacteria, the autophagy machinery may recognize damaged mitochondria as ROS‐producing invaders of the cell 136. Parkin was also reported to be recruited to Mycobacterium tuberculosis‐containing phagosomes 137. The key factors involved in recruiting cytosolic Parkin and unlocking autoinhibited Parkin E3 activity in this process remain to be identified.
Rab GTPase cycles are also important for mitochondrial encapsulation by the LC3‐labeled isolation membrane. Two Rab‐GAPs, TBC1D15 and TBC1D17, which associate with the mitochondrial outer membrane via interactions with Fis1, govern autophagosome morphology by modulating Rab7 activity during Parkin‐mediated mitophagy, but not starvation‐induced autophagy 138. Because TBC1D15 (and TBC1D17) can also directly interact with LC3 via the LIR motif, they control proper autophagic encapsulation of damaged mitochondria at the interface between mitochondria and the isolation membrane (Fig 4). Rab7 also localizes to isolation membranes in the early stage of xenophagy 139, suggesting another similarity between mitophagy and xenophagy as both involve a Rab7 activity of isolation membrane expansion.
Hierarchical analysis using several ATG‐KO cell lines during Parkin‐mediated mitophagy revealed that the recruitment of these autophagy proteins is not a linear cascade, but is rather a multi‐independent process 122. For example, lipidated LC3, the ULK1 complex, and Atg9A vesicles are independently recruited to damaged mitochondria, whose recruitment mechanisms, with the exception of lipidated LC3, are as of yet uncharacterized (Fig 4).
After Parkin translocation, damaged mitochondria are almost completely eliminated within 24–48 h. Therefore, the Parkin/PINK1 activation signal must quantitatively affect not only the ubiquitin system but also autophagy‐lysosome function. Under aberrant lysosomal storage or nutrient deprivation conditions, proteins involved in lysosomal and autophagic function are upregulated at the transcriptional levels by TFEB 140, 141, 142. In humans, TFEB together with MITF, TFE3, and TFEC comprises the MiT/TFE subfamily of basic helix‐loop‐helix leucine zipper transcription factors and has an ability to bind a specific 10‐bp palindromic motif found in the promoter sequence of genes encoding lysosomal and autophagic proteins to modulate their expression. Under normal conditions, TFEB transiently associates with the outer surface of the lysosome where it binds the heterodimeric Rag GTPase 143 and is phosphorylated on several residues by active mTORC1 144, 145, 146. Phosphorylation of Ser211, in particular, serves as a signal for the chaperone 14‐3‐3 proteins, which bind and sequester TFEB so that the inactivated form of TFEB stays in the cytosol. Upon starvation, TFEB dissociates from 14‐3‐3 allowing translocation to the nucleus where it induces the transcription of target genes. Recently, Nezich et al showed that Parkin‐/PINK1‐mediated mitophagy stimulation also induces TFEB (as well as MITF and TFE3) translocation to the nucleus and upregulates cathepsin B and p62 mRNAs, known TFEB target genes. Although Atg5 is dispensable for starvation‐induced TFEB translocation to the nucleus, Parkin‐mediated TFEB translocation requires Atg5 and Atg9A 147 (Fig 4). This strongly suggests that the molecular mechanism for TFEB translocation to the nucleus during mitophagy is different from that during starvation. Furthermore, KO of three MiT/TFE family members (TFEB, MITF, and TFE3) in HeLa cells causes moderate defects in Parkin‐mediated clearance of damaged mitochondria as well as p62 expression and lysosome morphology. It will be interesting, in the future, to test the physiological role of mitophagy‐dependent TFEB activation in vivo.
Mitophagy‐independent Parkin/PINK1 functions
Many autophagy‐independent Parkin functions have been reported. Parkin regulates the level of PARIS (ZNF746), a major transcriptional repressor of PGC‐1α expression, through the ubiquitin‐proteasome system 148. Conditional KO of Parkin in adult mice was shown to cause the progressive loss of dopamine neurons in a PARIS‐dependent manner via a decline of mitochondrial mass and respiration 149. Parkin also has been proposed to regulate mitosis and chromosome segregation 150. Recently, it was reported that Parkin complexes with Cdc20 and Cdh1, known APC/C co‐activators, to mediate the degradation of several mitosis regulators independent of APC/C. This pathway is PINK1 independent; however, Parkin E3 ligase activity is triggered following Plk1 phosphorylation of S378 151. Autophagy‐independent Parkin/PINK1 regulation of mitochondrial integrity maintenance has also been reported. Local oxidative damage of mitochondria induces small vesicular structures called mitochondrial‐derived vesicles (MDVs) that are transported to the late endosome and/or lysosomes. Both Parkin and PINK1 are required for the generation of MDVs, suggesting that Parkin‐/PINK1‐dependent MDVs are a faster response to mitochondrial damage than autophagic elimination 152, 153. Disruption of the mitochondrial membrane potential has also been reported to alter Rab8 and Rab11 functions following phosphorylation at a conserved Ser111 residue in a PINK1‐dependent manner 154. Although PINK1 does not directly phosphorylate these Rabs, this finding may suggest as of yet‐uncharacterized cascades of mitochondrial quality control.
Conclusion
Parkin‐mediated mitophagy research now spans multiple fields of active research including mitochondria, autophagy, the ubiquitin‐proteasome, neurology, and PD. This overlap in research interests and disciplines has in such a short time propelled the many advances in our understanding of Parkin‐mediated mitophagy. Based on studies using cultured cells (immortalized and non‐neuronal in most cases), we now know the detailed molecular mechanisms underlying Parkin recruitment to damaged mitochondria and how this impacts the subsequent steps leading to their selective engulfment by the autophagosome.
Although initially controversial, recent studies have confirmed Parkin/PINK1 involvement in autophagic clearance in neurons. Cai et al 155 showed that the mitochondria in the somatodendritic regions of mature cortical neurons are captured by an LC3‐labeled structure in a Parkin‐dependent manner. Similarly, Ashrafi et al 156 utilized mito‐KillerRed (see Box Box 1) to selectively damage a subset of mitochondria in hippocampal axons and observed autophagosome and axonal lysosome recruitment to the damaged mitochondria that was both Parkin and PINK1 dependent. Furthermore, mitochondrial delivery to the lysosome was observed in mouse primary neurons using mito‐Keima (see Box Box 1). This endogenous Parkin‐dependent mitophagy requires a certain period of time, but not any exposure of chemical depolarizing compounds 82. Despite being independently generated by many research groups, neither simple Parkin‐ nor simple Pink1‐KO mice exhibit severe PD‐like symptoms such as the loss of dopaminergic neurons, thus complicating Parkin/PINK1 mitophagy studies in vivo. However, Pickrell et al 157 recently created a new PD model mouse by crossing Parkin‐KO mice and Mutator mice that express a defective mitochondrial DNA polymerase. In the resulting Mutator Parkin‐KO mice, dopaminergic neurons degenerated and L‐DOPA (a metabolic precursor of dopamine widely used for PD patients) improved the motor deficit of the mice. They also found the accumulation of phosphorylated ubiquitin in neurons, but not in livers of the Mutator Parkin‐KO mice, strongly suggesting that PINK1 activation was caused by neuronal mitochondrial dysfunction.
In this review, we shed light on the biochemical and molecular aspects of Parkin‐/PINK1‐mediated mitophagy. Many questions remain to be answered before we can have a completely clear understanding of Parkin/PINK1 functions; however, we will continue to uncover the molecular basis linking the coordinated actions of Parkin/PINK1 and the ubiquitin signal with autophagy for clearing dysfunctional mitochondria.
Sidebar A: In need of answers.
Although many research papers have elucidated detailed molecular mechanisms of Parkin‐/PINK1‐dependent mitophagy, new discoveries always bring new questions. Here, we would like to outline several open questions that need to be answered in the future to further advance our understanding of mitophagy.
-
How does phosphorylated Parkin accelerate ubiquitin transthiolation from E2s to Cys431?
Kazlauskaite et al 92 demonstrated that purified recombinant Ser65‐phosphorylated Parkin exhibited maximal E3 ligase activity that no longer required Ser65‐phosphorylated ubiquitin. This is consistent with the results in which phosphorylated Parkin, but not Parkin bound with phosphorylated ubiquitin, had an “open” structure as evidenced by the accessibility of the catalytic Cys431 to ubiquitin‐vinyl sulfone 67, 87. In contrast, K161N and K211N mutations in a different phosphate‐binding pocket of the RING0 domain, which are not involved in binding phosphorylated ubiquitin, inhibited full activation of Ser65‐phosphorylated Parkin 67. This raises the possibility of another unknown step in the Parkin conformational change 93. Structural determination of the Parkin‐phosphorylated ubiquitin complex has revealed that the E2 binding region is too spatially removed from the catalytic Cys431 to allow for direct transfer of the ubiquitin molecule. As E2 association with phosphorylated Parkin has been suggested to induce further Parkin conformational changes, the ternary protein complex including phosphorylated Parkin, phosphorylated ubiquitin, and ubiquitin‐conjugated E2 enzyme needs to be solved either by biochemical or by structural approaches.
-
How are autophagy proteins recruited to the damaged mitochondria following Parkin‐/PINK1‐dependent ubiquitination?
Poly‐ubiquitinated chains on the surface of the outer membrane of the damaged mitochondria can recruit autophagy adaptors, which further recruit Atg8 homologues via LIR motifs. On the other hand, how essential upstream autophagy proteins such as the ULK1 complex and Atg9A vesicles are recruited to the mitochondria remains unknown. Although ectopic PINK1 targeting to mitochondria suggests that association of the ULK1 complex with the damaged mitochondria is optineurin/NDP52 dependent 131, precise cascades of the steps starting from the poly‐ubiquitinated chains for transfer of the signal to the autophagy machinery are largely unknown.
-
Do mitophagy defects directly affect the loss of dopaminergic neurons?
There is no doubt that elimination of the damaged mitochondria by mitophagy machinery is important for maintaining cellular homeostasis via healthy mitochondria. However, there is currently no direct evidence demonstrating how defects in mitophagy affect PD pathologies or degradation of dopaminergic neurons. To clarify these issues, an in vivo mitophagy assessment tool for dopaminergic neurons needs to be established.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
We thank Catherine L. Nezich, Michael Lazarou, Richard J. Youle, Lina Herhaus, and Ivan Dikic for valuable comments. We regret that space limitations prevented us from being able to cite the studies of numerous other colleagues, but hope the readers refer to the cited reviews for more information. This work was supported by JSPS PRESTO, KAKENHI Grant Number: 26650042, MEXT KAKENHI Grant Numbers 26111729 and 15H01196, and the Tomizawa Jun‐ichi and Keiko Fund for Young Scientist (to N.M.); by JSPS KAKENHI Grant Number 26000014 (to K.T.); and by the Takeda Science Foundation (to K.T. and N.M.).
EMBO Reports (2016) 17: 300–316
See the Glossary for abbreviations used in this article.
References
- 1. Okamoto K, Shaw JM (2005) Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu Rev Genet 39: 503–536 [DOI] [PubMed] [Google Scholar]
- 2. Tatsuta T, Langer T (2008) Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J 27: 306–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Birkmayer W, Hornykiewicz O (1961) The L‐3,4‐dioxyphenylalanine (DOPA)‐effect in Parkinson‐akinesia. Wien Klin Wochenschr 73: 787–788 [PubMed] [Google Scholar]
- 4. Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) Alpha‐Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 95: 6469–6473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bras J, Guerreiro R, Hardy J (2015) SnapShot: genetics of Parkinson's disease. Cell 160: 570–570 e571 [DOI] [PubMed] [Google Scholar]
- 6. Pickrell AM, Youle RJ (2015) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron 85: 257–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392: 605–608 [DOI] [PubMed] [Google Scholar]
- 8. Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K et al (2000) Familial Parkinson disease gene product, parkin, is a ubiquitin‐protein ligase. Nat Genet 25: 302–305 [DOI] [PubMed] [Google Scholar]
- 9. Imai Y, Soda M, Takahashi R (2000) Parkin suppresses unfolded protein stress‐induced cell death through its E3 ubiquitin‐protein ligase activity. J Biol Chem 275: 35661–35664 [DOI] [PubMed] [Google Scholar]
- 10. Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM (2000) Parkin functions as an E2‐dependent ubiquitin‐ protein ligase and promotes the degradation of the synaptic vesicle‐associated protein, CDCrel‐1. Proc Natl Acad Sci USA 97: 13354–13359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Valente EM, Bentivoglio AR, Dixon PH, Ferraris A, Ialongo T, Frontali M, Albanese A, Wood NW (2001) Localization of a novel locus for autosomal recessive early‐onset parkinsonism, PARK6, on human chromosome 1p35‐p36. Am J Hum Genet 68: 895–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Valente EM, Brancati F, Ferraris A, Graham EA, Davis MB, Breteler MM, Gasser T, Bonifati V, Bentivoglio AR, De Michele G et al (2002) PARK6‐linked parkinsonism occurs in several European families. Ann Neurol 51: 14–18 [PubMed] [Google Scholar]
- 13. Valente EM, Abou‐Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG et al (2004) Hereditary early‐onset Parkinson's disease caused by mutations in PINK1. Science 304: 1158–1160 [DOI] [PubMed] [Google Scholar]
- 14. Abou‐Sleiman PM, Muqit MM, Wood NW (2006) Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat Rev Neurosci 7: 207–219 [DOI] [PubMed] [Google Scholar]
- 15. Langston JW, Ballard P, Tetrud JW, Irwin I (1983) Chronic Parkinsonism in humans due to a product of meperidine‐analog synthesis. Science 219: 979–980 [DOI] [PubMed] [Google Scholar]
- 16. Liou HH, Tsai MC, Chen CJ, Jeng JS, Chang YC, Chen SY, Chen RC (1997) Environmental risk factors and Parkinson's disease: a case‐control study in Taiwan. Neurology 48: 1583–1588 [DOI] [PubMed] [Google Scholar]
- 17. Betarbet R, Sherer TB, MacKenzie G, Garcia‐Osuna M, Panov AV, Greenamyre JT (2000) Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci 3: 1301–1306 [DOI] [PubMed] [Google Scholar]
- 18. Funayama M, Ohe K, Amo T, Furuya N, Yamaguchi J, Saiki S, Li Y, Ogaki K, Ando M, Yoshino H et al (2015) CHCHD2 mutations in autosomal dominant late‐onset Parkinson's disease: a genome‐wide linkage and sequencing study. Lancet Neurol 14: 274–282 [DOI] [PubMed] [Google Scholar]
- 19. Gabriel K, Milenkovic D, Chacinska A, Muller J, Guiard B, Pfanner N, Meisinger C (2007) Novel mitochondrial intermembrane space proteins as substrates of the MIA import pathway. J Mol Biol 365: 612–620 [DOI] [PubMed] [Google Scholar]
- 20. Fischer M, Riemer J (2013) The mitochondrial disulfide relay system: roles in oxidative protein folding and beyond. Int J Cell Biol 2013: 742923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ (2003) Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA 100: 4078–4083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441: 1162–1166 [DOI] [PubMed] [Google Scholar]
- 23. Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM et al (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441: 1157–1161 [DOI] [PubMed] [Google Scholar]
- 24. Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, Yang L, Beal MF, Vogel H, Lu B (2006) Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci USA 103: 10793–10798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Komander D, Rape M (2012) The ubiquitin code. Annu Rev Biochem 81: 203–229 [DOI] [PubMed] [Google Scholar]
- 26. Weissman AM (2001) Themes and variations on ubiquitylation. Nat Rev Mol Cell Biol 2: 169–178 [DOI] [PubMed] [Google Scholar]
- 27. Pickart CM, Eddins MJ (2004) Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta 1695: 55–72 [DOI] [PubMed] [Google Scholar]
- 28. Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, Gygi SP (2003) A proteomics approach to understanding protein ubiquitination. Nat Biotechnol 21: 921–926 [DOI] [PubMed] [Google Scholar]
- 29. Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K, Nakagawa T, Kato M, Murata S, Yamaoka S et al (2009) Involvement of linear polyubiquitylation of NEMO in NF‐kappaB activation. Nat Cell Biol 11: 123–132 [DOI] [PubMed] [Google Scholar]
- 30. Olzmann JA, Li L, Chudaev MV, Chen J, Perez FA, Palmiter RD, Chin LS (2007) Parkin‐mediated K63‐linked polyubiquitination targets misfolded DJ‐1 to aggresomes via binding to HDAC6. J Cell Biol 178: 1025–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tan JM, Wong ES, Kirkpatrick DS, Pletnikova O, Ko HS, Tay SP, Ho MW, Troncoso J, Gygi SP, Lee MK et al (2008) Lysine 63‐linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet 17: 431–439 [DOI] [PubMed] [Google Scholar]
- 32. Pickart CM (2000) Ubiquitin in chains. Trends Biochem Sci 25: 544–548 [DOI] [PubMed] [Google Scholar]
- 33. Al‐Hakim A, Escribano‐Diaz C, Landry MC, O'Donnell L, Panier S, Szilard RK, Durocher D (2010) The ubiquitous role of ubiquitin in the DNA damage response. DNA Repair 9: 1229–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wenzel DM, Lissounov A, Brzovic PS, Klevit RE (2011) UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 474: 105–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183: 795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F et al (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189: 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sarraf SA, Raman M, Guarani‐Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW (2013) Landscape of the PARKIN‐dependent ubiquitylome in response to mitochondrial depolarization. Nature 496: 372–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191: 1367–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Santel A, Fuller MT (2001) Control of mitochondrial morphology by a human mitofusin. J Cell Sci 114: 867–874 [DOI] [PubMed] [Google Scholar]
- 40. Yoshii SR, Kishi C, Ishihara N, Mizushima N (2011) Parkin mediates proteasome‐dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem 286: 19630–19640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC (2011) Broad activation of the ubiquitin‐proteasome system by Parkin is critical for mitophagy. Hum Mol Genet 20: 1726–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G et al (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27: 433–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Meyer H, Weihl CC (2014) The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J Cell Sci 127: 3877–3883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Buchberger A, Schindelin H, Hanzelmann P (2015) Control of p97 function by cofactor binding. FEBS Lett 589: 2578–2589 [DOI] [PubMed] [Google Scholar]
- 45. Kimura Y, Fukushi J, Hori S, Matsuda N, Okatsu K, Kakiyama Y, Kawawaki J, Kakizuka A, Tanaka K (2013) Different dynamic movements of wild‐type and pathogenic VCPs and their cofactors to damaged mitochondria in a Parkin‐mediated mitochondrial quality control system. Genes Cells 18: 1131–1143 [DOI] [PubMed] [Google Scholar]
- 46. Kim NC, Tresse E, Kolaitis RM, Molliex A, Thomas RE, Alami NH, Wang B, Joshi A, Smith RB, Ritson GP et al (2013) VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron 78: 65–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Heo JM, Livnat‐Levanon N, Taylor EB, Jones KT, Dephoure N, Ring J, Xie J, Brodsky JL, Madeo F, Gygi SP et al (2010) A stress‐responsive system for mitochondrial protein degradation. Mol Cell 40: 465–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Narendra D, Walker JE, Youle R (2012) Mitochondrial quality control mediated by PINK1 and Parkin: links to parkinsonism. Cold Spring Harb Perspect Biol 4: a011338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL (2011) PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147: 893–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW (2010) Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin‐dependent manner upon induction of mitophagy. Hum Mol Genet 19: 4861–4870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L (2010) The mitochondrial fusion‐promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS ONE 5: e10054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liu S, Sawada T, Lee S, Yu W, Silverio G, Alapatt P, Millan I, Shen A, Saxton W, Kanao T et al (2012) Parkinson's disease‐associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet 8: e1002537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W (2010) PINK1/Parkin‐mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12: 119–131 [DOI] [PubMed] [Google Scholar]
- 54. Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP (2010) Disease‐causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6‐dependent mitophagy. J Cell Biol 189: 671–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vives‐Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS et al (2010) PINK1‐dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA 107: 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8: e1000298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lazarou M, Jin SM, Kane LA, Youle RJ (2012) Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell 22: 320–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Endo T, Yamano K (2009) Multiple pathways for mitochondrial protein traffic. Biol Chem 390: 723–730 [DOI] [PubMed] [Google Scholar]
- 59. Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N (2009) Importing mitochondrial proteins: machineries and mechanisms. Cell 138: 628–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hasson SA, Kane LA, Yamano K, Huang CH, Sliter DA, Buehler E, Wang C, Heman‐Ackah SM, Hessa T, Guha R et al (2013) High‐content genome‐wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature 504: 291–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Okatsu K, Uno M, Koyano F, Go E, Kimura M, Oka T, Tanaka K, Matsuda N (2013) A dimeric PINK1‐containing complex on depolarized mitochondria stimulates Parkin recruitment. J Biol Chem 288: 36372–36384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Okatsu K, Oka T, Iguchi M, Imamura K, Kosako H, Tani N, Kimura M, Go E, Koyano F, Funayama M et al (2012) PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat Commun 3: 1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Aerts L, Craessaerts K, De Strooper B, Morais VA (2015) PINK1 kinase catalytic activity is regulated by phosphorylation on serines 228 and 402. J Biol Chem 290: 2798–2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Okatsu K, Kimura M, Oka T, Tanaka K, Matsuda N (2015) Unconventional PINK1 localization to the outer membrane of depolarized mitochondria drives Parkin recruitment. J Cell Sci 128: 964–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lazarou M, Narendra DP, Jin SM, Tekle E, Banerjee S, Youle RJ (2013) PINK1 drives Parkin self‐association and HECT‐like E3 activity upstream of mitochondrial binding. J Cell Biol 200: 163–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zheng X, Hunter T (2013) Parkin mitochondrial translocation is achieved through a novel catalytic activity coupled mechanism. Cell Res 23: 886–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ordureau A, Sarraf SA, Duda DM, Heo JM, Jedrychowski MP, Sviderskiy VO, Olszewski JL, Koerber JT, Xie T, Beausoleil SA et al (2014) Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell 56: 360–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shiba‐Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, Hattori N (2012) PINK1‐mediated phosphorylation of the Parkin ubiquitin‐like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep 2: 1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Woodroof HI, Pogson JH, Begley M, Cantley LC, Deak M, Campbell DG, van Aalten DM, Whitworth AJ, Alessi DR, Muqit MM (2011) Discovery of catalytically active orthologues of the Parkinson's disease kinase PINK1: analysis of substrate specificity and impact of mutations. Open Biol 1: 110012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M et al (2012) PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol 2: 120080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Iguchi M, Kujuro Y, Okatsu K, Koyano F, Kosako H, Kimura M, Suzuki N, Uchiyama S, Tanaka K, Matsuda N (2013) Parkin‐catalyzed ubiquitin‐ester transfer is triggered by PINK1‐dependent phosphorylation. J Biol Chem 288: 22019–22032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T et al (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510: 162–166 [DOI] [PubMed] [Google Scholar]
- 73. Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MM (2014) Parkin is activated by PINK1‐dependent phosphorylation of ubiquitin at Ser65. Biochem J 460: 127–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205: 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wauer T, Swatek KN, Wagstaff JL, Gladkova C, Pruneda JN, Michel MA, Gersch M, Johnson CM, Freund SM, Komander D (2015) Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J 34: 307–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Herhaus L, Dikic I (2015) Expanding the ubiquitin code through post‐translational modification. EMBO Rep 16: 1071–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zheng X, Hunter T (2014) Pink1, the first ubiquitin kinase. EMBO J 33: 1621–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Okatsu K, Koyano F, Kimura M, Kosako H, Saeki Y, Tanaka K, Matsuda N (2015) Phosphorylated ubiquitin chain is the genuine Parkin receptor. J Cell Biol 209: 111–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ordureau A, Heo JM, Duda DM, Paulo JA, Olszewski JL, Yanishevski D, Rinehart J, Schulman BA, Harper JW (2015) Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc Natl Acad Sci USA 112: 6637–6642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Shiba‐Fukushima K, Arano T, Matsumoto G, Inoshita T, Yoshida S, Ishihama Y, Ryu KY, Nukina N, Hattori N, Imai Y (2014) Phosphorylation of mitochondrial polyubiquitin by PINK1 promotes Parkin mitochondrial tethering. PLoS Genet 10: e1004861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cunningham CN, Baughman JM, Phu L, Tea JS, Yu C, Coons M, Kirkpatrick DS, Bingol B, Corn JE (2015) USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat Cell Biol 17: 160–169 [DOI] [PubMed] [Google Scholar]
- 82. Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M (2014) The mitochondrial deubiquitinase USP30 opposes parkin‐mediated mitophagy. Nature 510: 370–375 [DOI] [PubMed] [Google Scholar]
- 83. Clague MJ, Urbe S (2006) Endocytosis: the DUB version. Trends Cell Biol 16: 551–559 [DOI] [PubMed] [Google Scholar]
- 84. Durcan TM, Tang MY, Perusse JR, Dashti EA, Aguileta MA, McLelland GL, Gros P, Shaler TA, Faubert D, Coulombe B et al (2014) USP8 regulates mitophagy by removing K6‐linked ubiquitin conjugates from parkin. EMBO J 33: 2473–2491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Cornelissen T, Haddad D, Wauters F, Van Humbeeck C, Mandemakers W, Koentjoro B, Sue C, Gevaert K, De Strooper B, Verstreken P et al (2014) The deubiquitinase USP15 antagonizes Parkin‐mediated mitochondrial ubiquitination and mitophagy. Hum Mol Genet 23: 5227–5242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Trempe JF, Sauve V, Grenier K, Seirafi M, Tang MY, Menade M, Al‐Abdul‐Wahid S, Krett J, Wong K, Kozlov G et al (2013) Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 340: 1451–1455 [DOI] [PubMed] [Google Scholar]
- 87. Wauer T, Komander D (2013) Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J 32: 2099–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Riley BE, Lougheed JC, Callaway K, Velasquez M, Brecht E, Nguyen L, Shaler T, Walker D, Yang Y, Regnstrom K et al (2013) Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat Commun 4: 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Chaugule VK, Burchell L, Barber KR, Sidhu A, Leslie SJ, Shaw GS, Walden H (2011) Autoregulation of Parkin activity through its ubiquitin‐like domain. EMBO J 30: 2853–2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kumar A, Aguirre JD, Condos TE, Martinez‐Torres RJ, Chaugule VK, Toth R, Sundaramoorthy R, Mercier P, Knebel A, Spratt DE et al (2015) Disruption of the autoinhibited state primes the E3 ligase parkin for activation and catalysis. EMBO J 34: 2506–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sauve V, Lilov A, Seirafi M, Vranas M, Rasool S, Kozlov G, Sprules T, Wang J, Trempe JF, Gehring K (2015) A Ubl/ubiquitin switch in the activation of Parkin. EMBO J 34: 2492–2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kazlauskaite A, Martinez‐Torres RJ, Wilkie S, Kumar A, Peltier J, Gonzalez A, Johnson C, Zhang J, Hope AG, Peggie M et al (2015) Binding to serine 65‐phosphorylated ubiquitin primes Parkin for optimal PINK1‐dependent phosphorylation and activation. EMBO Rep 16: 939–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wauer T, Simicek M, Schubert A, Komander D (2015) Mechanism of phospho‐ubiquitin‐induced PARKIN activation. Nature 524: 370–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Yamano K, Queliconi BB, Koyano F, Saeki Y, Hirokawa T, Tanaka K, Matsuda N (2015) Site‐specific interaction mapping of phosphorylated ubiquitin to uncover Parkin activation. J Biol Chem 290: 25199–25211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Aguileta MA, Korac J, Durcan TM, Trempe JF, Haber M, Gehring K, Elsasser S, Waidmann O, Fon EA, Husnjak K (2015) The E3 ubiquitin ligase parkin is recruited to the 26 S proteasome via the proteasomal ubiquitin receptor Rpn13. J Biol Chem 290: 7492–7505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Narendra DP, Wang C, Youle RJ, Walker JE (2013) PINK1 rendered temperature sensitive by disease‐associated and engineered mutations. Hum Mol Genet 22: 2572–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Beilina A, Van Der Brug M, Ahmad R, Kesavapany S, Miller DW, Petsko GA, Cookson MR (2005) Mutations in PTEN‐induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc Natl Acad Sci USA 102: 5703–5708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Stojanovski D, Bohnert M, Pfanner N, van der Laan M (2012) Mechanisms of protein sorting in mitochondria. Cold Spring Harb Perspect Biol 4: a011320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 191: 933–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Deas E, Plun‐Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SH, Renton AE, Harvey RJ, Whitworth AJ, Martins LM et al (2011) PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet 20: 867–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Shi G, Lee JR, Grimes DA, Racacho L, Ye D, Yang H, Ross OA, Farrer M, McQuibban GA, Bulman DE (2011) Functional alteration of PARL contributes to mitochondrial dysregulation in Parkinson's disease. Hum Mol Genet 20: 1966–1974 [DOI] [PubMed] [Google Scholar]
- 102. Meissner C, Lorenz H, Weihofen A, Selkoe DJ, Lemberg MK (2011) The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J Neurochem 117: 856–867 [DOI] [PubMed] [Google Scholar]
- 103. Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA (2012) Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep 13: 378–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Tasaki T, Sriram SM, Park KS, Kwon YT (2012) The N‐end rule pathway. Annu Rev Biochem 81: 261–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Yamano K, Youle RJ (2013) PINK1 is degraded through the N‐end rule pathway. Autophagy 9: 1758–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Gaume B, Klaus C, Ungermann C, Guiard B, Neupert W, Brunner M (1998) Unfolding of preproteins upon import into mitochondria. EMBO J 17: 6497–6507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Yamano K, Kuroyanagi‐Hasegawa M, Esaki M, Yokota M, Endo T (2008) Step‐size analyses of the mitochondrial Hsp70 import motor reveal the Brownian ratchet in operation. J Biol Chem 283: 27325–27332 [DOI] [PubMed] [Google Scholar]
- 108. Urban S, Freeman M (2003) Substrate specificity of rhomboid intramembrane proteases is governed by helix‐breaking residues in the substrate transmembrane domain. Mol Cell 11: 1425–1434 [DOI] [PubMed] [Google Scholar]
- 109. Meier S, Neupert W, Herrmann JM (2005) Proline residues of transmembrane domains determine the sorting of inner membrane proteins in mitochondria. J Cell Biol 170: 881–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Morais VA, Haddad D, Craessaerts K, De Bock PJ, Swerts J, Vilain S, Aerts L, Overbergh L, Grunewald A, Seibler P et al (2014) PINK1 loss‐of‐function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science 344: 203–207 [DOI] [PubMed] [Google Scholar]
- 111. Mizuno Y, Ohta S, Tanaka M, Takamiya S, Suzuki K, Sato T, Oya H, Ozawa T, Kagawa Y (1989) Deficiencies in complex I subunits of the respiratory chain in Parkinson's disease. Biochem Biophys Res Commun 163: 1450–1455 [DOI] [PubMed] [Google Scholar]
- 112. Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD (1989) Mitochondrial complex I deficiency in Parkinson's disease. Lancet 1: 1269 [DOI] [PubMed] [Google Scholar]
- 113. Thomas RE, Andrews LA, Burman JL, Lin WY, Pallanck LJ (2014) PINK1‐Parkin pathway activity is regulated by degradation of PINK1 in the mitochondrial matrix. PLoS Genet 10: e1004279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Mizushima N (2011) Autophagy in protein and organelle turnover. Cold Spring Harb Symp Quant Biol 76: 397–402 [DOI] [PubMed] [Google Scholar]
- 115. Kageyama S, Sou YS, Uemura T, Kametaka S, Saito T, Ishimura R, Kouno T, Bedford L, Mayer RJ, Lee MS et al (2014) Proteasome dysfunction activates autophagy and the Keap1‐Nrf2 pathway. J Biol Chem 289: 24944–24955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Okamoto K (2014) Organellophagy: eliminating cellular building blocks via selective autophagy. J Cell Biol 205: 435–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Stolz A, Ernst A, Dikic I (2014) Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 16: 495–501 [DOI] [PubMed] [Google Scholar]
- 118. Sica V, Galluzzi L, Bravo‐San Pedro JM, Izzo V, Maiuri MC, Kroemer G (2015) Organelle‐specific initiation of autophagy. Mol Cell 59: 522–539 [DOI] [PubMed] [Google Scholar]
- 119. Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147: 728–741 [DOI] [PubMed] [Google Scholar]
- 120. Feng Y, He D, Yao Z, Klionsky DJ (2014) The machinery of macroautophagy. Cell Res 24: 24–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Mizushima N, Yoshimori T, Ohsumi Y (2011) The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27: 107–132 [DOI] [PubMed] [Google Scholar]
- 122. Itakura E, Kishi‐Itakura C, Koyama‐Honda I, Mizushima N (2012) Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin‐mediated mitophagy. J Cell Sci 125: 1488–1499 [DOI] [PubMed] [Google Scholar]
- 123. Gilkerson RW, De Vries RL, Lebot P, Wikstrom JD, Torgyekes E, Shirihai OS, Przedborski S, Schon EA (2012) Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum Mol Genet 21: 978–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z (2009) Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1‐phosphatidylinositol‐3‐kinase complex. Nat Cell Biol 11: 468–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama‐Noda K, Ichimura T, Isobe T et al (2009) Two Beclin 1‐binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 11: 385–396 [DOI] [PubMed] [Google Scholar]
- 126. Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT (2008) Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3‐phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 182: 685–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Wong YC, Holzbaur EL (2014) Optineurin is an autophagy receptor for damaged mitochondria in parkin‐mediated mitophagy that is disrupted by an ALS‐linked mutation. Proc Natl Acad Sci USA 111: E4439–E4448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z (2010) LC3 and GATE‐16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J 29: 1792–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Okatsu K, Saisho K, Shimanuki M, Nakada K, Shitara H, Sou YS, Kimura M, Sato S, Hattori N, Komatsu M et al (2010) p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 15: 887–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ (2010) p62/SQSTM1 is required for Parkin‐induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6: 1090–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524: 309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW (2015) The PINK1‐PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell 60: 7–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C et al (2011) Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333: 228–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Matsumoto G, Shimogori T, Hattori N, Nukina N (2015) TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet 24: 4429–4442 [DOI] [PubMed] [Google Scholar]
- 135. Gleason CE, Ordureau A, Gourlay R, Arthur JS, Cohen P (2011) Polyubiquitin binding to optineurin is required for optimal activation of TANK‐binding kinase 1 and production of interferon beta. J Biol Chem 286: 35663–35674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Randow F, Youle RJ (2014) Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host Microbe 15: 403–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, Schneider DS, Nakamura K, Shiloh MU, Cox JS (2013) The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 501: 512–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Yamano K, Fogel AI, Wang C, van der Bliek AM, Youle RJ (2014) Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. eLife 3: e01612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Yamaguchi H, Nakagawa I, Yamamoto A, Amano A, Noda T, Yoshimori T (2009) An initial step of GAS‐containing autophagosome‐like vacuoles formation requires Rab7. PLoS Pathog 5: e1000670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS et al (2009) A gene network regulating lysosomal biogenesis and function. Science 325: 473–477 [DOI] [PubMed] [Google Scholar]
- 141. Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P et al (2011) TFEB links autophagy to lysosomal biogenesis. Science 332: 1429–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, Ballabio A (2011) Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet 20: 3852–3866 [DOI] [PubMed] [Google Scholar]
- 143. Martina JA, Puertollano R (2013) Rag GTPases mediate amino acid‐dependent recruitment of TFEB and MITF to lysosomes. J Cell Biol 200: 475–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC et al (2012) A lysosome‐to‐nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 31: 1095–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Martina JA, Chen Y, Gucek M, Puertollano R (2012) MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8: 903–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Roczniak‐Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM (2012) The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal 5: ra42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Nezich CL, Wang C, Fogel AI, Youle RJ (2015) MiT/TFE transcription factors are activated during mitophagy downstream of Parkin and Atg5. J Cell Biol 210: 435–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM (2011) PARIS (ZNF746) repression of PGC‐1alpha contributes to neurodegeneration in Parkinson's disease. Cell 144: 689–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Stevens DA, Lee Y, Kang HC, Lee BD, Lee YI, Bower A, Jiang H, Kang SU, Andrabi SA, Dawson VL et al (2015) Parkin loss leads to PARIS‐dependent declines in mitochondrial mass and respiration. Proc Natl Acad Sci USA 112: 11696–11701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Veeriah S, Taylor BS, Meng S, Fang F, Yilmaz E, Vivanco I, Janakiraman M, Schultz N, Hanrahan AJ, Pao W et al (2010) Somatic mutations of the Parkinson's disease‐associated gene PARK2 in glioblastoma and other human malignancies. Nat Genet 42: 77–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Lee SB, Kim JJ, Nam HJ, Gao B, Yin P, Qin B, Yi SY, Ham H, Evans D, Kim SH et al (2015) Parkin regulates mitosis and genomic stability through Cdc20/Cdh1. Mol Cell 60: 21–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA (2014) Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J 33: 282–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Sugiura A, McLelland GL, Fon EA, McBride HM (2014) A new pathway for mitochondrial quality control: mitochondrial‐derived vesicles. EMBO J 33: 2142–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Lai YC, Kondapalli C, Lehneck R, Procter JB, Dill BD, Woodroof HI, Gourlay R, Peggie M, Macartney TJ, Corti O et al (2015) Phosphoproteomic screening identifies Rab GTPases as novel downstream targets of PINK1. EMBO J 34: 2840–2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Cai Q, Zakaria HM, Simone A, Sheng ZH (2012) Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr Biol 22: 545–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL (2014) Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol 206: 655–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Pickrell AM, Huang CH, Kennedy SR, Ordureau A, Sideris DP, Hoekstra JG, Harper JW, Youle RJ (2015) Endogenous Parkin preserves dopaminergic substantia nigral neurons following mitochondrial DNA mutagenic stress. Neuron 87: 371–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Chen D, Chen X, Li M, Zhang H, Ding WX, Yin XM (2013) CCCP‐induced LC3 lipidation depends on Atg9 whereas FIP200/Atg13 and Beclin 1/Atg14 are dispensable. Biochem Biophys Res Commun 432: 226–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Padman BS, Bach M, Lucarelli G, Prescott M, Ramm G (2013) The protonophore CCCP interferes with lysosomal degradation of autophagic cargo in yeast and mammalian cells. Autophagy 9: 1862–1875 [DOI] [PubMed] [Google Scholar]
- 160. Burkhardt JK, Hester S, Argon Y (1989) The glycoprotein of VSV accumulates in a distal Golgi compartment in the presence of CCCP. J Cell Sci 92(Pt 4): 643–654 [DOI] [PubMed] [Google Scholar]
- 161. Llopis J, McCaffery JM, Miyawaki A, Farquhar MG, Tsien RY (1998) Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc Natl Acad Sci USA 95: 6803–6808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Iwata S, Lee JW, Okada K, Lee JK, Iwata M, Rasmussen B, Link TA, Ramaswamy S, Jap BK (1998) Complete structure of the 11‐subunit bovine mitochondrial cytochrome bc1 complex. Science 281: 64–71 [DOI] [PubMed] [Google Scholar]
- 163. Ziviani E, Tao RN, Whitworth AJ (2010) Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc Natl Acad Sci USA 107: 5018–5023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Ramsay RR, Singer TP (1992) Relation of superoxide generation and lipid peroxidation to the inhibition of NADH‐Q oxidoreductase by rotenone, piericidin A, and MPP+. Biochem Biophys Res Commun 189: 47–52 [DOI] [PubMed] [Google Scholar]
- 165. Bulina ME, Chudakov DM, Britanova OV, Yanushevich YG, Staroverov DB, Chepurnykh TV, Merzlyak EM, Shkrob MA, Lukyanov S, Lukyanov KA (2006) A genetically encoded photosensitizer. Nat Biotechnol 24: 95–99 [DOI] [PubMed] [Google Scholar]
- 166. Choubey V, Safiulina D, Vaarmann A, Cagalinec M, Wareski P, Kuum M, Zharkovsky A, Kaasik A (2011) Mutant A53T alpha‐synuclein induces neuronal death by increasing mitochondrial autophagy. J Biol Chem 286: 10814–10824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Wang Y, Nartiss Y, Steipe B, McQuibban GA, Kim PK (2012) ROS‐induced mitochondrial depolarization initiates PARK2/PARKIN‐dependent mitochondrial degradation by autophagy. Autophagy 8: 1462–1476 [DOI] [PubMed] [Google Scholar]
- 168. Yang JY, Yang WY (2011) Spatiotemporally controlled initiation of Parkin‐mediated mitophagy within single cells. Autophagy 7: 1230–1238 [DOI] [PubMed] [Google Scholar]
- 169. Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ (2002) A mitochondrial specific stress response in mammalian cells. EMBO J 21: 4411–4419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Pimenta de Castro I, Costa AC, Lam D, Tufi R, Fedele V, Moisoi N, Dinsdale D, Deas E, Loh SH, Martins LM (2012) Genetic analysis of mitochondrial protein misfolding in Drosophila melanogaster. Cell Death Differ 19: 1308–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Jin SM, Youle RJ (2013) The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin‐mediated mitophagy of polarized mitochondria. Autophagy 9: 1750–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A (2011) A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol 18: 1042–1052 [DOI] [PubMed] [Google Scholar]
- 173. Kinoshita E, Kinoshita‐Kikuta E, Takiyama K, Koike T (2006) Phosphate‐binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics 5: 749–757 [DOI] [PubMed] [Google Scholar]
- 174. Rogerson DT, Sachdeva A, Wang K, Haq T, Kazlauskaite A, Hancock SM, Huguenin‐Dezot N, Muqit MM, Fry AM, Bayliss R et al (2015) Efficient genetic encoding of phosphoserine and its nonhydrolyzable analog. Nat Chem Biol 11: 496–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Park HS, Hohn MJ, Umehara T, Guo LT, Osborne EM, Benner J, Noren CJ, Rinehart J, Soll D (2011) Expanding the genetic code of Escherichia coli with phosphoserine. Science 333: 1151–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Pirman NL, Barber KW, Aerni HR, Ma NJ, Haimovich AD, Rogulina S, Isaacs FJ, Rinehart J (2015) A flexible codon in genomically recoded Escherichia coli permits programmable protein phosphorylation. Nat Commun 6: 8130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Fiesel FC, Ando M, Hudec R, Hill AR, Castanedes‐Casey M, Caulfield TR, Moussaud‐Lamodiere EL, Stankowski JN, Bauer PO, Lorenzo‐Betancor O et al (2015) (Patho‐)physiological relevance of PINK1‐dependent ubiquitin phosphorylation. EMBO Rep 16: 1114–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Karbowski M, Youle RJ (2011) Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr Opin Cell Biol 23: 476–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y et al (2013) Autophagosomes form at ER‐mitochondria contact sites. Nature 495: 389–393 [DOI] [PubMed] [Google Scholar]