

Abstract
We have applied the CRISPR/Cas9 system in vivo to disrupt gene expression in neural stem cells in the developing mammalian brain. Two days after in utero electroporation of a single plasmid encoding Cas9 and an appropriate guide RNA (gRNA) into the embryonic neocortex of Tis21::GFP knock‐in mice, expression of GFP, which occurs specifically in neural stem cells committed to neurogenesis, was found to be nearly completely (≈90%) abolished in the progeny of the targeted cells. Importantly, upon in utero electroporation directly of recombinant Cas9/gRNA complex, near‐maximal efficiency of disruption of GFP expression was achieved already after 24 h. Furthermore, by using microinjection of the Cas9 protein/gRNA complex into neural stem cells in organotypic slice culture, we obtained disruption of GFP expression within a single cell cycle. Finally, we used either Cas9 plasmid in utero electroporation or Cas9 protein complex microinjection to disrupt the expression of Eomes/Tbr2, a gene fundamental for neocortical neurogenesis. This resulted in a reduction in basal progenitors and an increase in neuronal differentiation. Thus, the present in vivo application of the CRISPR/Cas9 system in neural stem cells provides a rapid, efficient and enduring disruption of expression of specific genes to dissect their role in mammalian brain development.
Keywords: CRISPR/Cas9, in utero electroporation, microinjection, neural stem cell, neurogenesis
Subject Categories: Methods & Resources, Stem Cells
Introduction
Manipulation of gene expression is a major way of assessing the function of individual genes in their physiological context. In the study of central nervous system development, as with other physiological systems, manipulation of gene expression is obtained usually either by transgenesis or by RNAi. For mammals, transgenesis, although the most comprehensive method, is laborious and time‐consuming, even in the case of the most amenable model organism, the mouse. Moreover, to ablate specific gene expression during brain development, the generation of conditional knockout lines, which constitutes additional effort, is often required, because ubiquitous deletion of relevant genes is often lethal already during early embryogenesis. As an alternative to transgenic knockout approaches, RNAi has been frequently used, but since it targets transcripts rather than the genomic locus, its effects are of a transient nature and are sometimes accompanied by off‐target effects.
The limitations of transgenic knockouts and RNAi‐mediated knockdowns in the study of mammalian neurodevelopment could potentially be overcome by fast and efficient disruption of gene expression using the recently described CRISPR/Cas9 technology 1, 2, 3. This technology originates from the type II bacterial CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 (CRISPR‐associated protein 9) system and is based on the recruitment of the Cas9 protein to the genomic locus complementary to the sequence of a chosen specific guide RNA (gRNA). Once at the target site, Cas9 generates a double‐strand DNA break that the cell attempts to repair, in most cases by non‐homologous end‐joining (NHEJ). Importantly, NHEJ can generate insertions and deletions (indels) that in turn can lead to permanent disruption of expression of the targeted gene, most often via the generation of a frameshift mutation (for recent reviews on CRISPR/Cas9 system, see 4, 5, 6, 7, 8).
Here, we sought to use the CRISPR/Cas9 system for the rapid yet permanent disruption of the expression of developmentally regulated genes in neural stem and progenitor cells in the neocortex of mouse embryos in vivo. To this end, we explored three distinct approaches. First, we in utero electroporated 9, 10 a single Cas9‐ and gRNA‐encoding plasmid into cortical stem cells of the developing brain. Second, to omit the steps of Cas9 and gRNA production and to accelerate the targeting process, we examined the direct delivery of a Cas9 protein/gRNA complex into these cells by in utero electroporation. Third, to dissect the effects of gene disruption in the immediate progeny of a targeted cortical stem cell, we explored the methodology of microinjection in organotypic slice culture 11, 12 to directly deliver a Cas9 protein/gRNA complex into single neural stem cells in developing brain tissue. Here, we report that these approaches can be successfully used to apply the CRISPR/Cas9 technology to efficiently disrupt the expression of developmentally regulated genes in the mouse brain and to dissect phenotypic consequences at the cell population as well as single cell level during embryonic development.
Results
Disruption of developmentally regulated gene expression in neural stem and progenitor cells upon in utero electroporation of Cas9/gRNA into embryonic mouse neocortex
To obtain proof of principle for the suitability of the CRISPR/Cas9 system to disrupt the expression of a neurodevelopmentally regulated gene, we decided to first target a gene for which one can safely assume that lack of its expression will not cause any phenotype. To this end, we used heterozygous Tis21::GFP knock‐in mice, in which GFP is under the control of the promoter of Tis21, a gene transiently expressed by a subset of neural stem and progenitor cells during brain development 13, 14. Specifically, Tis21 expression in the embryonic neocortex is induced in the ventricular zone (VZ) in those apical radial glial cells (aRGCs) that generate basal progenitors (BPs) destined for the subventricular zone (SVZ), in which Tis21 expression is sustained. BPs in turn generate neurons, which stop expressing Tis21 13, 14.
We initially used in utero electroporation of E13.5 Tis21::GFP mouse embryos (Fig 1A and E) to deliver Cas9 and gRNA into aRGCs. We first in utero electroporated plasmid DNA. For disruption of GFP expression, we used a single plasmid encoding both (i) a GFP‐targeting gRNA (gGFP) under a constitutive promoter (U6) and (ii) the Cas9 gene under a constitutive promoter (CAG) followed by a T2A self‐cleaving site and PaprikaRFP (Fig 1A).
Figure 1. CRISPR/Cas9‐induced disruption of GFP expression in the neocortex of Tis21::GFP mouse embryos upon in utero electroporation.
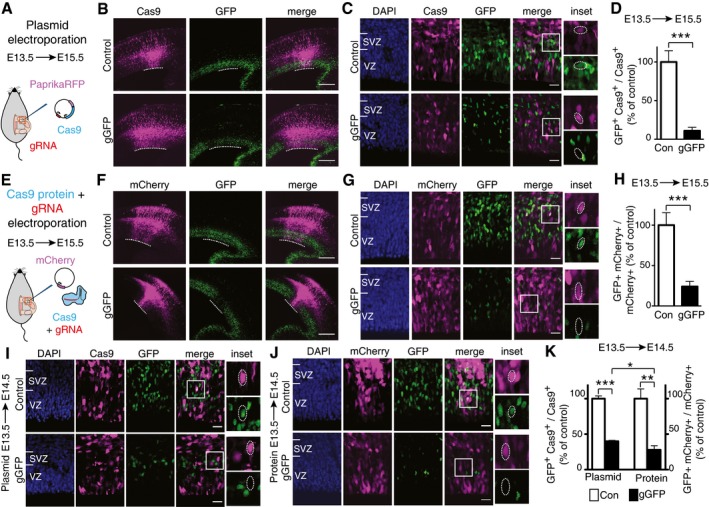
- Scheme of plasmid in utero electroporation.
- Overview of electroporated neocortices showing Cas9 expression as revealed by PaprikaRFP fluorescence (magenta) and the effects of Cas9 expression, together with control gRNA (top) or gGFP (bottom), on GFP expression (green, fluorescence). Dotted lines indicate the electroporated area of the VZ.
- Higher magnification of the VZ and SVZ of the electroporated area shown in (B), with DAPI staining (blue) depicted in addition to PaprikaRFP (Cas9) and GFP fluorescence. Boxes indicate areas shown at higher magnification in the insets (35 × 35 μm). Dotted lines indicate nuclei of progeny of electroporated aRGCs; note the presence of GFP fluorescence in the control (top) and its absence upon Cas9/gGFP electroporation (bottom).
- Quantification of the proportion of Cas9‐positive cells in the VZ plus SVZ that are GFP positive 48 h after control (Con, white) or gGFP (black) Cas9 plasmid electroporation. Data are the mean of four independent experiments (seven embryos per condition in total, from four litters).
- Scheme of Cas9/gRNA complex in utero electroporation.
- Overview of electroporated areas of neocortices as revealed by mCherry fluorescence (magenta) showing the effects of Cas9 protein together with either control gRNA (top) or gGFP (bottom) on GFP expression (green, fluorescence). Dotted lines indicate the electroporated area of the VZ.
- Higher magnification of the VZ and SVZ of the electroporated area shown in (F), with DAPI staining (blue) depicted in addition to mCherry and GFP fluorescence. Boxes indicate areas shown at higher magnification in the insets (35 × 35 μm). Dotted lines indicate nuclei of progeny of electroporated aRGCs; note the presence of GFP fluorescence in the control (top) and its absence upon Cas9/gGFP electroporation (bottom).
- Quantification of the proportion of mCherry‐positive cells in the VZ plus SVZ that are GFP positive 48 h after control (Con, white) or gGFP (black) Cas9 protein electroporation. Data are the mean of four independent experiments (five embryos per condition in total, from four litters).
- VZ and SVZ of the electroporated areas showing Cas9 expression as revealed by PaprikaRFP fluorescence (magenta) and the effects of Cas9 expression, together with control gRNA (top) or gGFP (bottom), on GFP expression (green); blue, DAPI staining. Boxes indicate areas shown at higher magnification in the insets (35 × 35 μm). Dotted lines indicate nuclei of progeny of electroporated aRGCs; note the presence of GFP fluorescence in the control (top) and its absence upon Cas9/gGFP electroporation (bottom).
- VZ and SVZ of the electroporated areas showing targeted cells as revealed by mCherry fluorescence (magenta) and the effects of Cas9 protein together with either control gRNA (top) or gGFP (bottom) on GFP expression (green); blue, DAPI staining. Boxes indicate areas shown at higher magnification in the insets (35 × 35 μm). Dotted lines indicate nuclei of progeny of electroporated aRGCs; note the presence of GFP fluorescence in the control (top) and its absence upon Cas9/gGFP electroporation (bottom).
- Quantification of the proportion of Cas9‐positive cells (left) and mCherry‐positive cells (right) in the VZ plus SVZ that are GFP positive 24 h after control (Con, white) or gGFP (black) Cas9 plasmid (left) or protein (right) electroporation. Data are the mean of three independent experiments each (three embryos per condition in total, from three litters).
To determine the efficiency of different gRNAs to target the GFP gene, we performed an in vitro assay with these in vitro transcribed gRNAs, recombinant Cas9 protein and an ≈800‐bp PCR product of GFP containing the various targeting sites. This led us to choose a gGFP, identical to a previously described one 15 and not hybridizing to the GFP mRNA, which elicited a virtually complete level of on‐target cutting and which was used in all future experiments concerning Tis21::GFP expression (Fig EV1A–C). A LacZ‐targeting gRNA (gLacZ) 16 was used as control.
Figure EV1. Determination of the on‐target Cas9 cutting efficiency directed by various gGFPs in vitro and of the disruption of GFP expression by the most appropriate gGFP in vivo .
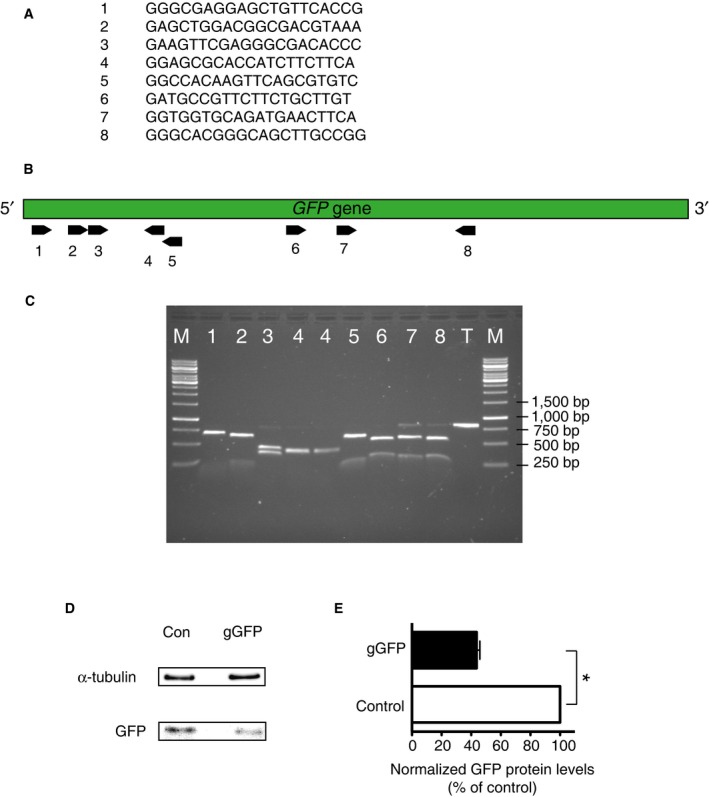
-
ASequences of the tested gRNAs targeting GFP
-
BOrientation and location of the targeting sites of the gRNAs 1–8 described in (A) (black arrows) within the GFP gene (green bar).
-
CAgarose gel showing the effects of the gRNAs 1–8 described in (A) to direct Cas9‐mediated cutting of an 800‐bp‐long GFP PCR product (lanes 1–8). T, template‐only control with no gRNA added; M, DNA size marker. As the targeting site of the gRNA‐1 was the one located most 5′ within the GFP gene, which increases the probability of obtaining disruption of gene expression, and the full‐size template was virtually undetectable upon the in vitro reaction with Cas9 and this gRNA, it was selected for all in vivo experiments and is referred to as gGFP.
-
D, ENeocortex of mouse E13.5 embryos was in utero electroporated with a plasmid encoding Cas9_T2A_PaprikaRFP and gRNA targeting either LacZ (Con) or GFP (gGFP) as in Fig 1I, followed at E14.5 by FACS isolation of PaprikaRFP‐positive cells and immunoblot analysis. (D) Representative immunoblots for α‐tubulin (top), verifying the analysis of equal amounts of total protein, and for GFP, showing its reduced levels upon Cas9/gGFP plasmid electroporation. (E) Quantification of the GFP protein level, normalized to α‐tubulin, 24 h after control (Con, white) or gGFP (black) Cas9 plasmid electroporation. Data are expressed as percentage of the respective control value (set to 100%) and are the mean of three immunoblots (six and eight embryos per condition, respectively, from three litters; gGFP 44%). Error bars indicate SEM; *P < 0.05 (Mann–Whitney U‐test).
As shown in Fig 1B–D, 48 h after the in utero electroporation, only ≈10% of the progeny of the aRGCs targeted with the Cas9/gGFP plasmid, as revealed by PaprikaRFP fluorescence in the VZ and SVZ, showed GFP fluorescence when compared to the control Cas9/gRNA electroporation. This lack of GFP expression in ≈90% of the Tis21::GFP‐positive progeny of the targeted aRGCs shows that in utero electroporation of a plasmid encoding both Cas9 and an appropriate gRNA can successfully disrupt gene expression in neural stem and progenitor cells in the embryonic brain.
In development changes in cell fate typically occur within a single cell cycle of the progenitor cell under study. In this regard, the in utero electroporation of a plasmid encoding both gRNA and Cas9 has the drawback that any genome editing can only occur after the gRNA and Cas9 have been transcribed and Cas9 has been translated, which may take up a substantial portion of interphase. In addition, with plasmid electroporation the number of gRNAs to be expressed is very limited and gRNA/Cas9 expression will continue to occur until the plasmid is diluted by cell division, which will increase the probability of off‐target effects. To overcome these limitations, we sought to directly electroporate the Cas9 protein in a complex with gRNA. Cas9 has a predicted isoelectric point of 9 (ExPASy) and hence is cationic at physiological pH, but gRNA/Cas9 complexes, due to their nucleic acid component, are known to be anionic at physiological pH 17 and thus will migrate towards the anode upon application of an electric field. Indeed, it has recently been shown 17, 18, 19 that gRNA/Cas9 complexes can be delivered into mammalian cell lines by electroporation.
We therefore prepared complexes consisting of either gGFP or control gRNA (gLacZ) and recombinant Cas9 protein and in utero electroporated these, along with a pCAGGS‐mCherry plasmid to identify the targeted aRGCs and their progeny, into the neocortex of E13.5 Tis21::GFP embryos (Fig 1E). When compared to control, > 75% of the Tis21::GFP‐positive progeny of the targeted aRGCs (as indicated by mCherry fluorescence) lacked GFP fluorescence after 48 h (Fig 1F–H). Collectively, these data demonstrate that Cas9 protein together with an appropriate gRNA can be successfully delivered into neural stem cells of developing neocortex by means of in utero electroporation and can efficiently induce disruption of gene expression in these cells and the progeny derived therefrom.
We next investigated how fast Cas9/gGFP plasmid and Cas9 protein/gGFP complexes electroporated into embryonic mouse neocortex were able to disrupt gene expression. To this end, we performed the analysis at an early time point of 24 h after the electroporation (Fig 1I–K). Upon Cas9 protein/gGFP complex electroporation, we detected disruption of GFP expression in 72% of the progeny of the targeted cells when compared to control. Electro‐poration of the Cas9/gGFP plasmid, instead, showed disruption of GFP expression in ≈60% of the progeny when compared to control. The magnitude of this reduction in GFP‐positive cells was corroborated by immunoblotting, which revealed a 56% decrease in the GFP level in electroporated cells (Fig EV1D and E). These findings show that the magnitude of disruption of gene expression upon Cas9/gGFP plasmid electroporation is greater after 48 h when compared to 24 h (Fig 1D vs. K left), whereas electroporation of Cas9 protein/gGFP complexes yields nearly the same magnitude of gene disruption after 24 h or 48 h (Fig 1H vs. K right). This suggests that electroporated Cas9 protein/gRNA complexes exert their effects earlier than electroporated Cas9/gRNA plasmids, presumably because in the latter case additional time is required for transcription of the gRNA and transcription and translation of the Cas9 protein.
Next, we sought to compare the in vivo efficiency of the CRISPR/Cas9 system, delivered as either Cas9/gRNA plasmid or Cas9 protein/gRNA complexes (Fig 1), to that of RNAi, using esiRNA electroporation 9, 10. To this end, we in utero electroporated esiRNAs targeting GFP (esiGFP) or luciferase (Control), together with a pCAGGS‐mCherry plasmid to identify the targeted aRGCs and their progeny, into the neocortex of E13.5 Tis21::GFP embryos (Fig EV2). Our results show that at 24 h after electroporation, the disruption of GFP expression by esiRNA (61%, Fig EV2B) is less than that by Cas9 protein/gRNA complexes (72%, Fig 1K right), but comparable to that by Cas9/gRNA plasmid (60%, Fig 1K left). At 48 h after electroporation, the disruption of GFP expression by esiRNA (70%, Fig EV2D) was found to be less than that by Cas9/gRNA plasmid (90%, Fig 1D), but almost comparable to Cas9 protein/gRNA complexes (76%, Fig 1H). These results indicate that the CRISPR/Cas9 system, when applied to the developing brain in vivo, is at least comparable, if not superior, to RNAi in terms of the efficiency of disruption of gene expression.
Figure EV2. RNAi‐induced disruption of GFP expression in the neocortex of Tis21::GFP mouse embryos upon in utero electroporation.
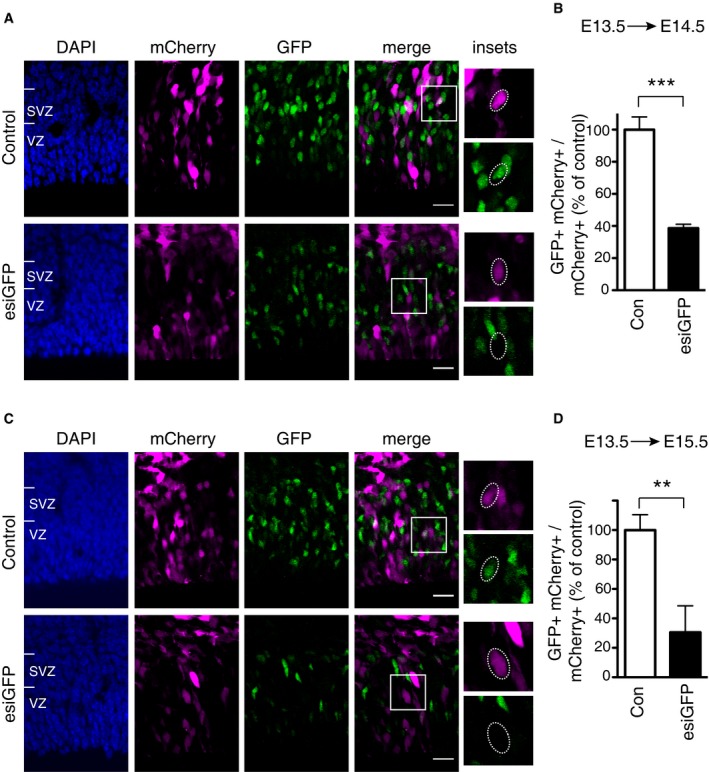
- VZ and SVZ of the electroporated areas showing targeted cells as revealed by mCherry fluorescence (magenta) and the effects of either control esiRNAs (top) or esiGFP (bottom) on GFP expression (green) 24 h after electroporation; blue, DAPI staining. Boxes indicate areas shown at higher magnification in the insets (35 × 35 μm). Dotted lines indicate nuclei of progeny of electroporated aRGCs; note the presence of GFP fluorescence in the control (top) and its absence upon esiGFP electroporation (bottom).
- Quantification of the proportion of mCherry‐positive cells in the VZ plus SVZ that are GFP positive 24 h after control (Con, white) or esiGFP (black) electroporation. Data are the mean of three independent experiments (five embryos per condition in total, from three litters).
- VZ and SVZ of the electroporated areas showing targeted cells as revealed by mCherry fluorescence (magenta) and the effects of either control esiRNAs (top) or esiGFP (bottom) on GFP expression (green) 48 h after electroporation; blue, DAPI staining. Boxes indicate areas shown at higher magnification in the insets (35 × 35 μm). Dotted lines indicate nuclei of progeny of electroporated aRGCs; note the presence of GFP fluorescence in the control (top) and its absence upon esiGFP electroporation (bottom).
- Quantification of the proportion of mCherry‐positive cells in the VZ plus SVZ that are GFP positive 48 h after control (Con, white) or esiGFP (black) electroporation. Data are the mean of three independent experiments (three embryos per condition in total, from three litters).
Microinjection of Cas9 protein/gRNA into single neural stem cells in embryonic mouse neocortex allows analysis of disruption of gene expression in the immediate daughter cells
In the light of these data, we examined whether disruption of gene expression by Cas9 protein can be achieved in single neural stem cells and affects their immediate daughter cells. To this end, we made use of a recently developed system 11, 12 in which nucleic acids and/or proteins are microinjected into single neocortical aRGCs in organotypic slice culture and the fate of the daughter cell pair arising from an aRGC division is examined (see also 20). Specifically, aRGCs in slices of telencephalon of heterozygous E14.5 Tis21::GFP mice were microinjected with recombinant Cas9 protein together with either gGFP or control gRNA (gLacZ), along with dextran 10,000‐Alexa 555 (Dx‐A555) as a fluorescent tracer to identify the daughter cells of the microinjected aRGCs (Fig 2A).
Figure 2. CRISPR/Cas9‐induced disruption of GFP expression in the daughter cells of single microinjected aRGCs in organotypic slices of telencephalon of Tis21::GFP mouse embryos.
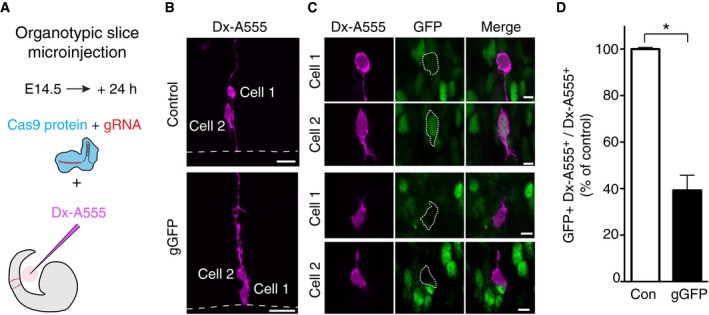
- Scheme of the Cas9/gRNA complex microinjection.
- Daughter cells of single aRGCs microinjected with either Cas9/control gRNA (top) or Cas9/gGFP (bottom) revealed by Dx‐A555 immunofluorescence (magenta); cell 1, aRGC daughter; cell 2, BP daughter. Dashed lines indicate ventricular surface. Images are maximum intensity projections of 20 (Control) and 24 (gGFP) optical sections. Scale bars, 20 μm.
- Single optical sections of cells 1 and 2 depicted in (B), showing the effects of Cas9 and control gRNA (top) or gGFP (bottom) on GFP expression (as revealed by immunofluorescence, green). Cells 1 and 2 are indicated by dotted lines; note the presence of GFP immunofluorescence in the daughter cells in control and its absence upon Cas9/gGFP microinjection. Scale bars, 5 μm.
- Quantification of the proportion of daughter cells (Dx‐A555+) of microinjected cells that show GFP expression 24 h after control (Con, white) or gGFP (black) microinjection. Control is set to 100% and the gGFP condition expressed relative to control (39%). Data are the mean of two independent experiments (three embryos and one litter per experiment, total number of daughter cells scored: control 142, gGFP 88); bars indicate the variation of the two individual values from the mean (*P < 0.05, Fisher's test).
Figure 2B shows two examples of daughter cell pairs after 24 h of organotypic slice culture. In both Cas9 protein/control gRNA and Cas9 protein/gGFP microinjection, the two daughter cells had adopted a different fate as revealed by their morphology. One daughter (cell 1) had remained attached at the ventricular surface and exhibited the characteristic bipolar shape of an aRGC, whereas the other daughter (cell 2) had delaminated from the ventricular surface and showed the typical multipolar shape of a newborn basal intermediate progenitor (bIP). These two cases of daughter cell pair fate were indicative of an asymmetric, self‐renewing bIP‐genic division of the microinjected aRGC mother cells, which in embryonic neocortex of Tis21::GFP mice are known to be Tis21::GFP positive 14, 21.
Consistent with this, both daughter cells of the aRGC microinjected with Cas9 protein/control gRNA showed GFP fluorescence (Fig 2C top). In contrast, the daughter cells of the aRGC microinjected with Cas9 protein/gGFP lacked GFP fluorescence (Fig 2C bottom), indicative of disruption of Tis21::GFP expression. In fact, as the cell bodies of both the self‐renewed aRGC daughter and the newborn bIP daughter were still observed at and near the ventricular surface, respectively (Fig 2C bottom), the division of the microinjected aRGC mother cell must have occurred relatively late within the 24‐h culture period following the microinjection, which presumably targeted an aRGC in G1. Given a half‐life of the GFP in the progeny of Tis21::GFP‐expressing cells of approximately 12 h 14, we conclude that disruption of gene expression upon microinjection of Cas9 protein/gGFP into the aRGC occurred very rapidly thereafter and before the cell underwent asymmetric division.
Quantification of the magnitude of disruption of Tis21::GFP expression upon Cas9 protein/gGFP microinjection into aRGCs revealed a 61% reduction in GFP expression in their daughter cells (Fig 2D). Of note, the magnitude of this reduction in GFP‐immunoreactive aRGC progeny 24 h after Cas9 protein/gGFP microinjection was very similar to that observed 24 h after microinjection of GFP esiRNA (58%, Fig EV3). Taken together, these data demonstrate that upon microinjection into single aRGCs in organotypic slice culture of developing neocortex, Cas9 protein together with an appropriate gRNA can rapidly and efficiently induce disruption of gene expression in these neural stem cells and their progeny.
Figure EV3. RNAi‐induced disruption of GFP expression in the daughter cells of single microinjected aRGCs in organotypic slices of telencephalon of Tis21::GFP mouse embryos.
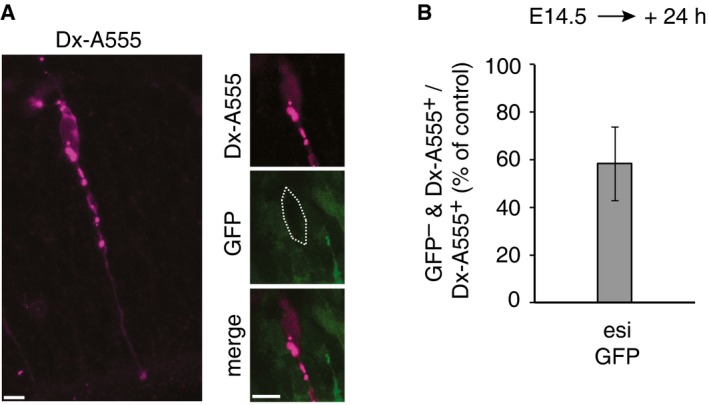
- Maximum intensity projection (left, stack of 5 optical sections) of a daughter cell of a single aRGC microinjected with esiGFP (as revealed by Dx‐A555 immunofluorescence, magenta), and a single optical section (right) of the cell body (dotted line) of that cell, showing an example of the lack of Tis21::GFP expression (as revealed by GFP immunofluorescence, green). Scale bars, 5 μm.
- Quantification of the proportion of Dx‐A555+ cells that lack Tis21::GFP expression (GFP –) 24 h after esiGFP microinjection. Data are expressed as percentage of the value obtained 24 h after control esiRNA microinjection, which corresponded to that of dextran‐only microinjected aRGCs 12, and are the mean of two independent experiments (21 and 23 Dx‐A555+ daughter cells of esiGFP‐microinjected aRGCs scored, respectively); bar indicates the variation of the two individual values from the mean.
CRISPR/Cas9‐mediated disruption of Eomes/Tbr2 expression in embryonic neocortex affects neurogenesis
To demonstrate that the present system of delivering Cas9/gRNA into neural stem cells and of analysing the fate of their progeny can be used to dissect a neurodevelopmental phenotype, we sought to target the gene Eomes, which encodes the transcription factor Tbr2 (for the sake of simplicity, we shall use the term Tbr2 for both gene and protein from here onwards). Tbr2 has a fundamental role in neocortical development. Homozygous silencing of Tbr2 in humans leads to neurodevelopmental disorders such as microcephaly 22. Conditional ablation of Tbr2 expression in the embryonic mouse neocortex causes reduction in BPs and an increase in direct neurogenesis from aRGCs 23, 24. During mouse corticogenesis, Tbr2 mRNA levels are highest in Tis21‐positive aRGCs, which however lack Tbr2 protein (presumably due to microRNA‐mediated translational repression), followed by bIPs, which characteristically contain the Tbr2 protein, while newborn neurons stop expressing Tbr2 20, 25, 26, 27.
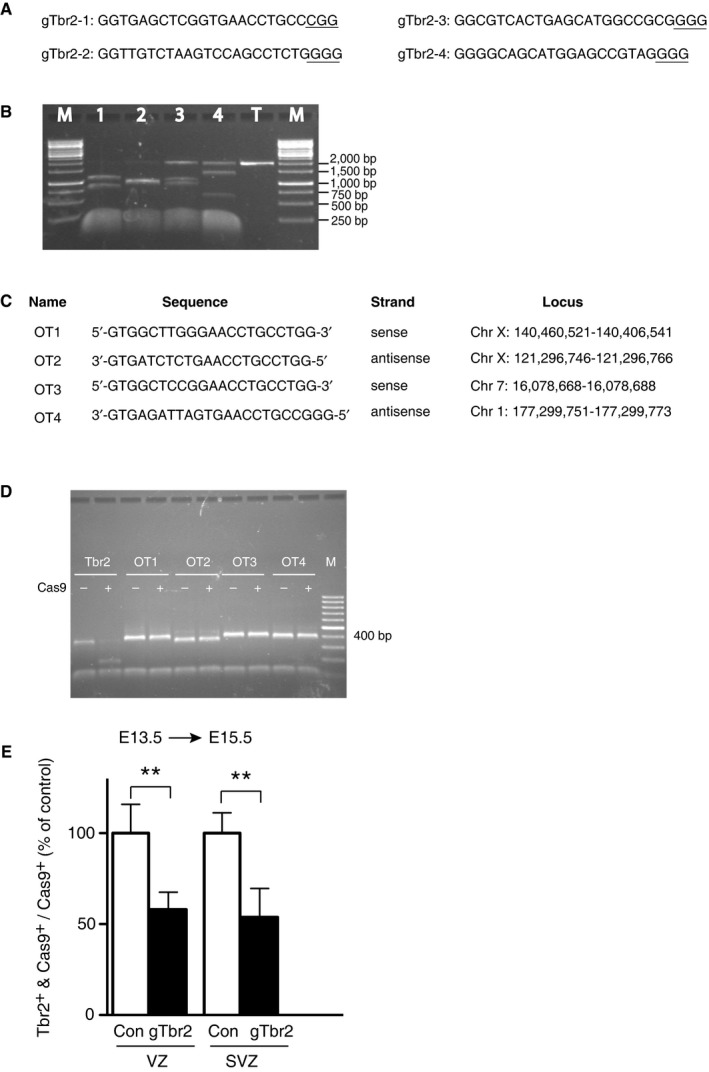
To disrupt Tbr2 expression, we chose 4 gRNAs targeting different sites in exon 1 of the mouse Tbr2 gene (Fig EV4A) and assessed in vitro the efficiency of these gRNAs to induce cutting by recombinant Cas9 of a ≈2‐kb genomic PCR product containing exon 1 (Fig EV4B). Importantly, the gRNA with the highest on‐target efficiency (gTbr2‐1) showed no off‐target activity towards four major off‐target sites (Fig EV4C and D). This gRNA (from here onwards referred to as gTbr2), which did not hybridize to the Tbr2 mRNA, was used in all future experiments concerning Tbr2 expression and function.
Figure EV4. Determination of the on‐target Cas9 cutting efficiency and specificity when directed by various gTbr2s in vitro, and of the in vivo disruption of Tbr2 expression in the VZ and SVZ by the most appropriate gTbr2.
-
A, BIn vitro assay of on‐target Cas9 cutting efficiency directed by various gTbr2s. (A) Sequences of the 4 tested gRNAs targeting Tbr2, referred to as gTbr2‐1 to gTbr2‐4, all of which target exon 1. PAM (protospacer adjacent motif) is underlined. (B) Agarose gel showing the effect of the gTbr2‐1 to gTbr2‐4 described in (A) to direct Cas9‐mediated cutting of a 2‐kb‐long Tbr2 PCR product (lanes 1–4). T, template‐only control with no gTbr2 added; M, DNA size marker. The predicted size of the PCR product fragments after Cas9/gTbr2‐mediated cutting was as follows: gTbr2‐1 (850 bp + 1,150 bp), gTbr2‐2 (1,000 bp + 1,000 bp), gTbr2‐3 (1,100 bp + 900 bp) and gTbr2_4 (1,400 bp + 600 bp). Note that the size of the fragments observed matched the predictions. As the targeting site of the gTbr2‐1 was the one located most 5′ within exon 1 of the Tbr2 gene, and as this gTbr2 exhibited the highest efficiency to direct Cas9‐mediated cutting as assessed by the lack of detectable full‐size template, it was selected for all in vivo experiments and for simplicity is referred to just as gTbr2.
-
C, DIn vitro assay of the specificity of Cas9 cutting when directed by gTbr2. (C) Names, sequences, strands and genomic loci of the four major off‐target sites (labelled OT1–4) of the gTbr2. (D) Agarose gel showing the lack of effect of gTbr2 to direct Cas9‐mediated cutting of the ≈400‐bp PCR products of the off‐target loci (lanes OT1–OT4) as compared to that of the on‐target locus (lane Tbr2). M, DNA size marker.
-
ECRISPR/Cas9‐mediated reduction in Tbr2 immunoreactivity in both VZ and SVZ. Neocortex of mouse E13.5 embryos was in utero electroporated with a plasmid encoding Cas9_T2A_PaprikaRFP and either control gRNA (Con) or gTbr2, and analysed at E15.5 as in Fig 3A–D. Quantification of the proportion of Cas9+ cells that are Tbr2+ in the VZ (left) and SVZ (right) 48 h after control (Con, white) or gTbr2 (black) Cas9 plasmid electroporation. Data are the mean of four independent experiments (four embryos per condition, from four litters). Controls were set to 100% and the gTbr2 condition expressed relative to control (VZ, 58%; SVZ, 54%). Error bars indicate SD; **P < 0.01 (Student's t‐test).
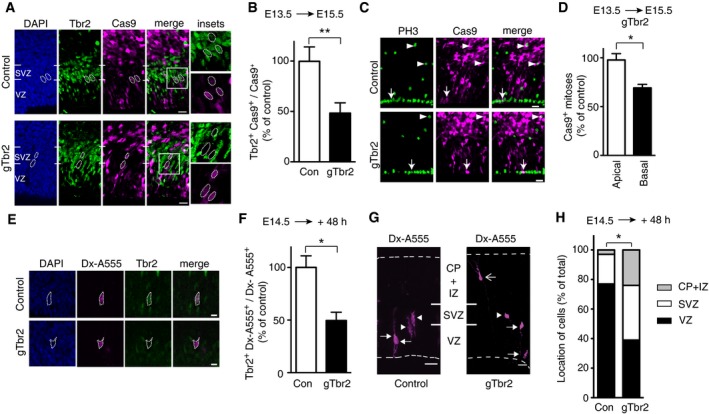
Next, a plasmid encoding constitutively expressed Cas9_T2A_PaprikaRFP and either gTbr2 or control gRNA (gLacZ) (Fig 1A) was in utero electroporated into E13.5 wild‐type mouse embryos. Analysis of PaprikaRFP‐positive cells in the VZ plus SVZ revealed that 48 h post‐electroporation, the Cas9/gTbr2‐targeted cells that showed Tbr2 immunoreactivity were reduced by half when compared to control (Fig 3A and B). The magnitude of the reduction in Tbr2‐immunoreactive cells in the VZ plus SVZ did not increase further at later time points post‐electroporation and was in the same range as the in vivo efficiency of the gTbr2 to induce indels at the target site in exon 1 of the Tbr2 gene upon electroporation of the Cas9/gTbr2 plasmid (see Fig 4A below). Separate analysis of the VZ vs. SVZ 48 h after electroporation of the control Cas9/gRNA plasmid resulted in an essentially identical ≈50% reduction in Tbr2‐immunoreactive cells in both VZ and SVZ (Fig EV4E). We conclude that: (i) expression in aRGCs of Cas9, together with the gTbr2 used here, causes bi‐allelic disruption of the Tbr2 gene in about half of these cells and their progeny and (ii) this suffices, despite the presence of Tbr2 mRNA in BP‐genic aRGCs 20, to render half of the aRGC‐derived BPs Tbr2‐negative, both newborn ones transiting the VZ and mature ones still found in the SVZ 48 h post‐electroporation.
Figure 3. CRISPR/Cas9‐mediated disruption of Tbr2 expression affects neurogenesis in embryonic mouse neocortex.
-
A–DNeocortex of mouse E13.5 embryos was in utero electroporated with a plasmid encoding, under constitutive promoters, Cas9_T2A_PaprikaRFP and gRNA targeting either LacZ (Control, Con) or Tbr2 (gTbr2), followed by analysis at E15.5. (A) Electroporated area showing the effects of Cas9 expression (revealed by PaprikaRFP fluorescence, magenta) together with either control gRNA (top) or gTbr2 (bottom) on Tbr2 expression (green); blue, DAPI staining. Boxes indicate areas shown at higher magnification in the insets (45 × 45 μm). Dotted lines indicate nuclei of progeny of electroporated aRGCs; note the presence of Tbr2 immunoreactivity in the control (top) and its absence upon Cas9/gTbr2 plasmid electroporation (bottom). (B) Quantification of the proportion of Cas9+ cells that are Tbr2+ 48 h after control (Con, white) or gTbr2 (black) plasmid electroporation. Data are the mean of four independent experiments (six embryos per condition, from four litters). (C) Electroporated area showing the effects of Cas9 expression (revealed by PaprikaRFP fluorescence, magenta) together with either control gRNA (top) or gTbr2 (bottom) on the abundance of mitotic progenitors as revealed by phosphohistone H3 (PH3) immunofluorescence (green). Apical and basal mitoses are indicated by arrows and arrowheads, respectively; note the reduction in mitotic Cas9+ BPs upon Cas9/gTbr2 plasmid electroporation. (D) Quantification of apical and basal Cas9+ mitoses (as revealed by PH3 immunofluorescence) in a unit area 48 h after Cas9/gTbr2 plasmid electroporation. Data are expressed as percentage of the data obtained upon control plasmid electroporation and are the mean of three independent experiments (three embryos per condition in total, from three litters); note the reduction in basal (black, 68%) but not apical (white) mitoses.
-
E–HSingle aRGCs in neocortex in organotypic slices of telencephalon of E14.5 mouse embryos were microinjected with recombinant Cas9 protein together with gRNA targeting either LacZ (Control, Con) or Tbr2 (gTbr2) and with dextran 10,000‐Alexa 555 (Dx‐A555), followed by analysis after 48 h of culture. (E) Progeny of single microinjected aRGCs (revealed by Dx‐A555 immunofluorescence, magenta, dotted lines) showing the effects of Cas9 together with either control gRNA (top) or gTbr2 (bottom) on Tbr2 expression (green); blue, DAPI staining. Note the presence of Tbr2 immunoreactivity in the control (top) and its absence upon Cas9/gTbr2 (bottom) protein microinjection. Images are single optical sections. Scale bars, 5 μm. (F) Quantification of the proportion of the progeny (Dx‐A555+) of microinjected aRGCs that show Tbr2 expression 48 h after control (Con, white) or gTbr2 (black) microinjection. Data are the mean of 3 independent experiments (3 embryos per condition; total number of cells scored: control 45, gTbr2 41). (G) Distribution across the cortical wall of the progeny (revealed by Dx‐A555 immunofluorescence, magenta; arrows, aRGC; arrowheads, BPs; open arrow, neuron) of single control (left) or gTbr2 (right)‐microinjected aRGCs. Dashed lines indicate ventricular surface (bottom) and basal lamina (top). Images are maximum intensity projections of 67 (Control) and 94 (gTbr2) single optical sections. (H) Quantification of the distribution of the progeny (Dx‐A555+) of microinjected aRGCs in the VZ, SVZ and intermediate zone/cortical plate (CP+IZ) 48 h after control (Con, white) or gTbr2 (black) microinjection (total number of cells scored: control 37, gTbr2 51, pooled from at least 3 independent experiments). Note that upon gTbr2 microinjection, the distribution of cells across the various cortical zones is significantly different (VZ: control 77%, gTbr2 39%; SVZ: control 20%, gTbr2 37%; IZ/CP: control 3%, gTbr2 24%; *P < 0.05, Kolmogorov–Smirnov test).
Figure 4. Sequencing analysis of the CRISPR/Cas9‐induced disruption of the Tbr2 locus.
-
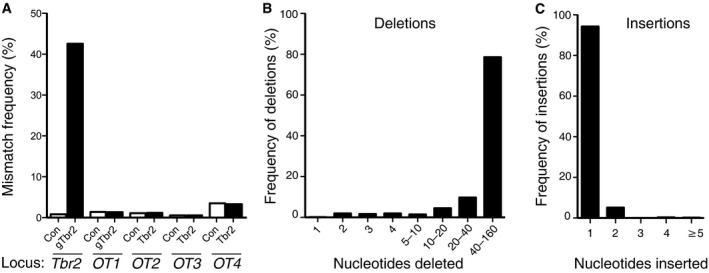
AQuantification of the frequency of mismatches to the mouse genome sequence spanning 30 bp upstream and downstream of the Tbr2 target site and the off‐target sites (OT1‐4). Data are expressed as percentage of the total number of reads and are the mean of two independent experiments (8 and 5 embryos per condition, respectively).
-
B, CAnalysis of one of the experiments of (A) for deletions (B) and insertions (C) in the region spanning 80 bp upstream and downstream of the Tbr2 target site upon Cas9/gTbr2 plasmid electroporation.
Complete ablation of the Tbr2 gene in the developing mouse neocortex has previously been shown to result in a reduction in the level of BPs of up to ≈50% 23, 24. Given the disruption of the Tbr2 gene in about half of the targeted neocortical aRGCs and their progeny upon in utero electroporation of the Cas9/gTbr2 plasmid, one would therefore expect a reduction in the level of derivative BPs by ≈25% if the absence of Tbr2 protein were to exert a similar effect as previously observed. Indeed, upon Cas9/gTbr2 plasmid electroporation, we observed exactly this extent of reduction in the abundance of mitotic BPs as compared to control, without any effect on the abundance of apical mitoses (Fig 3C and D). Thus, CRISPR/Cas9‐mediated disruption of a major determinant of BP generation by in utero electroporation of neural stem cells in developing neocortex results in the expected phenotype in their progeny.
We further dissected the phenotype of CRISPR/Cas9‐mediated disruption of Tbr2 gene expression by microinjecting single neocortical aRGCs in organotypic slice culture at E14.5 with recombinant Cas9 protein together with gTbr2 and examining the fate of their progeny 48 h later. In agreement with the in utero electroporation data (Fig 3A and B), the progeny of aRGCs microinjected with Cas9 protein and gTbr2 showed a ≈50% reduction in Tbr2 expression compared to that of aRGCs microinjected with Cas9 protein and control gRNA (Fig 3E and F). This disruption of Tbr2 expression had a major effect on the fate of the progeny. Upon Cas9 protein/control gRNA microinjection, 77% of the progeny were cells remaining in the VZ and 23% were cells that had delaminated and were located basally (SVZ and IZ+CP combined, Fig 3G and H). In striking contrast, upon Cas9 protein/gTbr2 microinjection, 61% of the progeny were cells in basal locations, including cells in the intermediate zone and cortical plate exhibiting neuronal morphology (Fig 3G and H). These data suggest that upon loss of Tbr2 mRNA from BP‐genic aRGCs and of Tbr2 protein from the aRGC progeny, aRGC self‐renewal is decreased and neuronal differentiation is increased.
Collectively, our results show that CRISPR/Cas9‐mediated disruption, upon either in utero electroporation or microinjection into neural stem cells in developing neocortex, of a gene fundamentally important for neocortex development allows mechanistic dissection of the resulting phenotype in the immediate progeny of the targeted cells.
In utero electroporation of Cas9/gTbr2 does not induce detectable off‐target effects
To corroborate that the neurodevelopmental phenotype observed upon Cas9/gTbr2 electroporation (Fig 3) indeed resulted from the disruption of the Tbr2 locus rather than another locus, we performed next‐generation sequencing of amplicons containing either the target site or one of four major off‐target sites (Fig EV4C). To this end, we prepared a cell suspension from E15.5 neocortex 48 h after in utero electroporation of Cas9‐T2A‐PaprikaRFP/gTbr2 or Cas9‐T2A‐PaprikaRFP/gLacZ plasmids (as in Fig 3A–D), isolated the PaprikaRFP‐expressing cells by FACS, and used their DNA to generate ≈300‐bp amplicons.
Upon control Cas9/gRNA electroporation, < 1% of the ≈100,000 reads of the Tbr2 locus analysed showed mismatches to the mouse genome reference sequence (Fig 4A), suggesting that this was the level of PCR error. In contrast, upon Cas9/gTbr2 electroporation, mismatches were found on average in about half of all reads (≈43%, Fig 4A), indicating an indel frequency in vivo that was consistent with the disruption of Tbr2 gene expression in about half of the progeny of the targeted aRGCs as revealed by Tbr2 immunofluorescence (Fig 3B).
Importantly, no increase in the low frequency of mismatches compared to control was found for the four major off‐target sites upon Cas9/gTbr2 electroporation (Fig 4A). Taken together, these data strongly suggest that the neurodevelopmental phenotype observed upon targeting with Cas9/gTbr2 (Fig 3) was indeed caused by the selective disruption of the Tbr2 locus.
We further analysed the types of indels caused by Cas9/gTbr2. Almost all indels either started at the targeted site or encompassed it (see Fig EV5 for a few examples), underlining the specificity of the effect of Cas9/gTbr2 on the targeted site. The majority (73%) of the mutations detected at the targeted site and in its immediate proximity (80 bp downstream to 80 bp upstream) were deletions. Amongst them, deletions longer than 40 nucleotides were the most frequent (Fig 4B). With regard to the insertions detected, by far the most frequent were single‐nucleotide insertions at the targeted site (Fig 4C). These data provide a mechanistic explanation for the lack of Tbr2 protein, as the gTbr2 target site in the Tbr2 gene is located at the codon in exon 1 encoding leucine‐14 and three quarters of all indels (two‐thirds of all deletions plus the single nucleotide insertion) will cause a premature stop codon.
Figure EV5. Examples of types of indels caused by electroporation of Cas9/gTbr2 plasmid.
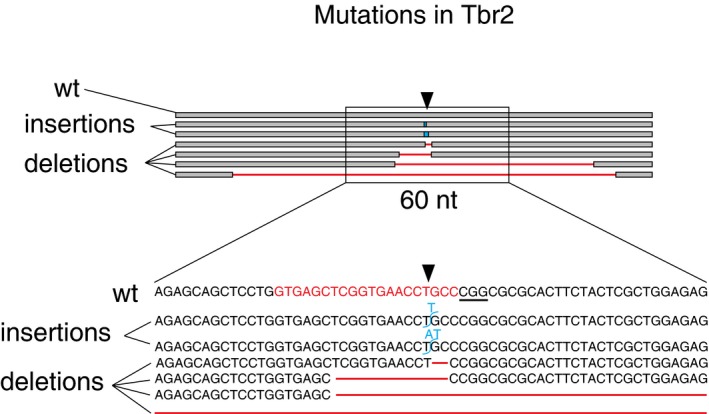
Illustration of the genomic locus 100 bp upstream and downstream (top) of the gTbr2 target site (indicated by black arrowheads) in the 5′ region of exon 1 of the Tbr2 gene, showing the wild‐type read (wt) and six examples of mutated reads with indels (two insertions: blue; four deletions: red). Bottom: the region 30 bp upstream and downstream of the target site (rectangle, 60 nt). gTbr2 targeting sequence is labelled in red in the wild‐type sequence. Underlined nucleotides indicate PAM.
Discussion
We have established CRISPR/Cas9‐mediated disruption of gene expression for investigating the role of gene products in mammalian brain development in vivo. Specifically, we achieved the ablation of developmentally regulated proteins in embryonic mouse neocortex by application of two approaches that allow for rapid and efficient delivery of the relevant components of the CRISPR/Cas9 system, in utero electroporation of the primary germinal zone of the neocortex and microinjection into single neural stem cells in neocortical tissue. The present methodology considerably advances previous approaches of acutely interfering with gene expression in the developing brain, which were essentially based on RNAi 10, 28, 29, in that it rapidly results in permanent gene disruption in the targeted neural stem cells and their progeny. Our study also substantially extends previous applications of the CRISPR/Cas9 technology to investigate the function of genes with key roles in the brain 30, 31 in that we have dissected the effects of gene disruption in neural stem cells and their immediate progeny during embryonic development. Three aspects of the present methodology deserve particular discussion.
First, the efficiency with which a rapid‐onset phenotype is observed. We demonstrate that upon in utero electroporation of a single plasmid encoding Cas9 and an appropriate gRNA, we were able to detect a phenotype, caused by the resulting disruption of gene expression, already in the immediate progeny of the targeted neural stem cells. The efficiency of disruption was such that it resulted in a 50% to 90% loss of the protein product 48 h after gene targeting, as revealed by (immuno)fluorescence. In fact, as exemplified for Tbr2, the lack of detectable protein product in the progeny of the neural stem cells matched the occurrence of indels at the genomic target site. This not only implies that bi‐allelic gene disruption rapidly occurred in most, if not all, targeted neural stem cells with indels at the Tbr2 locus, but also that existing Tbr2 protein disappeared from the progeny during the 48 h time period between gene targeting and immunofluorescence analysis.
Second, the direct in vivo delivery of a recombinant Cas9 protein/gRNA complex into neural stem cells of embryonic neocortex by in utero electroporation. Electroporating a protein/gRNA complex instead of a plasmid in order to deliver Cas9 and gRNA into cells in vivo has been shown 17, 18 to reduce off‐target effects and to facilitate delivering multiple gRNAs at the same time. Moreover, and of importance for neurodevelopmental studies, electroporation of a protein/gRNA complex instead of a plasmid reduces the time needed for genome editing to take place, as shown in the present study (Fig 1). Moreover, this mode of introducing the Cas9 protein/ gRNA complex into neural stem cells constitutes, to the best of our knowledge, the first case of directly delivering a protein into the cytoplasm of VZ cells in the mammalian brain by electroporation. It should be noted that it may be worthwhile to explore extending this approach to proteins other than Cas9 that carry a sufficient net charge at physiological pH, such as appropriate antibodies, or when studying an acute action of a protein in the targeted VZ cells is desired.
Third, the single cell resolution for analysing phenotypes that is obtained upon microinjection of a recombinant Cas9 protein/gRNA complex into neural stem cells in neocortical tissue. As microinjection predominantly targets single aRGCs in the G1 phase of the cell cycle 11, it enables us to assess the effects of CRISPR/Cas9‐mediated disruption of expression of the gene under study already within the same cell cycle of the microinjected neural stem cell. This in turn is of particular importance in developmental biology, in which fate‐changing events that occur within the life cycle of a given single stem or progenitor cell constitute a major area of research.
In conclusion, we have shown that the CRISPR/Cas9 system can be successfully applied to dissect the function of specific genes during embryonic brain development. In the light of the advantages of the CRISPR/Cas9 system over other modes of gene inactivation, the present approaches to deliver the components of this system rapidly and efficiently into neural stem cells in vivo provide new avenues for future functional screenings and loss‐of‐function studies.
Materials and Methods
Animals
All experimental procedures were conducted in agreement with the German Animal Welfare Legislation after approval by the Landesdirektion Sachsen. Animals used for this study were kept pathogen‐free at the Biomedical Services Facility (BMS) of the MPI‐CBG. Embryonic day E0.5 was set at noon of the day of vaginal plug identification. All experiments were performed in the dorsolateral telencephalon of mouse embryos, at a medial position along the rostro‐caudal axis. Mouse lines used were Tis21::GFP 14 and C57Bl/6J wild‐type mice (Janvier).
Plasmids, gRNAs and recombinant protein
Previously published gRNAs targeting GFP (for sequences, see Fig EV1) and LacZ (5′‐TGCGAATACGCCCACGCGATCGG; underlined nucleotides, PAM) were used 15, 16. Genomic sequence of mouse Eomes/Tbr2 was analysed for CRISPR/Cas9 target sites by Geneious 8.1.6 software (Biomatters), and four gRNAs targeting exon 1 were selected (for sequences, see Fig EV4). All gRNAs used in vivo were identical in sequence to the DNA sense strand and not complementary to the mRNA sequence. pD1321‐AP plasmids containing gRNAs under the control of the human U6 promoter were obtained from DNA 2.0. The same plasmid contained the Cas9 gene under the CAG promoter, followed by sequences encoding the proteolytic self‐cleavage 2A peptide from Thosea asigna virus (T2A) and PaprikaRFP, which was used as a marker of Cas9 expression. Cas9 was flanked with two nuclear localization signals (5′ and 3′). gRNAs were either purchased (Eupheria Biotech) or produced by in vitro transcription from a PCR product containing the T7 promoter followed by the gRNA sequence and the optimized gRNA scaffold 32 using the esiSCRIBE In Vitro Transcription Kit (Eupheria Biotech) following the manufacturer's instructions. Recombinant Cas9 protein (ToolGen, or MPI‐CBG) from Streptococcus pyogenes was used. esiRNAs used in Figs EV2 and EV3 have been previously described 10, 33.
In utero electroporation
In utero electroporations were performed as previously described 9, 10. Briefly, E13.5 mouse embryos were anesthetized with isofluorane and subsequently injected subcutaneously with an analgesic. The peritoneal cavity was then surgically opened and the uterus exposed. For the electroporation of plasmids (Figs 1B–D, I and K, 3A–D and 4), embryos were injected intraventricularly with a solution containing 0.1% Fast Green (Sigma) in sterile PBS, containing 2.5 μg/μl of one of the Cas9‐PaprikaRFP‐gRNA plasmids, with the gRNA being either gLacZ, gGFP or gTbr2.
For the recombinant Cas9 protein/gRNA in utero electroporation (Fig 1E–H, J and K), embryos were prepared in the same way as described above. Prior to injection, 1 μg/μl recombinant Cas9 protein in protein buffer (20 mM Hepes pH 7.5, 150 mM KCl) and reaction mixture containing 0.33 μg/μl of the in vitro transcribed gRNA of interest in a total volume of 15 μl were incubated for 15 min at 37°C to form a functional complex, followed by a centrifugation (1 min, 13,000 g) through a Durapore PVDF 0.22‐μm filter (Merck Millipore). Upon addition of Fast Green and the pCAGGS‐mCherry plasmid, the mixture to be injected contained, in final concentrations, 0.5 μg/μl recombinant Cas9 protein in complex with 0.167 μg/μl of the gRNA of interest, together with 0.3 μg/μl pCAGGS‐mCherry plasmid and 0.1% of Fast Green.
For the electroporation of esiRNAs (Fig EV2), embryos were prepared in the same way as described above and esiRNAs (0.18 μg/μl) were electroporated together with 0.3 μg/μl pCAGGS‐mCherry plasmid and 0.1% of Fast Green.
All electroporations were performed with six 50‐ms pulses of 30–36 V (for plasmid electroporation) or 36 V (for Cas 9 protein/gRNA electroporation, esiRNA) at 1‐s intervals. Embryos were harvested 24 or 48 h post‐electroporation and PFA‐fixed for immunofluorescence analysis.
Microinjection
E14.5 telencephalon was dissected at room temperature in Tyrode solution pre‐warmed at 37°C. After removal of meninges, ~300‐μm slices were cut using a micro‐knife. Slices were transferred to 3.5‐cm dishes containing 37°C warm slice culture medium (SCM; neurobasal medium (Gibco) supplemented with 10% rat serum (Charles River Japan), 2 mM L‐glutamine (Gibco), Penstrep (Gibco), N2 supplement (17502‐048, Invitrogen), B27 supplement (17504‐044, Invitrogen) and 10 mM Hepes–NaOH pH 7.3). Microinjection was performed as previously described 11, 12. Briefly, 4‐5 telencephalon slices were transferred to 3.5‐cm dishes containing 37°C warm CO2‐independent microinjection medium (CIMM; DMEM‐F12 (Sigma D2906) containing 2 mM L‐glutamine, Penstrep, N2 and B27 supplements, and 25 mM (final concentration) Hepes–NaOH pH 7.3). Slices were kept at 37°C on a heated stage during microinjection, which was performed manually using a compensation pressure of 100–200 hPa.
The microinjection solution contained either recombinant Cas9 protein in complex with the gRNA of interest or esiRNAs as above, together with dextran 10,000‐Alexa 555 as a tracer (2 μg/μl, Molecular Probes). Microinjection solutions were centrifuged at 16,000 g for 30 min at 4°C. The supernatant was collected, kept on ice and used for microinjection. After microinjection, slices were embedded in collagen and kept in culture for either 24 or 48 h in a slice culture incubator at 37°C, with 40% O2 (Air Liquide). At the end of the slice culture, SCM was removed and slices were fixed by adding 2 ml of 4% PFA directly to the dish. After 30 min at room temperature followed by 12 h at 4°C, slices were removed from the collagen and processed for immunostaining.
Immunofluorescence
Immunofluorescence was performed on 50‐ to 70‐μm vibratome sections as previously described 20. Antibodies used were rabbit pAb anti‐Tbr2 (Abcam, ab23345), rat pAb anti‐phosphohistone (Abcam, ab10543), goat pAb anti‐EGFP (MPI‐CBG), chicken pAb anti‐Tbr2 (Millipore, AB15894), and mouse mAb anti‐dextran (clone DX1, Stem Cell Technologies #60026). The secondary antibodies used were coupled to Alexa Fluor 488, 555 or 647 and were from donkey. DNA staining was performed with DAPI (Sigma). PaprikaRFP and mCherry direct fluorescence was detected without antibody staining. GFP fluorescence was detected either directly (Figs 1 and EV2) or using the aforementioned antibody (Figs 2 and EV3). All images were acquired using a Zeiss LSM 510 Duoscan laser‐scanning confocal microscope or a Zeiss LSM 780 NLO multiphoton laser‐scanning microscope. Images were analysed and processed with ImageJ (http://imagej.nih.gov/ij/).
Immunoblotting
RFP‐positive cells were isolated by FACS as described below, and proteins were analysed by immunoblotting using mouse mAb anti‐α‐tubulin (Sigma, T5168) and rabbit pAb anti‐GFP (Abcam, ab6556) as primary antibodies and HRP‐donkey anti‐mouse and anti‐rabbit (Jackson immunoresearch) as secondary antibodies. Immunoreactive bands were detected by using SuperSignal West Dura (Thermo Scientific) and quantified using ImageJ.
Cas9 in vitro reactions
Individual gRNAs were first tested in vitro using recombinant Cas9 protein and a PCR product of the target locus. The GFP PCR product was obtained using the following primers: forward 5′‐CTGAGTCCGGACTTGTACAG and reverse 5′‐GAGTGGTATGAAAGGCGCAGC; for the 2‐kb Eomes/Tbr2 PCR product, 5′‐GGGACCTGCCAAACTAGACC (forward) and 5′‐GCGATTTGTGGGCTGCATAG (reverse) primers were used (Fig EV4); for the 400‐bp Eomes/Tbr2 and off‐target PCR products, we used the same primers as for sequencing (see below and Table EV1). Cutting by Cas9 of the target sequence in vitro was performed according to the manufacturer's instructions (ToolGen). Briefly, an in vitro reaction contained 12 ng/μl of the PCR product, 50 ng/μl of recombinant Cas9 protein and 35 ng/μl of the indicated in vitro transcribed gRNA, in a total volume of 10 μl. Reactions were carried out at 37°C for 1 h. Samples were then treated with 4 μg of RNAse (Qiagen) for 15 min at 37°C, followed by addition of 2.5 μg of proteinase K (Merck) and further incubation for 10 min at 55°C. After 5 min at room temperature, samples received 1 μl of STOP solution (30% glycerol, 1.2% SDS, 250 mM EDTA pH 8.0; ToolGen) and were incubated for further 15 min at 37°C, followed by analysis on a 1% agarose gel.
Tbr2 and off‐target loci DNA sequence analysis
Cas9‐PaprikaRFP‐gRNA plasmid‐electroporated neocortices (3–4) were pooled and cells dissociated using the MACS Neural Tissue Dissociation kit as described previously 20. PaprikaRFP‐positive cells were isolated by FACS using a BD FACSAria II (BD Bioscience) and collected at 5,000 cells per well in a 96‐well PCR plate containing 5 μl of water per well. Plates were stored at −80°C until further processed.
To detect mismatches to the mouse genome sequence, we used a nested PCR set‐up to specifically amplify the Tbr2 target region and direct PCR amplification of off‐target regions OT1, OT2, OT3 and OT4 with a dual‐barcoding Illumina paired‐read sequencing approach 34. All gene‐specific primers were universally tailed to allow subsequent dual‐barcoding PCR. An overview of tailed gene‐specific primers, annealing temperatures and product sizes is provided in Table EV1. PCRs used as template ≈1,500 heat‐treated cells (10 min, 95°C) in 20 μl PCR mix containing Phusion High‐Fidelity DNA polymerase (NEB, Frankfurt, Germany), appropriate buffer, 5% DMSO, dNTPs and tailed gene‐specific primers. PCR conditions were as follows: denaturation at 95°C for 30 s, followed by 40 cycles of amplification (95°C for 10 s, amplicon‐specific annealing for 20 s and extension at 72°C for 2 min), final extension was performed for 10 min at 72°C followed by an infinite cooling step. For Tbr2, the nested PCR was performed from 1 μl diluted PCR fragment as described above with 20 amplification cycles. Non‐incorporated primers were removed by size selection using the E‐Gel® Clone well system (0.8%, Thermo Fisher Scientific Inc., Waltham, USA).
Primers containing Illumina adapters, 10‐bp barcodes and the universal tail region were added to gene‐specific and size‐selected amplicons by PCR. Sample ID and barcode assignment are described in Table EV2. PCRs contained 20 μl PCR mastermix with appropriate PCR buffer, 5% DMSO, dNTP, Phusion High‐Fidelity DNA polymerase (NEB, Frankfurt, Germany), barcoded primers (2 μM equimolar mix of index 1 and index 2) and 2–6 ng template PCR fragment. PCR conditions were as follows: 10‐min initial denaturation at 95°C, followed by 10 cycles (Tbr2, OT1, OT3) and 15 cycles (OT4, OT5) of 95°C for 25 s, 60°C for 30 s and 72°C for 90 s, a final extension step for 5 min at 72°C and incubation at 15°C. Products of the dual‐barcoding PCR were checked electrophoretically and quantified (Qubit Fluorimetric DNA quantification, Thermo Fisher Scientific Inc., Waltham, USA). Equimolar concentrations of five amplicon‐specific pools were generated (Tbr2, OT1‐OT4), which were purified using AMPure XP (Beckman Coulter, Brea, USA) with a ratio of 0.7:1 beads to DNA. Purified pools were quantified using the Library Quant Illumina Kit (KAPA Biosystems, Boston, USA). The resulting five amplicon‐specific pools were mixed in equimolar amounts, and the sequencing library was prepared as recommended by Illumina (MiSeq Reagent kit v3). About 12.8 million 300‐bp read pairs with 10% PhiX spiked‐in were generated on an Illumina MiSeq sequencer. Initial data processing was performed with Illumina RTA version 1.18.54 and, after filtering of the raw data on the Galaxy platform, only reads with 90% of bases with a FASTAQ > 20 were used.
Data analysis was performed with Geneious 8.1.6 software (Biomatters) 35. Reads were aligned to the reference sequence, and indel analysis was performed by inspecting the area 30 bp upstream and 30 bp downstream of the gTbr2 targeting site for mismatches. To analyse indel types in the reads obtained from gTbr2‐targeted cells, a 160‐bp‐long sequence around the targeting site was analysed (80 bp upstream and 80 bp downstream). Sequences that had combined mutations (both insertions and deletions) were not analysed.
Quantifications and statistics
For each in utero electroporation experiment, 1–3 litters with 1–4 embryos per condition (control or gene disruption) were analysed, and three sections per embryo were used for quantifications. All cell counts were performed in standardized microscopic fields and processed using Excel (Microsoft), and results were plotted using Prism (GraphPad Software). For each condition, data from one experiment were pooled, and the mean of the indicated number of experiments was calculated. Statistical significance was determined using either unpaired Student's t‐test (Figs 1D, H and K, EV2B and D, 3B, and EV4E), Mann–Whitney U‐test (Figs EV1E, and 3D and F), Fisher's test (Fig 2D) or the Kolmogorov–Smirnov test (Fig 3H).
Author contributions
NK co‐designed the study and experiments, co‐wrote the manuscript and performed experiments; ET co‐designed and performed experiments; ST, FKW, DS and SW performed experiments; MS co‐designed and co‐supervised the study; WBH co‐supervised the study and co‐wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Acknowledgements
We are grateful to the Services and Facilities of the Max Planck Institute of Molecular Cell Biology and Genetics for the outstanding support provided, notably J. Helppi and his team of the Animal Facility, J. Jarrells and her team of the Gene Expression Facility, J. Peychl and his team of the Light Microscopy Facility, D. Drechsel and his team of the Protein Expression and Purification Facility and I. Nüsslein and her team of the FACS Facility. We also thank Kathrin Lang and Vinzenz Lange (DKMS Life Science Lab, Dresden, Germany) for discussions on the barcoding strategy, for providing barcoding primers and for performing the MiSeq sequencing run. We would like to thank all members of the Huttner group for helpful discussions, especially M. Albert and M. Wilsch‐Bräuninger for advice and K. Long for critical reading of the manuscript. NK was supported by an EMBO long‐term fellowship (ALTF 861‐2013). WBH was supported by grants from the DFG (SFB 655, A2) and the ERC (250197), by the DFG‐funded Center for Regenerative Therapies Dresden and by the Fonds der Chemischen Industrie.
EMBO Reports (2016) 17: 338–348
References
- 1. Cong L, Ran FA, Cox D, Lin SL, Barretto R, Habib N, Hsu PD, Wu XB, Jiang WY, Marraffini LA et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mali P, Yang LH, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM (2013) RNA‐guided human genome engineering via Cas9. Science 339: 823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual‐RNA‐guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Doudna JA, Charpentier E (2014) Genome editing. The new frontier of genome engineering with CRISPR‐Cas9. Science 346: 1258096 [DOI] [PubMed] [Google Scholar]
- 5. Harrison MM, Jenkins BV, O'Connor‐Giles KM, Wildonger J (2014) A CRISPR view of development. Genes Dev 28: 1859–1872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hsu PD, Lander ES, Zhang F (2014) Development and applications of CRISPR‐Cas9 for genome engineering. Cell 157: 1262–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Peng Y, Clark KJ, Campbell JM, Panetta MR, Guo Y, Ekker SC (2014) Making designer mutants in model organisms. Development 141: 4042–4054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Charpentier E (2015) CRISPR‐Cas9: how research on a bacterial RNA‐guided mechanism opened new perspectives in biotechnology and biomedicine. EMBO Mol Med 7: 363–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takahashi M, Sato K, Nomura T, Osumi N (2002) Manipulating gene expressions by electroporation in the developing brain of mammalian embryos. Differentiation 70: 155–162 [DOI] [PubMed] [Google Scholar]
- 10. Calegari F, Haubensak W, Yang D, Huttner WB, Buchholz F (2002) Tissue‐specific RNA interference in postimplantation mouse embryos with endoribonuclease‐prepared short interfering RNA. Proc Natl Acad Sci USA 99: 14236–14240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Taverna E, Haffner C, Pepperkok R, Huttner WB (2012) A new approach to manipulate the fate of single neural stem cells in tissue. Nat Neurosci 15: 329–337 [DOI] [PubMed] [Google Scholar]
- 12. Wong FK, Haffner C, Huttner WB, Taverna E (2014) Microinjection of membrane‐impermeable molecules into single neural stem cells in brain tissue. Nat Protoc 9: 1170–1182 [DOI] [PubMed] [Google Scholar]
- 13. Iacopetti P, Michelini M, Stuckmann I, Oback B, Aaku‐Saraste E, Huttner WB (1999) Expression of the antiproliferative gene TIS21 at the onset of neurogenesis identifies single neuroepithelial cells that switch from proliferative to neuron‐generating division. Proc Natl Acad Sci USA 96: 4639–4644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Haubensak W, Attardo A, Denk W, Huttner WB (2004) Neurons arise in the basal neuroepithelium of the early mammalian telencephalon: a major site of neurogenesis. Proc Natl Acad Sci USA 101: 3196–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG et al (2014) Genome‐scale CRISPR‐Cas9 knockout screening in human cells. Science 343: 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M et al (2014) CRISPR‐Cas9 knockin mice for genome editing and cancer modeling. Cell 159: 440–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, Maeder ML, Joung JK, Chen ZY, Liu DR (2015) Cationic lipid‐mediated delivery of proteins enables efficient protein‐based genome editing in vitro and in vivo. Nat Biotechnol 33: 73–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim S, Kim D, Cho SW, Kim J, Kim JS (2014) Highly efficient RNA‐guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 24: 1012–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liang XQ, Potter J, Kumar S, Zou YF, Quintanilla R, Sridharan M, Carte J, Chen W, Roark N, Ranganathan S et al (2015) Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J Biotechnol 208: 44–53 [DOI] [PubMed] [Google Scholar]
- 20. Florio M, Albert M, Taverna E, Namba T, Brandl H, Lewitus E, Haffner C, Sykes A, Wong FK, Peters J et al (2015) Human‐specific gene ARHGAP11B promotes basal progenitor amplification and neocortex expansion. Science 347: 1465–1470 [DOI] [PubMed] [Google Scholar]
- 21. Attardo A, Calegari F, Haubensak W, Wilsch‐Bräuninger M, Huttner WB (2008) Live imaging at the onset of cortical neurogenesis reveals differential appearance of the neuronal phenotype in apical versus basal progenitor progeny. PLoS One 3: e2388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baala L, Briault S, Etchevers HC, Laumonnier F, Natiq A, Amiel J, Boddaert N, Picard C, Sbiti A, Asermouh A, et al (2007) Homozygous silencing of T‐box transcription factor EOMES leads to microcephaly with polymicrogyria and corpus callosum agenesis. Nat Genet 39: 454–456 [DOI] [PubMed] [Google Scholar]
- 23. Sessa A, Mao CA, Hadjantonakis AK, Klein WH, Broccoli V (2008) Tbr2 directs conversion of radial glia into basal precursors and guides neuronal amplification by indirect neurogenesis in the developing neocortex. Neuron 60: 56–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Arnold SJ, Huang GJ, Cheung AF, Era T, Nishikawa S, Bikoff EK, Molnar Z, Robertson EJ, Groszer M (2008) The T‐box transcription factor Eomes/Tbr2 regulates neurogenesis in the cortical subventricular zone. Genes Dev 22: 2479–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, Hevner RF (2005) Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci 25: 247–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Arai Y, Pulvers JN, Haffner C, Schilling B, Nusslein I, Calegari F, Huttner WB (2011) Neural stem and progenitor cells shorten S‐phase on commitment to neuron production. Nat Commun 2: 154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vasistha NA, Garcia‐Moreno F, Arora S, Cheung AF, Arnold SJ, Robertson EJ, Molnar Z (2014) Cortical and clonal contribution of Tbr2 expressing progenitors in the developing mouse brain. Cereb Cortex 25: 3290–3302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bai J, Ramos RL, Ackman JB, Thomas AM, Lee RV, LoTurco JJ (2003) RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat Neurosci 6: 1277–1283 [DOI] [PubMed] [Google Scholar]
- 29. LoTurco J, Manent JB, Sidiqi F (2009) New and improved tools for in utero electroporation studies of developing cerebral cortex. Cereb Cortex 19: I120–I125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Straub C, Granger AJ, Saulnier JL, Sabatini BL (2014) CRISPR/Cas9‐mediated gene knock‐down in post‐mitotic neurons. PLoS One 9: e105584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zuckermann M, Hovestadt V, Knobbe‐Thomsen CB, Zapatka M, Northcott PA, Schramm K, Belic J, Jones DT, Tschida B, Moriarity B et al (2015) Somatic CRISPR/Cas9‐mediated tumour suppressor disruption enables versatile brain tumour modelling. Nat Commun 6: 7391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW, Park J, Blackburn EH, Weissman JS, Qi LS et al (2013) Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155: 1479–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang D, Buchholz F, Huang Z, Goga A, Chen CY, Brodsky FM, Bishop JM (2002) Short RNA duplexes produced by hydrolysis with Escherichia coli RNase III mediate effective RNA interference in mammalian cells. Proc Natl Acad Sci USA 99: 9942–9947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lange V, Bohme I, Hofmann J, Lang K, Sauter J, Schone B, Paul P, Albrecht V, Andreas JM, Baier DM et al (2014) Cost‐efficient high‐throughput HLA typing by MiSeq amplicon sequencing. BMC Genom 15: 63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kearse M, Moir R, Wilson A, Stones‐Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C et al (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28: 1647–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File