

Abstract
E2F transcription factors control the oscillating expression pattern of multiple target genes during the cell cycle. Activator E2Fs, E2F1–3, induce an upswing of E2F targets, which is essential for the G1‐to‐S phase transition, whereas atypical E2Fs, E2F7 and E2F8, mediate a downswing of the same targets during late S, G2, and M phases. Expression of atypical E2Fs is induced by E2F1–3, but it is unknown how atypical E2Fs are inactivated in a timely manner. Here, we demonstrate that E2F7 and E2F8 are substrates of the anaphase‐promoting complex/cyclosome (APC/C). Removal of CDH1, or mutating the CDH1‐interacting KEN boxes, stabilized E2F7/8 from anaphase onwards and during G1. Expressing KEN mutant E2F7 during G1 impairs S phase entry and eventually results in cell death. Furthermore, we show that E2F8, but not E2F7, interacts also with APC/CC dc20. Importantly, atypical E2Fs can activate APC/CC dh1 by repressing its inhibitors cyclin A, cyclin E, and Emi1. In conclusion, we discovered a feedback loop between atypical E2Fs and APC/CC dh1, which ensures balanced expression of cell cycle genes and normal cell cycle progression.
Keywords: anaphase‐promoting complex, CDH1, cell cycle, DNA replication, E2F
Subject Categories: Cell Cycle
Introduction
Eukaryotic cell division is tightly controlled by transcriptional and posttranslational regulation of genes that drive progression through the different phases of the cell cycle. The decision to enter S phase is of critical importance, because unscheduled entry leads to replication stress, which may result in mutations through errors during DNA synthesis, and eventually could lead to genetic instability and cancer 1, 2. The activity of E2F transcription factors plays a central role in the ability of cells to enter S phase. Vertebrate species have no less than eight different E2F family members (E2F1–8), three dimerization partners (DP1–3), and 3 pocket proteins (RB, P107, P130), whose interplay determines transcription of hundreds of target genes in mammalian cells.
Dissecting the specific and unique functions of the E2F family members in various cellular processes, including S phase entry, has proven a difficult task 3. For instance, E2Fs 1–3 switch from activators to repressors during differentiation of intestinal progenitor cells, depending on pocket protein binding 4. However, the actions of the atypical E2F family members, E2F7 and E2F8, seem almost exclusively repressive, as they lack transactivation domains and are not regulated by pocket proteins 5, 6, 7, 8. After the start of S phase, E2F7 represses many genes that drive the G1/S transition, via occupation of E2F motifs on proximal promoters 8. Based on their high extent of homology and redundant functions in vivo, E2F8 is expected to act in the same manner 9. In addition, E2F7/8 drive the switch from mitotic cell cycles to endocycles in placenta and liver, and can suppress apoptosis during embryonic development, demonstrating their central role in cell cycle regulation 9, 10, 11.
Expression of atypical E2Fs is comparatively high in tissues with high rates of cell division 5, 6. Thus, given that E2F7 and E2F8 are potent cell cycle regulators, their activities must be tightly controlled. During late G1, a feed‐forward loop is triggered through activation of E2F1–3 resulting in a rise of cyclin‐dependent kinase (Cdk) activity and rapid transcriptional activation of E2F target genes, including cyclins and E2Fs themselves 12. Since E2F7 and E2F8 are also E2F target genes, we proposed the requirement for additional control mechanisms to inhibit their preliminary activation in late G1.
Another important mechanism that coordinates progression from G1 to S phase is the inactivation of the anaphase‐promoting complex/cyclosome APC/CCdh1. This permits accumulation of proteins that are required to progress through a single round of S phase properly, such as cyclin A, geminin, and CDC6 13, 14. Inactivation of APC/CCdh1 also relies on rising Cdk activity, leading to inactivation of CDH1, as well as E2F‐mediated transcription of Emi1, a direct APC/CCdh1 inhibitor 15, 16, 17. APC/CCdh1 maintains a necessary time window in G1 to prepare the cell for replication, illustrated by the fact that loss of CDH1 can lead to premature S phase entry, and induces replication stress and the accumulation of DNA damage 18, 19, 20.
Here, we show that E2F7 and E2F8 are targeted for ubiquitin‐mediated proteasomal degradation via APC/CCdh1 during the late stages of mitosis and G1 phase. Remarkably, E2F7 and E2F8 in turn can activate the APC/CCdh1 via repression of Emi1 and cyclins A and E, which are all known to inhibit APC/C activity. Thus, during the course of G1 phase, APC/CCdh1 eventually stimulates its own inactivation: Targeting E2F7 and E2F8 for destruction permits accumulation of Emi1, and cyclins A and E. Collectively these data show that atypical E2Fs and APC/CCdh1 are engaged in a feedback loop that tightly coordinates S phase entry and proper DNA replication. Importantly, we show that blocking the APC/CCdh1‐mediated destruction of ectopically expressed E2F7 and to a lesser extent E2F8 prohibits S phase and results in cell death.
Results
E2F7 and E2F8 are targeted for proteasomal degradation during G1
We first explored whether atypical E2Fs are subjected to posttranslational regulation. We treated the hTERT‐immortalized retinal pigment epithelium cell line RPE1‐TERT (RPE) with the protein translation inhibitor cycloheximide (CHX) and found a robust decrease in E2F7/8 protein levels within 1 h of treatment (Fig 1A and B). The disappearance of E2F7 and E2F8 was inhibited by co‐treatment with the proteasome inhibitor MG132. Then, we arrested RPE1 cells in G1 with the selective CDK4/6 inhibitor PD0332991 (PD). Protein expression of E2F7 and E2F8 was markedly reduced after 16 h of treatment (Fig 1C). U2OS osteosarcoma cells with intact RB function 21 also showed a reduction in E2F7/8 protein levels after 16 h of PD treatment. Arresting these cells in G2 phase with the CDK2 inhibitor NU6140 did not decrease E2F7/8 expression, suggesting that this effect is cell cycle phase‐specific (Fig 1C). We found that the disappearance E2F7/8 by PD0332991 treatment could be rescued by adding the proteasome inhibitor MG132 2 h prior to harvesting (Fig 1D). Together, these results strongly point toward a high turnover of E2F7 and E2F8 via proteasomal degradation, particularly during the G1 phase of the cell cycle.
Figure 1. High turnover of E2F7 and E2F8 via proteasomal degradation during G1 phase.
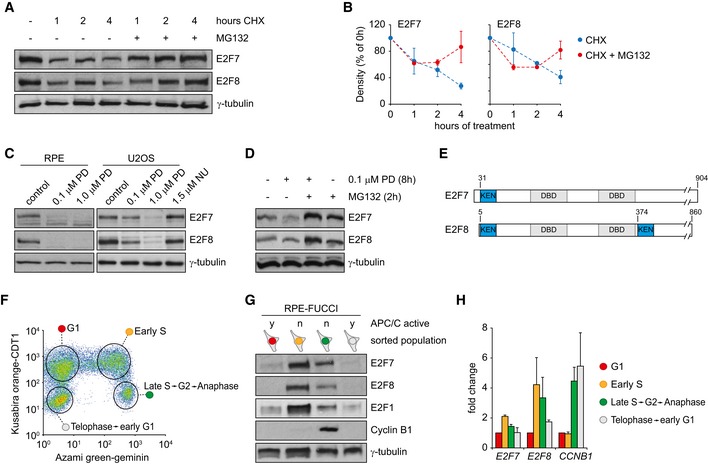
- Protein expression E2F7 and E2F8 in RPE cells treated with 100 μg/ml cycloheximide (CHX) and 10 μM MG132. The drugs were added simultaneously.
- Quantification of (A). E2F/tubulin density ratios were calculated for n = 3 independent experiments, and 0 h was set to 100%. Error bars indicate s.e.m.
- Protein levels of E2F7 and E2F8 in RPE and U2OS cells after 16 h of treatment with the CDK4/6 inhibitor PD0332991, or the CDK2 inhibitor NU6140.
- Protein expression of E2F7 and E2F8 after 8 h of PD0332991 treatment, in the presence or absence of the proteasome inhibitor MG132 (10 μM) for 2 h prior to harvesting.
- Schematic overview of conserved KEN motifs in human/mouse E2F7 and E2F8 proteins.
- FACS profile showing expression of cell cycle markers in RPE cells with stable expression of the FUCCI system. Encircled areas indicate the gates used to sort cell cycle‐specific populations.
- Immunoblots of FACS‐sorted RPE‐FUCCI cells. Cells were sorted based on expression of truncated versions of and Azami green‐tagged geminin (amino acids 1–130) and Kusabira orange‐tagged CDT1 (amino acids 30–120), respectively. Blots are representative examples of four independent replicates derived from two different stable RPE‐FUCCI clones.
- Normalized transcript levels of atypical E2Fs and cyclin B1 in sorted RPE‐FUCCI cells measured by qPCR. Bars represent average ± s.e.m. of fold change, relative to expression in G1 (n = 3).
One likely candidate to mediate proteasomal degradation early in G1 phase is APC/CCdh1. Using the ELM protein sequence analysis resource (http://elm.eu.org), we found that atypical E2Fs contain evolutionary conserved KEN domains, which are the canonical substrate recognition motifs for APC/CCdh1 (Fig 1E) 22. Furthermore, observations in a cell free system suggested that atypical E2Fs may be substrates of the APC/C 23. We then took advantage of the Fluorescent Ubiquitination‐based Cell Cycle Indicator (FUCCI) system, which is based on the activities of APC/CCdh1 and SCFSkp2 24. Using FACS sorting, we isolated cell populations in different phases of the cell cycle as indicated to determine protein and mRNA levels of atypical E2Fs (Fig 1F). From the onset of anaphase until the next S phase the APC/C is active, and Azami green‐tagged geminin1‐110 is absent. Notably, E2F7 and E2F8 proteins were nearly undetectable in these G1 cells (Fig 1G). The protein levels of E2F1 and cyclin B1, which are also APC/C substrates 25, 26, 27, showed expression patterns consistent with APC/C activity (Fig 1G). Interestingly, CCNB1 transcript levels were not decreased in cells labeled as telophase‐to‐early G1, confirming that this sharp drop in cyclin B1 protein was entirely caused by APC/C‐mediated proteasomal degradation (Fig 1H). Although protein and transcript levels of E2F7 and E2F8 in sorted cells showed a similar trend, transcripts were only mildly regulated in the cell cycle, while protein levels fluctuated considerably (Fig 1H). This confirms the important contribution of posttranslational regulation mechanisms. Collectively, these data show that E2F7 and E2F8 are relatively unstable proteins during G1 phase and that their degradation coincides with high APC/C activity.
E2F7 and E2F8 are APC/CCdh1 substrates
To determine whether E2F7 and E2F8 are bona fide APC/CCdh1 substrates in human cells, we transfected 293T cells with Flag‐tagged CDH1. We observed a robust reduction of endogenous E2F7/8 proteins after overexpression of CDH1 similar to the known APC/CCdh1 substrates CDC6 and aurora kinase A (Fig 2A and B). To rule out an indirect transcriptional effect of CDH1 overexpression on E2F7/8, we then expressed EGFP‐tagged E2F7 or E2F8 (transcriptionally controlled by a CMV promoter) in combination with CDH1. We observed a near‐complete reduction in exogenous E2F7/8 by CDH1 overexpression (Fig 2C). Reciprocally, depleting CDH1 in cells with stable doxycycline‐inducible expression of E2F7 or E2F8 using RNAi, stabilized exogenous E2F7 and E2F8 proteins (Fig 2D). We then performed co‐immunoprecipitation experiments and found that E2F7/8 and CDH1 physically interacted with each other (Fig 2E).
Figure 2. Atypical E2Fs are targeted for degradation by APC/CC dh1 .
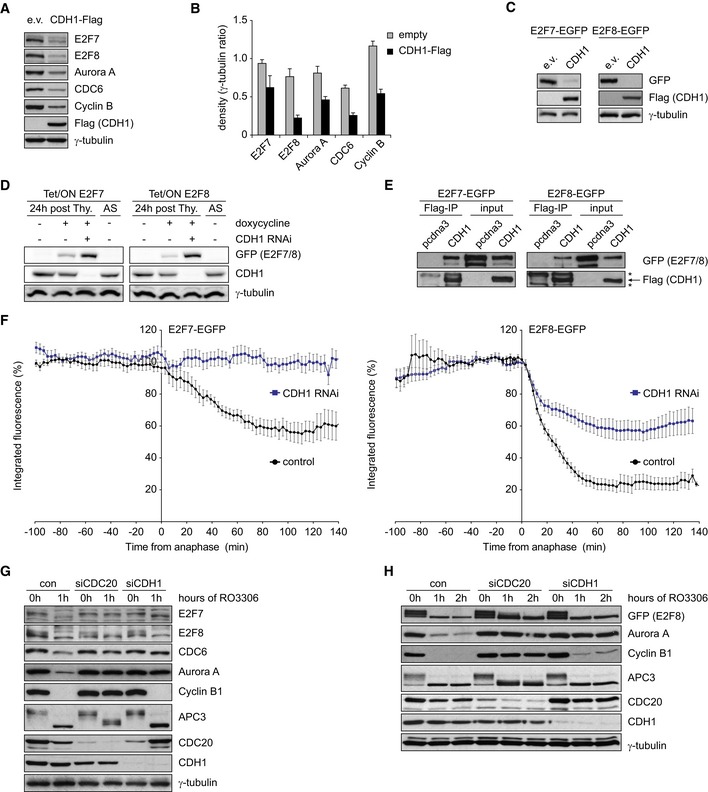
- Protein levels of E2F7/8 and two known APC/CC dh1 substrates in 293T cells 48 h after transfection with CDH1‐Flag. Immunoblots are representative examples of two independent replicates.
- Quantification of (A). Band density of indicated proteins was corrected for loading differences by calculating the ratio over tubulin. Bars represent mean ± s.e.m. of n = 2.
- Expression of EGFP‐tagged E2F7/8 in 293T cells 48 h after transfection of Flag‐tagged CDH1 or empty vector.
- Effect of CDH1 depletion on protein levels of E2F7/8 in HeLa cells with stable expression of inducible E2F7/8‐EGFP. Overexpression of E2F7 was induced using doxycycline at the onset of release from a thymidine block.
- Co‐immunoprecipitation of EGFP‐tagged E2F7/8 with CDH1‐Flag after 48 h of co‐expression in 293T cells. Cells were treated with 10 μM MG132 for 5 h prior to harvesting to limit immediate proteasomal degradation of E2F7/8 after binding to CDH1. Asterisks indicate IgG bands; arrow indicates the CDH1‐Flag band.
- HeLa cells with stable inducible E2F7/8‐EGFP were imaged by fluorescence and differential contrast (DIC) microscopy. Cells were treated with CDH1 siRNA for 10 h, synchronized at the G1‐S border by 16‐h thymidine treatment, followed by thymidine release and induction of E2F7/8‐EGFP by doxycycline. Mean integrated fluorescence of the cells was measured and normalized to the intensity in the frame of nuclear envelope breakdown (NEBD) (set at 100%), as determined by cytoplasmic dispersal of the fluorescent signal. The x‐axis is set to 0 at the onset of anaphase, as observed in the DIC channel. Graphs shown are mean ± s.e.m. Left graph: control n = 15, Cdh1 RNAi n = 14 both from three independent experiments. Right graph: control n = 13, Cdh1 RNAi n = 13 both from two independent experiments.
- Expression of indicated proteins during mitotic exit of RPE cells treated with siRNA against CDC20 or CDH1. CDK1 inhibitor RO3306 and MPS1 inhibitor reversine were added to force mitotic exit. A scheme of the experimental procedure is shown in Fig EV1B. Blots are representative of two independent experiments.
- Expression of indicated proteins during mitotic exit of HeLa cells expressing inducible E2F8‐EGFP, treated with siRNA against CDC20 or CDH1. RO3306 and reversine were added to force mitotic exit. Doxycycline was added 12 h prior to mitotic shake‐off. Blots are representative of two independent experiments.
Next, we followed the levels of E2F7 and E2F8 when cells progressed through mitosis and G1 phase. We used time‐lapse microscopy experiments in HeLa cell lines with stable inducible overexpression of EGFP‐tagged E2F7 and E2F8. These experiments revealed that both proteins are degraded during telophase and the subsequent G1 phase, coinciding with APC/CCdh1 activation (Figs 2F and EV1A). Importantly, we could completely prevent the degradation of E2F7 by treating cells with siRNA oligos targeting CDH1. CDH1 RNAi also caused a marked, but incomplete stabilization of E2F8, indicating that additional E3 ligase activity, such as CDC20, could be partially responsible for E2F8 degradation (Fig 2F). To evaluate the contribution of CDC20 in degrading atypical E2Fs and to verify that the APC/C targets endogenous E2F7 and E2F8 proteins, we transfected RPE cells with CDC20 or CDH1 RNAi and measured protein levels during mitosis. The RNAi‐treated cells were synchronized in prophase with nocodazole and then released by washing out the drug and forced into mitotic exit with the CDK1 inhibitor RO3306 and the MPS1 inhibitor reversine (Fig EV1B). E2F7 was almost completely degraded within 1 h after release, and CDH1 RNAi completely prevented this degradation (Fig 2G), demonstrating that endogenous E2F7 protein levels are regulated by CDH1. Surprisingly, CDC20 RNAi also prevented degradation of E2F7, as well as CDC6 and aurora kinase A, which were previously shown to be regulated by CDH1, but not by CDC20 28, 29. Most likely, CDH1 activity was indirectly compromised by CDC20 RNAi. This is illustrated by incomplete dephosphorylation of the APC/C subunit APC3, which is heavily phosphorylated during the prophase arrest and rapidly dephosphorylated after the release (Fig 2G). In contrast, cells treated with CDH1 RNAi showed complete degradation of the APC/CCdc20 substrate cyclin B1, demonstrating that CDC20 is highly active under CDH1 RNAi conditions. Nevertheless, E2F7 as well as CDC6 and aurora kinase A were completely stabilized, making it highly unlikely that E2F7 is targeted by CDC20. We could not detect major differences in the expression levels of endogenous E2F8 protein levels after release from the mitotic arrest or under CDC20 RNAi or CDH1 RNAi conditions. Since the protein expression of E2F8 was very low during nocodazole treatment, we used HeLa cells with inducible overexpression of E2F8‐EGFP to further evaluate the effects of CDC20 RNAi. Consistent with the time‐lapse imaging, CDH1 RNAi resulted in a partial stabilization of E2F8‐EGFP during mitotic exit (Fig 2H). Remarkably, this stabilization was more pronounced in cells treated with CDC20 RNAi, suggesting that CDC20 can target E2F8 for degradation.
Figure EV1. Detection of E2F7/8 during mitosis and mitotic exit.
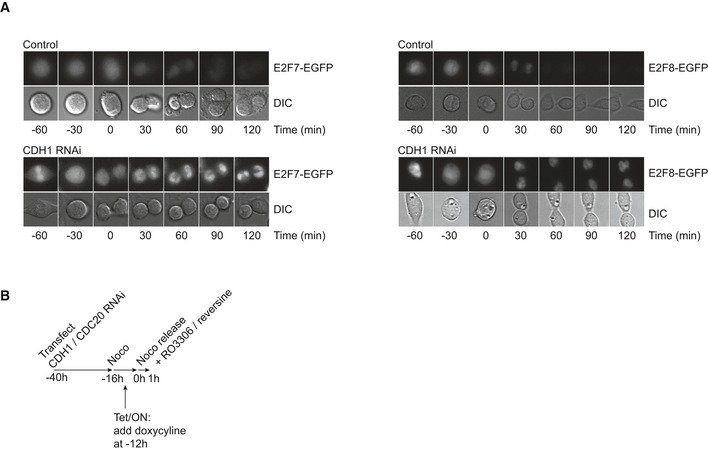
- Montages of representative cells from HeLa cells with inducible expression of E2F7/8‐EGFP. Time (min) indicates time from onset of anaphase.
- Schematic overview of mitotic release experiments shown in Fig 2G and H.
These results show that E2F7 and E2F8 are targeted for degradation by APC/CCdh1 during mitotic exit. Furthermore, E2F7 appears to be more tightly regulated by APC/CCdh1 than E2F8. APC/CCdc20 and most likely a yet‐unidentified additional E3 ligase activity account for additional E2F8 degradation.
Mutation of KEN domains in E2F7 and E2F8 prevent their APC/CCdh1‐mediated destruction
To evaluate whether the KEN domains on E2F7 and E2F8 are indeed true APC/CCdh1‐interacting sites, we performed site‐directed mutagenesis experiments. E2F7 contains a conserved KEN domain at amino acid positions 31–33. E2F8 has two conserved KEN motifs, located at amino acids 5–7 and 374–376, respectively. The KEN motifs were replaced with three consecutive alanine residues (Fig EV2A). For E2F8 we created single‐ as well as double‐mutant plasmids (E2F8KEN5mut and E2F8KEN374mut, and E2F8K/K, respectively). We co‐transfected 293T cells with equal amounts of these constructs in combination with CDH1‐Flag or empty vector. Notably, protein levels of KEN mutant versions of E2F7 and E2F8 were clearly higher than their wild‐type counterparts, particularly in the presence of CDH1‐Flag (Fig 3A). Because both KEN mutations in E2F8 had a similar effect, we concluded that both domains are functional and decided to perform all subsequent experiments with the double mutant (E2F8K/K). We then performed stable transfection of these constructs in HeLa cells expressing the Tet repressor. We confirmed that the KEN mutants are not misfolded or otherwise dysfunctional, by showing repression of the known E2F target genes CDC6 and cyclin A2 after 16 h of doxycycline treatment (Fig 3B). In fact, cell lines expressing KEN mutants showed stronger repression, notwithstanding comparable overall expression levels. Using FACS analysis to plot levels of E2F7/8 (EGFP) against DNA content (propidium iodide) we noted that stable cell lines expressing the wild‐type versions of E2F7/8 contain only few EGFP‐positive cells with 2C DNA content (G1 phase, Fig 3C). However, induction of E2F7/8KEN showed a much higher percentage of EGFP‐positive cells with 2C DNA content (Figs 3C and EV2B). Importantly, CDH1 RNAi during doxycycline induction caused a massive increase in the number of strongly EGFP‐positive E2F7WT‐ or E2F8WT‐expressing cells with 2C DNA content, comparable to the effect of KEN mutation, confirming CDH1‐dependent degradation of E2F7/8 during G1 (Figs 3C and EV2B).
Figure EV2. Generation of KEN mutant E2F7 and E2F8.
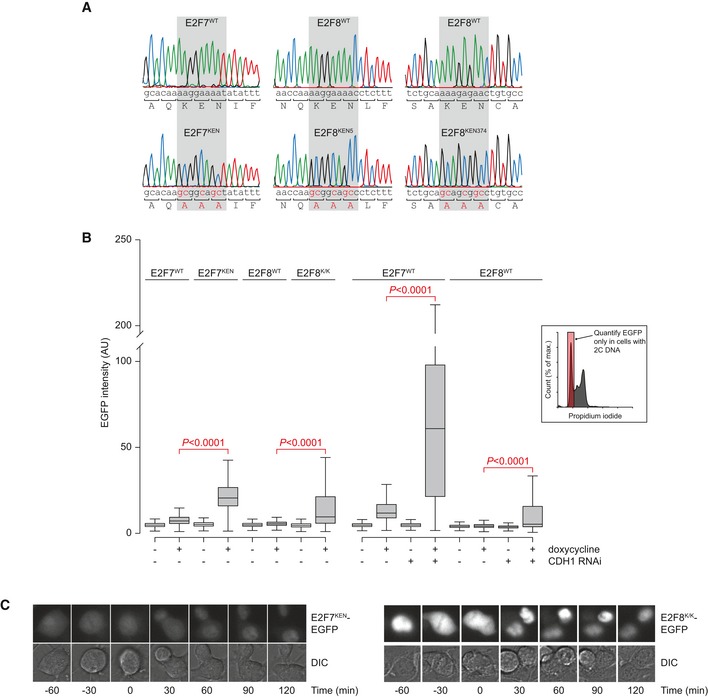
- Sanger sequencing result after site‐directed mutagenesis of the nucleotides encoding the KEN domains in E2F7 and E2F8. The middle panel shows the N‐terminal KEN sequence starting at amino acid 5, the right panel shows the KEN domain starting at amino acid 374 in E2F8.
- Quantification of EGFP levels determined with FACS analysis in HeLa cell lines with stable inducible expression of indicated constructs, with or without CDH1 RNAi transfection. Boxes indicate 25th and 75th percentiles, whiskers indicate 5th and 95th percentiles. For every condition, 10,000 cells were measured. Statistics were performed with Kruskal‐Wallis tests followed by Dunn's individual group comparison method. The inlay shows how we first gated the G1 cells (2C DNA content) prior to EGFP measurement.
- Montages of representative HeLa cells expressing KEN mutant versions of E2F7/8. Time (min) indicates time from onset of anaphase.
Figure 3. Mutation of KEN domains results in marked stabilization of E2F7 and E2F8.
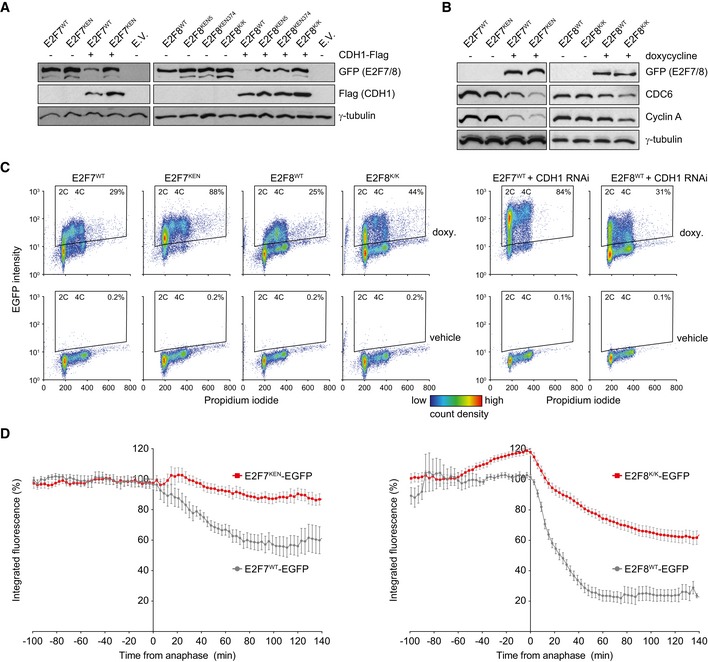
- Protein expression of wild‐type and KEN mutant E2F7 and E2F8 in 293T cells in the presence or absence of CDH1‐Flag 48 h after transfection. For E2F8, KEN motifs starting at amino acid 5 (KEN5) or 374 (KEN374) were mutated separately into three consecutive alanines or in combination (K/K). Blots are representative of two independent experiments.
- Immunoblots showing repression of the E2F target genes CDC6 and cyclin A in HeLa cells with stable doxycycline‐inducible expression of wild‐type and KEN double‐mutant E2F7/8 after 16 h of doxycycline treatment.
- FACS plots showing DNA content on the x‐axis (propidium iodide) of HeLa cells with stable expression of wild‐type or KEN mutant E2F7, after 24 h of doxycycline. Thresholds for EGFP positivity were set by applying the same gate to vehicle‐ and doxycycline‐treated cells.
- HeLa cells expressing inducible E2F7‐EGFP (left destruction graph) or E2F8‐EGFP (right destruction graph) were blocked with thymidine for 16 h and then released in fresh medium with doxycycline. Imaging was performed as in Fig 2E. The x‐axis is set to 0 at the frame of anaphase onset. Graphs show mean ± s.e.m., wild‐type (wt) shown from Fig 2E in gray. E2F7KEN‐EGFP: n = 13 from two independent experiments, E2F8K/K‐EGFP: n = 26 from three independent experiments.
Time‐lapse imaging showed that E2F7KEN and E2F8K/K degradation after anaphase is blocked, in sharp contrast to their wild‐type counterparts (Figs 3D and EV2C). We found that E2F8K/Kmut degradation was not completely prevented, unlike E2F7KEN. In line with this, co‐immunoprecipitations showed a robust KEN‐domain‐dependent interaction between E2F7 and endogenous CDH1, but a much weaker interaction with E2F8, which was not affected by KEN mutation (Fig EV3A). The residual degradation of KEN mutant E2F8 may be explained by APC/C binding via CDC20. Indeed, we found three putative D‐boxes in E2F8 (Fig EV3B). Accordingly, we found that both wild‐type and KEN mutant E2F8 co‐immunoprecipitated with CDC20‐Flag (Fig EV3C and D). In contrast, CDC20 did not co‐immunoprecipitate with E2F7, again suggesting that E2F7 is only targeted by CDH1 (Fig EV3D). These data show that E2F7 and E2F8 have functional KEN domains, and mutation of these domains results in stabilization of E2F7 and E2F8 during mitotic exit and G1.
Figure EV3. E2F8 binds CDC20.
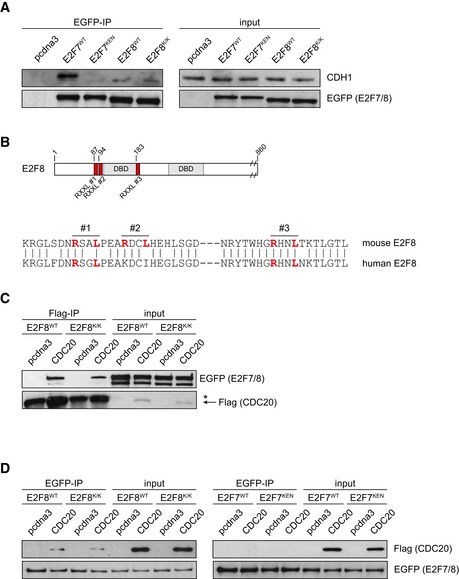
- Co‐immunoprecipitation of EGFP‐tagged E2F7 and E2F8 with endogenous CDH1. GFP‐binding agarose beads were used to pull down the fusion proteins. Cells were treated with 10 μM MG132 for 5 h prior to harvesting to prevent potential immediate proteasomal degradation of E2F7/8 after binding to CDH1.
- Schematic overview of putative D‐boxes (RXXL) in mouse E2F8, and alignment of human and mouse E2F8 sequences.
- Co‐immunoprecipitation of EGFP‐tagged E2F8WT or E2F8K/Kmut with CDC20‐Flag after 48 h of co‐expression in 293T cells. Cells were treated with 10 μM MG132 for 5 h prior to harvesting to prevent potential immediate proteasomal degradation of E2F7/8 after binding to CDC20.
- Co‐immunoprecipitation as in (B), performed with GFP‐binding agarose beads. Note that CDC20 only interacted with E2F8, in a KEN‐independent manner.
Stabilization of E2F7/8 during G1 inhibits cell proliferation by reduced S phase entry and increased cell death
We previously showed that inducible E2F7WT overexpression inhibits proliferation by delaying S phase progression, while not affecting S phase entry 8. This could indicate that during unperturbed cell cycles, cells very efficiently degrade E2F7/8 for normal progression through G1 phase. We therefore hypothesized that stabilization of E2F7 or E2F8 in G1 by preventing its APC/CCdh1‐mediated degradation would prevent cells from starting S phase. To test this hypothesis, we quantified incorporation of the thymidine‐analog BrdU in cells with stable inducible expression of KEN mutant and wild‐type E2F7/8. Both E2F7WT‐ and E2F8WT‐expressing cells continued to incorporate BrdU at high levels after 24 and even 48 h of doxycycline treatment (Figs 4A and EV4A). Nevertheless, proliferation was inhibited by induced overexpression of the wild‐type versions of E2F7 and E2F8 (Fig 4B). However, induction of E2F7KEN severely reduced BrdU incorporation, indicating that interfering with APC/C‐mediated degradation of E2F7 in G1 negatively impacts S phase entry. The overexpression of E2F8K/K also demonstrated a significant reduction in BrdU incorporation, although the effect was less pronounced than that of E2F7KEN (Fig 4A). To further investigate whether cells overexpressing KEN mutant E2F7 arrest in G1, we performed a time‐course experiments by releasing cells from a mitotic arrest. The control cells showed a very steep induction of classic target genes, such as E2F1 and CCNE1, at 8 h and 12 h after release indicating S phase entry, but this induction was completely blocked by E2F7KEN expression (Fig 4C). Remarkably, the cell line expressing E2F8K/K only showed a minor inhibition of target gene expression. The difference between E2F8 and E2F7 could be potentially explained by differences in the percentages of cells expressing detectable levels of the constructs. To correct for this, we traced cell cycle fates at the individual cell level. We quantified S phase entry using PCNA‐mCherry as a marker and followed its subcellular distribution with time‐lapse imaging (Fig 4D, Movie EV1). The onset of S phase is characterized by formation of PCNA dots, known as replication factories 30. Whereas the bulk of cells without expression of KEN mutant E2F7 and E2F8 (EGFP‐negative) entered S phase during the 40 h of imaging, only very few cells expressing E2F7KEN did so (Figs 4E and G, and EV4B). E2F8K/K‐expressing cells also showed a reduced percentage of cells entering S phase, but again the effect was less pronounced. Instead of entering S phase, 50% of the E2F7KEN‐expressing cells died within 40 h of live cell imaging, as detected by leakage of PCNA into the cytoplasm, followed by cell blebbing (Figs 4F and EV4B, Movie EV2). Most of these cells died during G1 defined by observation of a normal mitosis, but no evidence of PCNA nuclear dot formation prior to cell death (Fig 4G).
Figure 4. Mutating the KEN domain of E2F7 inhibits S phase entry and progression.
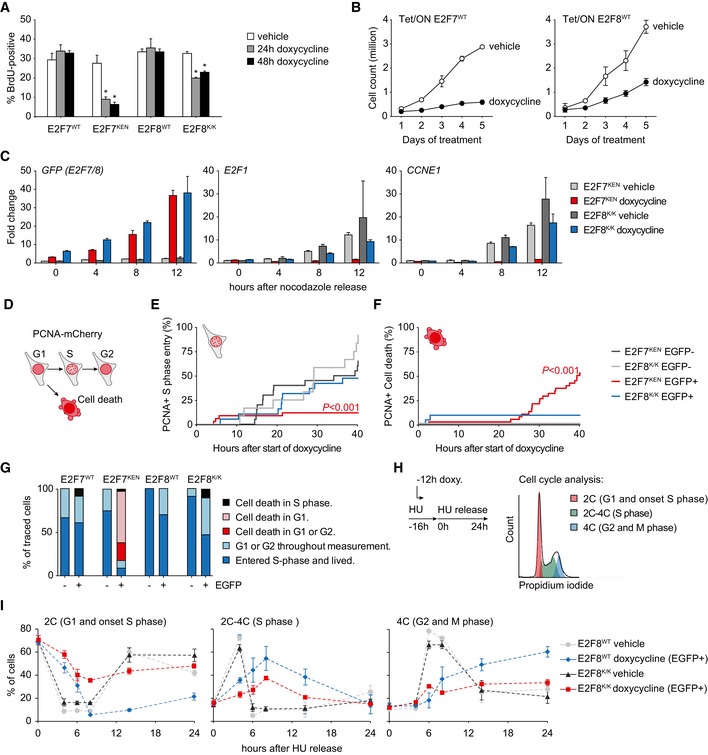
- BrdU incorporation in HeLa cells with inducible wild‐type and KEN mutant E2F7/8 expression, measured by flow cytometry. Bars represent mean ± s.e.m. (n = 3). *P < 0.05 versus vehicle, determined by one‐way ANOVA followed by Holm‐Sidak pairwise comparisons.
- Proliferation curves of HeLa cells with stable inducible wild‐type E2F7‐EGFP and E2F8‐EGFP expression after doxycycline treatment. Dots represent average ± s.e.m. of two replicates.
- Quantitative PCRs showing expression of KEN mutant E2F7/8 and two classic E2F target genes in inducible HeLa cell lines after a mitotic shake‐off. Doxycycline was added 4 h prior to the nocodazole release. Bars represent average ± s.d. of two independent replicates, which were both measured in duplo.
- Schematic overview of live microscopy using mCherry‐tagged PCNA to monitor cell cycle progression. Onset of S phase is marked by nuclear dots formation, which disappear when S phase is completed. PCNA leakage into the cytosol was always followed by apoptosis.
- Cumulative progression into S phase of HeLa cells expressing KEN mutant E2F7/8‐EGFP, monitored by nuclear PCNA dot formation. P‐value indicates significant change of EGFP‐positive versus negative cells from the same cell line, determined by log‐rank (Mantel‐Cox) tests.
- Cumulative cell death of HeLa cells expressing KEN mutant E2F7/8‐EGFP, monitored by cell blebbing and cytosolic PCNA‐mCherry. Statistical analysis was performed as described in (E).
- Quantification of cell fates in PCNA live imaging experiments from (C–E), specified per cell cycle phase. Explanation of the legend: cell death in S phase, cell dies after appearance of PCNA dots; cell death in G, cell death after mitosis, but prior to PCNA dot formation; cell death in G1 or G2, cell death without observing mitosis or PCNA dot formation.
- Schematic overview of synchronization experiment. HeLa cells were arrested at the onset of S phase with 2 mM hydroxyurea (HU), and induction of E2F8WT or E2F8K/Kmut was started 12 h prior to release from HU. FACS plat shows a representative example of fitting the different phases with a Watson exact model.
- Quantification of DNA content of synchronized cells described under (H) measured by flow cytometry. Data points represent average ± s.e.m. of two independent experiments. In case of doxycycline‐treated cells, only EGFP‐positive cells were counted. The gating strategy and quantification of EGFP in both cell lines is shown in Fig EV5B and C.
Figure EV4. Stabilization of KEN mutant E2F7/8 inhibits DNA replication and triggers cell death.
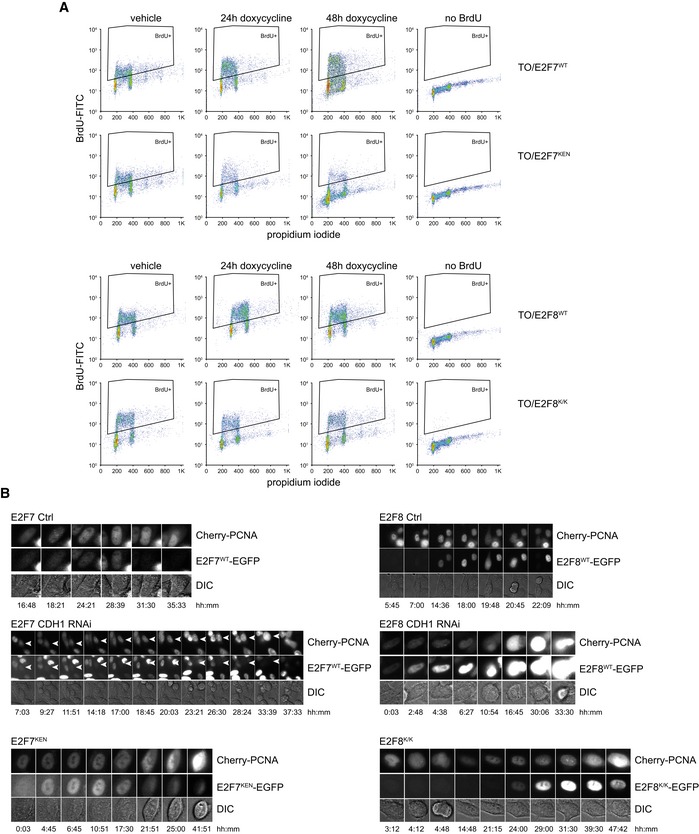
- FACS data showing scatterplots of DNA synthesis (anti‐BrdU‐FITC) versus DNA content (propidium iodide) in cells with stable expression of indicated inducible constructs. Gates were used to calculate the percentage of BrdU‐positive cells shown in Fig 4A.
- Montages of PCNA dot formation and cell death in cells with inducible expression of indicated constructs, and effect of CDH1 RNAi. Arrowheads indicate a cell that enters S phase, but undergoes cell death. Time in hh:mm from the onset of imaging and doxycycline induction.
The KEN mutations in E2F8 caused a much less severe S phase entry defect than in E2F7, and while many E2F7KEN cells died during imaging, E2F8K/K were mostly still alive at the end of analysis (Figs 4F and G, and EV4B). To verify this, we stained cells with annexin V and again found that only the E2F7KEN‐expressing cells showed a robust increase of apoptosis (Fig EV5A). We then asked whether the E2F8K/K‐expressing cells were able progress through S phase normally. To this end, we released E2F8‐expressing cells from a HU arrest and quantified cell cycle progression of doxycycline‐treated cells and their vehicle‐treated counterparts (Fig 4H). Although the percentages of positive cells and average EGFP intensity were very similar between E2F8WT and E2F8K/K, we gated out EGFP‐negative cells to avoid bias (Fig EV5B and C). We observed a marked delay in S phase progression of cells expressing E2F8K/K compared to E2F8WT‐expressing cells, suggesting that unscheduled expression of E2F8 in G1 causes problems during the subsequent S phase (Figs 4I and EV5D).
Figure EV5. Stabilization of KEN mutant E2F7/8 inhibits DNA replication and triggers cell death; continued.
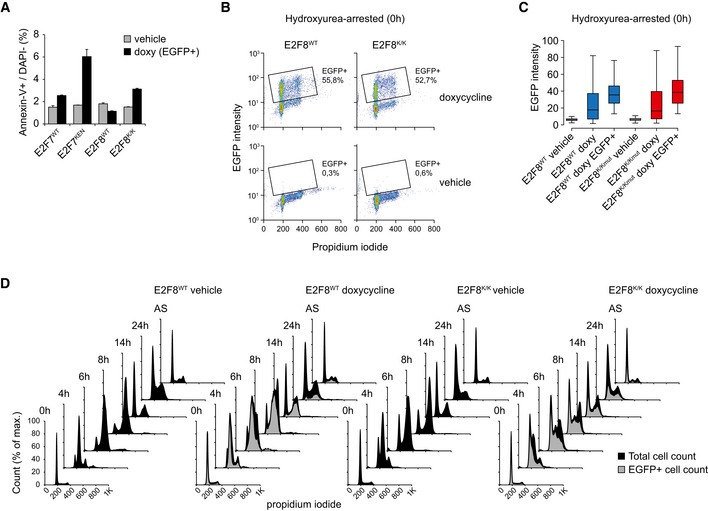
- Apoptosis, measured by FACS analysis of annexin V‐stained HeLa cells expressing the indicated inducible constructs. Apoptosis staining was done using the annexin V‐APC detection kit (Thermo Scientific), according to the manufacturer's instructions. Doxycycline was added 24 h prior to harvesting, and only EGFP‐positive cells were measured to avoid bias from differences in percentages of E2F‐expressing cells. Bars represent average ± s.e.m. of four independent replicates.
- FACS plots of DNA content versus E2F8‐EGFP intensity in HU‐arrested cells, 12 h after doxycycline induction.
- Quantification of EGFP levels in cells with 2C DNA content, determined with FACS analysis in HeLa cell lines with stable inducible expression of indicated constructs. Boxes indicate 25th and 75th percentiles, whiskers indicate 5th and 95th percentiles. For every condition, 10,000 cells were measured.
- Representative FACS DNA content plots of HeLa cells with stable inducible expression of indicated E2F8 constructs. Gray overlays indicate the fraction of E2F8‐EGFP‐positive cells within each doxycycline‐treated cell line.
Next, we tested whether depletion of CDH1 would also impair S phase entry in E2F7/8WT‐expressing cells, using BrdU incorporation and live PCNA imaging. Treatment with CDH1 RNAi slightly reduced the percentage of BrdU‐positive E2F7/8WT‐expressing cells (Fig 5A). An explanation for this modest effect is that CDH1 depletion causes the accumulation of cyclin A and other proteins that drive S phase entry, which could in part override the effect of E2F7/8 stabilization 19. However, despite modest effects on BrdU incorporation, time‐lapse microscopy of individual cells showed that CDH1 RNAi completely prevented normal PCNA dot formation in the E2F7WT‐ or E2F8WT‐EGFP‐expressing cells (Fig 5B). Instead, almost all cells with combined CDH1 depletion and overexpression of E2F7/8 underwent cell death, as seen by leakage of PCNA‐mCherry into the cytosol followed by cell blebbing (Figs 5B and C, and EV4B). Notably, CDH1 RNAi did not cause a discernible effect on S phase entry or cell death in EGFP‐negative control cells, except for a minor delay in S phase entry of EGFP‐negative cells in the E2F8WT cell line. These data demonstrate that APC/CCdh1‐dependent degradation of ectopic E2F7 or E2F8 during G1 is required for the initiation and progression of DNA replication.
Figure 5. Stabilization of ectopic E2F7/8 in G1 by CDH1 depletion causes massive cell death.
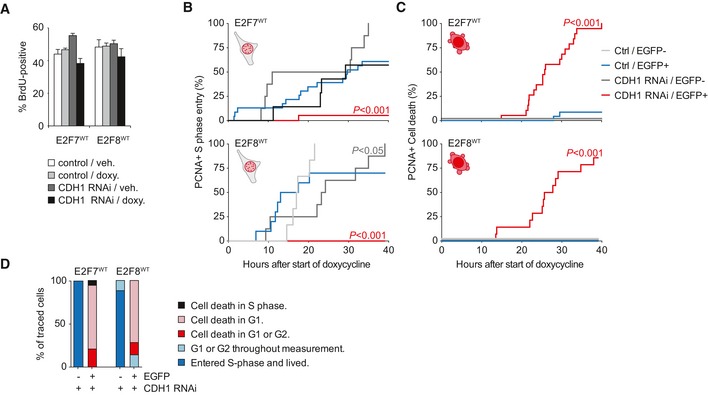
- Effect of CDH1 RNAi on percentages of BrdU‐positive HeLa cells after induction of E2F7/8 expression, measured by flow cytometry. Bars represent mean ± s.e.m. (n = 4).
- Effect of CDH1 RNAi on cumulative S phase entry of cells with inducible expression of E2F7/8WT, measured by co‐expressed PCNA‐mCherry. P‐values indicate significant change of CDH1 RNAi versus control. Font colors of P‐values match with the line colors of the significantly changed conditions.
- Effect of CDH1 RNAi on cumulative death of cells with inducible expression of E2F7/8WT. P‐values indicate significant change of CDH1 RNAi versus control.
- Quantification of cell fates in PCNA live imaging experiments from (B) and (C), specified per cell cycle phase. Explanation of the legend: cell death in S phase, cell dies after appearance of PCNA dots; cell death in G1, cell death after mitosis, but prior to PCNA dot formation; cell death in G1 or G2, cell death without observing mitosis or PCNA dot formation.
A feedback loop between E2F7/8 and APC/CCdh1
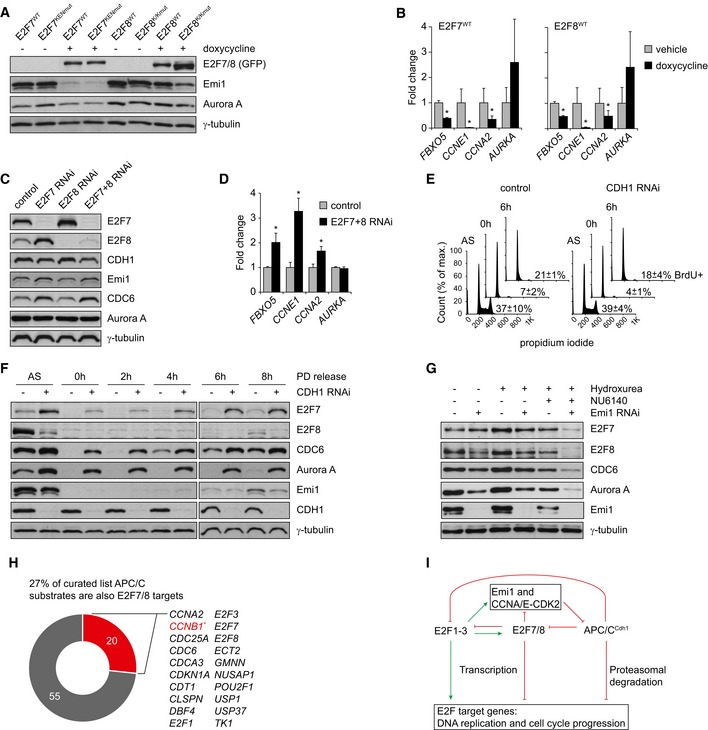
During late G1, APC/CCdh1 is inhibited by two mechanisms. First, phosphorylation of CDH1 by cyclin A/CDK2 or cyclin E/CDK2 prevents its interaction with APC/C, and second, the endogenous APC/C inhibitor Emi1 starts to be expressed 31, 32. Both mechanisms are at least in part dependent on E2F activity, although the role of E2F7/8 has not been explored yet 16, 33. We found that doxycycline‐induced expression of E2F7 and E2F8 caused a severe reduction in Emi1 protein levels (Fig 6A). This reduction was stronger with KEN mutants, particularly in the case of E2F8. To show that this is a direct effect, we then performed quantitative PCR on FACS‐sorted, HU‐synchronized cells (Fig EV6A and B). Quantitative PCR showed that 8 h of doxycycline treatment was sufficient to cause a marked repression of FBXO5, the gene encoding Emi1 in cells expressing E2F7WT or E2F8WT (Fig 6B). Cyclin E1 and A2, two other APC/C inhibitors, were also transcriptionally repressed by E2F7/8 (Fig 6B). Aurora kinase A was not repressed at the transcriptional level, but proteins levels were reduced upon overexpression of E2F7/8 (Fig 6A and B). These findings suggest that E2F7/8‐mediated repression of the APC/C inhibitors Emi1, cyclin E, and cyclin A results in enhanced APC/C activity as observed by the enhanced degradation of the APC/C substrate aurora kinase A.
Figure 6. Atypical E2Fs can activate APC/CC dh1 via Emi1‐ and CDK2‐associated cyclins.
- Immunoblots showing Emi1 protein levels in HeLa cells with stable inducible expression of E2F7 or E2F8 after 16 h of doxycycline treatment.
- Quantitative PCR of FBXO5 transcripts in FACS‐sorted cells with doxycycline‐induced E2F7/8 expression, after 8 h of doxycycline treatment. To avoid bias from cell cycle defects, cells were released from HU arrest at the onset of doxycycline treatment, resulting in a strong enrichment of cells in late S or G2 of both vehicle‐ and doxycycline‐treated cells (see Fig EV3C). Bars in indicate mean ± s.e.m. (n = 3); asterisks indicate P < 0.05 versus vehicle and scrambled siRNA, respectively.
- Immunoblots of protein lysates from HeLa cells treated with siRNA against E2F7 and E2F8. Cells were harvested 6 h after release from HU block to enrich for cells in mid‐ to late S phase, where E2F7/8 expression was previously found to be high.
- Quantitative PCR of FBXO5 transcripts in HeLa cells treated with siRNA against E2F7 and E2F8. Cells were harvested 6 h after release from HU block to enrich for cells in mid‐ to late S phase. Bars indicate mean ± s.e.m. (n = 3); asterisks indicate P < 0.05 versus vehicle and scrambled siRNA, respectively.
- Cell cycle profiles of RPE cells determined by FACS analysis, during PD0332991 treatment, or 6 h after release. Percentages indicate the numbers of BrdU‐positive cells for each condition in, or 6 h after removing the drug (n = 3). BrdU was added to the cell culture medium 1 h prior to harvesting. A schematic overview of the experiment is shown in Fig EV6C.
- Immunoblots showing increased levels of E2F7 and other APC/CC dh1 substrates after CDH1 RNAi in RPE cells, during late G1 and early S phase.
- Immunoblots showing the effect of Emi1 RNAi on E2F7 and E2F8 expression in RPE cells. Hydroxyurea (2 nM) was added to arrest cells in S phase, the cyclin‐dependent kinase 2 inhibitor NU6140 (100 nM) was added to block APC/C inhibition by cyclin‐dependent kinase 2. Emi1 RNAi transfections were performed 48 h prior to harvesting, and drugs were added 24 h prior to harvesting.
- Overlap between E2F target genes and APC/C substrates. The APC/C substrates are taken from a curated list 36, 37 with the addition of E2F7 and E2F8 based on the current paper; E2F7/8 targets from 8, 10. Asterisk: cyclin B1 is indicated in red, because we only found it to be directly regulated by E2F7/8 in a hepatoblastoma cell line, but not HeLa cells.
- Simplified working model of interaction between atypical E2Fs and APC/C to coordinate DNA replication, based on the work described in this paper. Asterisks indicate indirect effect via transcriptional regulation of Emi1 and cyclin A/E.
Figure EV6. Schematic overviews of experiments to show feedback between atypical E2Fs and APC/CCdh1 .
- Experimental scheme of FACS sorting in synchronized cells with stable expression of E2F7/8 in the presence or absence of doxycycline induction.
- Example of gating strategy to sort late S/G2 cells with detectable E2F7/8‐EGFP.
- Schematic overview of synchronization using the CDK4/6 inhibitor PD0332991. RPE cells were transfected with CDH1 RNAi, then treated overnight with 0.5 μM of the CDK4/6 inhibitor PD0332991, and released by washing away the drug.
- Schematic overview of Emi1 RNAi experiments in RPE cells. Hydroxyurea and the CDK2 inhibitor NU6140 were added simultaneously.
To test whether endogenous E2F7/8 repress Emi1, we transfected HeLa cells with RNAi directed against E2F7 and E2F8. We harvested the cells in late S phase, 6 h after HU release, where E2F7 and E2F8 are expected to be highly expressed. Because E2F7 and E2F8 can transcriptionally repress each other, and can compensate for each other's function, we analyzed target gene expression in cells where both atypical E2Fs were knocked down (Fig 6C and D). We found a significant increase in FBXO5 transcript and Emi1 protein levels upon inactivation of atypical E2Fs, demonstrating that atypical E2Fs functions as transcriptional repressors of APC/C inhibitors (Fig 6C and D).
Next, we tested how APC/C impacts endogenous E2F7/8 expression during G1 and S phase. We transfected RPE cells with CDH1 RNAi, arrested them in G1 with the CDK4/6 inhibitor PD0332991, and released them by removing the drug (Fig EV6C). We verified that this synchronization approach was successful by FACS analysis of propidium iodide/BrdU‐stained cells (Fig 6E). We found no discernible effect of CDH1 RNAi on cell cycle progression under these experimental conditions, although it was previously reported that G1 is shortened after CDH1 depletion after nocodazole release 34, 35. However, knockdown of CDH1 resulted in enhanced protein expression of E2F7 during PD0332991 treatment and after release, as well as the APC/CCdh1 substrates CDC6 and aurora kinase A (Fig 6F). Importantly, 8 h after release, when Emi1 expression became clearly detectable, CDH1 RNAi caused a reduction in Emi1 levels, consistent with unscheduled E2F7 expression. E2F8 expression was again very low during G1 and surprisingly not enhanced after CDH1 RNAi. In fact E2F8 was further reduced, following a pattern similar to Emi1 expression (Fig 6F). To more conclusively study the effects of APC/C activity on endogenous E2F8, we performed an inverse experiment. Emi1 is critical for inactivation of the APC/C during S phase; therefore, we reasoned that Emi1 RNAi would result in decreased E2F7/8 expression. We arrested cells in S phase using HU, and indeed E2F7 and E2F8, as well as CDC6 and aurora kinase A, were partially degraded (Fig 6G). Because CDK2 can also inhibit APC/CCdh1, we also added the CDK2 inhibitor NU6140. This approach resulted in a near‐complete degradation of E2F7/8 as well as the other substrates after Emi1 RNAi, providing further support for a direct feedback loop between E2F7/8 and APC/C activity.
We noticed that many APC/C substrates are also target genes of E2F7/8. CDC6 and cyclin A2 are prominent examples, but in fact 27% of the genes from a published curated list of APC/C substrates 36, 37 are E2F7 and E2F8 target genes we have previously identified 8, 10 (Fig 6H). Based on this observation and the data presented here, we propose a model where the interplay between atypical E2Fs and APC/C controls the levels of important cell cycle genes on the transcriptional and posttranslational level (Fig 6I).
Discussion
Using a combination of time‐lapse fluorescence imaging experiments, in which we followed cells as they progress through the cell cycle, we found that E2F7 and E2F8 are degraded during late mitosis via APC/CCdh1. Furthermore, we demonstrate that APC/CCdh1 plays an important role in keeping E2F7/8 levels low throughout G1, because mutations in the KEN boxes or knockdown of CDH1 stabilized atypical E2Fs during this phase of the cell cycle. Combining E2F7/8 overexpression levels with ablation of CDH1 or expressing atypical E2F‐KEN mutants resulted in severely impaired S phase entry. These findings suggest that degradation of E2F7/8 is required for cells to enter S phase. Since atypical E2Fs function as transcriptional repressors of genes involved in DNA replication, inactivation of atypical E2F activity would allow the initiation of the DNA synthesis machinery 5, 6, 7, 8. The activator E2F1, E2F2, and E2F3 are required for transcriptional activation of S phase genes, and RB needs to be phosphorylated by cyclin/CDK complexes to release its inhibitory effect on the activator E2Fs. Similar to RB, atypical E2Fs can repress transcription of activator E2Fs as well, and we now provide a potential mechanism for keeping E2F7/8 levels low during G1 through APC/CCdh1‐mediated degradation to allow an upswing of E2F activator activity and transcription of S phase genes.
In many cellular contexts, the activator E2Fs and atypical E2Fs counterbalance each other: first, common target genes can be regulated in opposite manner, and secondly, E2F1–3 can directly activate E2F7/8, whereas E2F7/8 can also repress E2F1–3 9, 10, 11, 38. Thus, it seems counterintuitive that both classes of E2Fs would be regulated in the same manner at the posttranslational level. Nevertheless, activator E2Fs were found to be targeted for destruction by the APC/C under specific conditions. E2F3 degradation via APC/CCdh1 was linked to cell cycle exit and differentiation 39. E2F1 is degraded via APC/CCdc20 in prometaphase, but also by APC/CCdh1 26, 27. Interestingly the interaction with the dimerization partner DP protects E2F1 from destruction via the APC/C 26. Thus, a significant pool of E2F1 could be insensitive to degradation in G1 cells. In contrast, E2F7/8 do not interact with DP, and thus cannot be protected against degradation in this manner 5, 6, 7, 40. Also, the substrate binding by CDC20 and CDH1 appeared to occur via non‐canonical recognition motifs in E2F1, and E2F3, because the mutation of putative D‐boxes in E2F1 and E2F3 did not affect their degradation 26, 39. This indicates that their degradation follows different kinetics than the degradation of E2F7/8. Thus, APC/C activity can control the balance between activator and atypical E2F activity.
We provide evidence that E2F8, but not E2F7 is targeted for degradation by APC/CCdc20. However, the physiological relevance is yet unknown, because our time‐lapse imaging experiments showed that cells expressing stable, KEN mutant E2F8 did not have any apparent mitosis defects. We also observed that E2F8 levels already decrease during G2 and prophase (nocodazole), to rather low levels; furthermore, E2F8 was extremely low during G1, and contrary to E2F7, CDH1 depletion did not enhance its expression. Thus, it is highly likely that additional, yet‐unidentified E3 ligases can mediate E2F8 degradation. This could possibly be SKP1‐CUL1‐F‐box‐protein (SCF) complex, given its important role in cell cycle regulation 31 and its role in regulating activator E2F expression 41.
We noted a substantial overlap between activator E2Fs, atypical E2Fs, and APC/CCdh1 in regulating the expression of cell cycle genes (Fig 6I). Whereas E2F1–3 function as transcriptional activators and E2F7/8 as transcriptional repressors, APC/CCdh1 induces the degradation of a common set of E2F targets. The majority of the E2F target genes are essential for cell cycle progression, suggesting that the interaction between E2Fs and APC/CCdh1 coordinates the transcription and degradation of these cell cycle genes to allow proper cell cycle progression. Moreover, inactivation of the inhibitors of these cell cycle genes, the atypical E2Fs and APC/CCdh1, resulted in similar in vivo phenotypes. For example in mice, deletion of the CDH1‐encoding gene Fzr1, or E2f7/8 loss, both cause defects in trophoblast giant cell polyploidization 11, 19. In contrast, inactivation of the activators E2F1, E2F2, and E2F3, resulted in an enhanced trophoblast giant cell polyploidization 11. These findings provide further support that E2F‐ APC/CCdh1 interaction is critical for regulating common cell cycle genes.
Many of these common E2F target genes display an oscillating expression pattern during the cell cycle, characterized by low expression during M/G1, an upswing in expression during G1/S and downswing during G2/M 42. The orchestration of this oscillating pattern is partially mediated through the high APC/CCdh1 activity during the M and G1 phases, the induction of activator E2F1–3 during G1/S, and the increase of E2F7/8 during S/G2. Since APC/CCdh1 can degrade E2Fs as well as E2F targets, its activity needs to be downregulated during S/G2. Previous studies have demonstrated that Emi1 and CDK2 are the main inhibitors of APC/CCdh1 15, 16. Importantly, we show here that all these APC/CCdh1 inhibitors are directly repressed by the atypical E2Fs, providing strong evidence for a novel feedback loop between APC/CCdh1 and E2F7/8. Thus, E2F7/8 can activate its own degradation as well as the degradation of some of its targets by transcriptionally repressing the APC/CCdh1 inhibitors Emi1 and cyclin A/E‐CDK2. This regulatory mechanism is most likely important during late S, G2, and M phases to coordinate the downregulation of E2F target genes. In addition, it might be also relevant during DNA damage, because previous studies have shown that E2F7/8 expression increases during DNA damage 43, 44, 45 and APC/CCdh1 activity becomes reactivated in G2 cells with DNA damage 46, 47. Further support for an APC/CCdh1‐E2F feedback loop is provided by the fact that the Emi1‐encoding gene FBXO5 and the CDK2‐activating genes CCNA2 and CCNE1/2 are known target genes of the activator E2F1–3 33, 48, 49. Since E2F1–3 are induced during the G1/S phase transition, the transcriptional activation of Emi1 and CDK2‐activating cyclins most likely mediates the inhibition of APC/CCdh1 activity, which is crucial for S and G2 phase progression and mitotic entry 16, 50, 51.
In summary, we show a novel and important role for atypical E2Fs in determining the balance between synthesis and repression of E2F target genes, and balancing APC/C activity.
Materials and Methods
Generation of cell lines and cell culture
Mouse E2F8 cDNA (Reference sequence: NM_001013368.5) was amplified with primers that introduced a NotI site at the 5′ and a XhoI site at the 3′ end, using Pfu polymerase (New England Biolabs). The cDNA was then cloned into the pEGFP‐N3 plasmid (Invitrogen) using a double digestion with these two enzymes, followed by ligation with T4 ligase (New England Biolabs), in such a way that a C‐terminal E2F8‐EGFP fusion protein is transcribed. Subsequently, E2F8‐EGFP was digested from the pEGFP plasmid by an enzymatic digestion with NotI and subsequently ligated into the pcDNA4/TO plasmid (Invitrogen) using T4 ligase. Correct orientation of E2F8‐EGFP in the plasmid was verified by running XhoI‐digested plasmids in an agarose gel. Plasmids containing E2F7 were generated as described before 8.
Site‐directed mutagenesis against sequences encoding KEN domains was performed by PCR amplification with Pfu polymerase of the E2F7/8 plasmids with primers encoding the required mutations. To ensure complete inhibition of these motifs, the KEN sequences were replaced by three consecutive alanines. Successful cloning and mutagenesis were confirmed with Sanger sequencing (Macrogen).
Tet repressor‐expressing HeLa cells (T‐REx HeLa; Invitrogen) were transfected with these constructs, and stable clones were established by Zeocin selection (Invitrogen, 300 μg/ml). The cells were cultured in DMEM containing 10% Tet System Approved fetal bovine serum (Clontech). Overexpression was induced by adding 0.2 μg/ml doxycycline (Sigma) to the cell culture medium. Cells were synchronized by adding 2 mM hydroxyurea (Sigma‐Aldrich) to the medium for 16 h, or 2.5 mM thymidine to arrest cells at the onset of S phase. Cells were released from the block by washing three times with PBS and adding fresh medium containing 10% FBS. Other drugs used were as follows: MPS1 inhibitor reversine (50 nm, Cayman Chemicals 10004412), CDK1 inhibitor RO‐3306 (10 μM, Calbiochem #217699), Cdk4/6 inhibitor PD0332991 (0.5 μM, Selleck S116), nocodazole (830 nM Sigma‐Aldrich M1404), and CDK2 inhibitor NU6140 (10 μM, Tocris 3301).
Flow cytometry and FACS sorting
For measurement of DNA contents, cells were trypsinized, washed with PBS, fixed with 70% ethanol, and stored at 4°C up to 1 week. Cells were washed twice with TBS and then reconstituted in PBS containing 20 μg/ml propidium iodide, 250 μg/ml RNase A, and 0.1% bovine serum albumin (BSA).
DNA synthesis was measured by adding 100 μM 5‐bromo‐2′‐deoxyuridine (BrdU; Sigma) to the culture medium for 2 h prior to harvesting. Ethanol‐fixed samples were washed once with PBS, cells were incubated with 0.1 N HCl/0.5 mg/ml pepsin for 20 min. After washing with TBS/0.5% Tween/0.1% BSA, 2 N HCl was added for 12 min, followed by pH neutralization with sodium tetraborate buffer (pH 8.5). After washing twice with TBS‐0.1% Tween, cells were incubated for 1 h with a FITC‐conjugated antibody against BrdU (Becton Dickinson 347583). Cells were washed with TBS, and labeled with propidium iodide as indicated above. All samples were measured on a BD FACSCanto II (BD Biosciences) and further analyzed using FlowJo software.
Two‐way FACS sorting based on FUCCI markers was performed on a BD Influx system by a senior operator. Cells were collected in cold PBS and briefly centrifuged at 800 g. After removing the PBS, lysis buffer was added to the cell pellet. For RNA isolation, 50,000 cells were collected, and for protein 300,000 cells.
Transfections
HEK293T cells were seeded at a density of 3 million in 10‐cm petri dishes. The next morning cells were transfected with 10 μg of E2F7/8 plasmid and/or 2 μg of CDH1‐Flag using the calcium phosphate method. After 48 h, transfected cells were harvested. For co‐immunoprecipitation experiments, 10 μM of MG132 (Cayman Chemicals) was added to the culture medium 5 h prior to harvesting.
For siRNA experiments, RPE or HeLa cells were plated in 6‐well dishes and transfected with 10 nM of CDC20, CDH1, Emi1, E2F7, E2F8, or scrambled siRNA (all SmartPool; Dharmacon) using RNAiMAX or Lipofectamine according to the manufacturer's instructions, with the modification that transfection complexes were added to the cells for 6 h in basic medium containing no serum or antibiotics. Afterward, cells were washed and fresh medium containing 10% FBS and pen/strep was added.
To create inducible E2F7/8‐EGFP cell lines co‐expressing Cherry‐PCNA, cells were infected with the viral plasmid pLIB‐Cherry‐PCNA as described before 14. Virus carrying Cherry‐PCNA was created by calcium phosphate transfection of HEK293T cells, and double infection of target cells in the presence of polybrene.
Time‐lapse fluorescence microscopy
Acquisition of differential interference contrast (DIC) and fluorescence images started 24 h after transfection on a microscope (Axio Observer Z1; Carl Zeiss) in a heated culture chamber (5% CO2 at 37°C) using DMEM with 8% FCS and antibiotics. The microscope was equipped with an LD 0.55 condenser and 40× NA 1.40 Plan Apochromat oil DIC objective and CFP/YFP and GFP/HcRed filter blocks (Carl Zeiss) to select specific fluorescence. Images were taken using ZEN 2012 acquisition software (Carl Zeiss) with a charge‐coupled device camera [ORCA R2 Black and White CCD (Hamamatsu Photonics)] at 50‐ms exposure time for EGFP excitation and 200‐ to 300‐ms exposure time for mCherry excitation at 30% LED intensity. For quantitative analysis of degradation, ImageJ (National Institutes of Health) and Excel (Microsoft) were used. Captured images were processed using Photoshop and Illustrator software (Adobe). Plots were created by GraphPad Prism version 6.0f, for Mac OS X (GraphPad Software).
Quantitative PCR
Isolation of RNA, cDNA synthesis, and qPCR were performed as previously described 8. Gene expression was calculated using a ΔΔC t method adapted for multiple‐reference gene correction 52. All samples were corrected for the two reference genes β‐actin and GAPDH. Primer sequences are provided in Table EV1.
Co‐IP and immunoblotting
Cells were harvested by washing twice with PBS, and scraped in a lysis buffer containing 50 mM Tris–HCl, 1 mM EDTA, 150 mM NaCl, 0.25% deoxycholic acid, 1% Nonidet P‐40, 1 mM NaF, 1 mM NaV3O4, and protease inhibitor cocktail (Roche). Cells were lysed on ice for 20 min, and centrifuged for 10 min at 12,000 g. The supernatants were then immunoblotted with standard SDS–PAGE techniques. Antibodies used throughout this paper are listed in Table EV2. Visualization was done by ECL (GE Healthcare RPN2106) and exposure to a film (GE Healthcare). All blot photos are representative examples of three independent experiments, unless stated otherwise.
Co‐immunoprecipitations were performed as follows: cells in one near‐confluent 10‐cm dish were harvested in 1 ml of protein lysis buffer as described above. Then, BSA‐blocked prot G beads (Fastflow; Millipore) were added and incubated for 30 min at 4°C to preclear the lysates. One 1% of the total input was kept separately, and immunoprecipitation was done by rotating samples at 4°C for 1 h in the presence of 2 μg of anti‐Flag and BSA‐blocked prot G beads. EGFP‐tagged fusion proteins were immunoprecipitated with GFP‐Trap (Chromotek). After washing the beads with lysis buffer, protein was eluted using Laemmli loading buffer and immunoblots were performed as described above.
Data analysis
All immunoblots, co‐immunoprecipitations, FACS data, and qPCR results were replicated three times, unless stated otherwise in the figure legends. Statistical analysis on qPCR and FACS data was performed by ANOVA followed by Holm‐Sidak post hoc individual group comparisons. Individual curves for S phase entry and cell death were compared as pairs by log‐rank (Mantel–Cox) test.
Author contributions
MB, RY, APW, HAS, EAvL, NA, IJ, and BW performed experiments. MB, RMFW, AdB, and BW conceived experiments, analyzed and interpreted the data, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Movie EV1
Movie EV2
Review Process File
Acknowledgements
We thank Dr. Daniele Guardavaccaro (Hubrecht Institute, Utrecht, NL) for kindly providing CDH1 and CDC20 constructs and helpful discussion. The stable RPE‐FUCCI cell lines were provided by Prof. Rene Medema (Netherlands Cancer Institute, NL). Ger Arkesteijn (Utrecht University, NL) provided expert assistance with FACS sorting experiments. This work was financially supported by a Dutch Cancer Society grant (KWF: UU2013‐5777) to BW and AdB, Netherlands Organization for Scientific Research (NWO: ALW‐IN11‐28) to AdB, and a HFSP project grant to MB and RMFW (RGP0053/2010).
EMBO Reports (2016) 17: 414–427
References
- 1. Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller MC, Shaikh N, Domingo E, Kanu N, Dewhurst SM, Gronroos E et al (2013) Replication stress links structural and numerical cancer chromosomal instability. Nature 494: 492–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lecona E, Fernandez‐Capetillo O (2014) Replication stress and cancer: it takes two to tango. Exp Cell Res 329: 26–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen HZ, Tsai SY, Leone G (2009) Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer 9: 785–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chong JL, Wenzel PL, Saenz‐Robles MT, Nair V, Ferrey A, Hagan JP, Gomez YM, Sharma N, Chen HZ, Ouseph M et al (2009) E2f1‐3 switch from activators in progenitor cells to repressors in differentiating cells. Nature 462: 930–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. de Bruin A, Maiti B, Jakoi L, Timmers C, Buerki R, Leone G (2003) Identification and characterization of E2F7, a novel mammalian E2F family member capable of blocking cellular proliferation. J Biol Chem 278: 42041–42049 [DOI] [PubMed] [Google Scholar]
- 6. Maiti B, Li J, de Bruin A, Gordon F, Timmers C, Opavsky R, Patil K, Tuttle J, Cleghorn W, Leone G (2005) Cloning and characterization of mouse E2F8, a novel mammalian E2F family member capable of blocking cellular proliferation. J Biol Chem 280: 18211–18220 [DOI] [PubMed] [Google Scholar]
- 7. Di Stefano L, Jensen MR, Helin K (2003) E2F7, a novel E2F featuring DP‐independent repression of a subset of E2F‐regulated genes. EMBO J 22: 6289–6298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Westendorp B, Mokry M, Groot Koerkamp MJ, Holstege FC, Cuppen E, de Bruin A (2012) E2F7 represses a network of oscillating cell cycle genes to control S‐phase progression. Nucleic Acids Res 40: 3511–3523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li J, Ran C, Li E, Gordon F, Comstock G, Siddiqui H, Cleghorn W, Chen HZ, Kornacker K, Liu CG et al (2008) Synergistic function of E2F7 and E2F8 is essential for cell survival and embryonic development. Dev Cell 14: 62–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pandit SK, Westendorp B, Nantasanti S, van Liere E, Tooten PC, Cornelissen PW, Toussaint MJ, Lamers WH, de Bruin A (2012) E2F8 is essential for polyploidization in mammalian cells. Nat Cell Biol 14: 1181–1191 [DOI] [PubMed] [Google Scholar]
- 11. Chen HZ, Ouseph MM, Li J, Pecot T, Chokshi V, Kent L, Bae S, Byrne M, Duran C, Comstock G et al (2012) Canonical and atypical E2Fs regulate the mammalian endocycle. Nat Cell Biol 14: 1192–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bertoli C, Skotheim JM, de Bruin RA (2013) Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol 14: 518–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mailand N, Diffley JF (2005) CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C‐dependent proteolysis. Cell 122: 915–926 [DOI] [PubMed] [Google Scholar]
- 14. Clijsters L, Ogink J, Wolthuis R (2013) The spindle checkpoint, APC/C(Cdc20), and APC/C(Cdh1) play distinct roles in connecting mitosis to S phase. J Cell Biol 201: 1013–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sorensen CS, Lukas C, Kramer ER, Peters JM, Bartek J, Lukas J (2001) A conserved cyclin‐binding domain determines functional interplay between anaphase‐promoting complex‐Cdh1 and cyclin A‐Cdk2 during cell cycle progression. Mol Cell Biol 21: 3692–3703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hsu JY, Reimann JD, Sorensen CS, Lukas J, Jackson PK (2002) E2F‐dependent accumulation of hEmi1 regulates S phase entry by inhibiting APC(Cdh1). Nat Cell Biol 4: 358–366 [DOI] [PubMed] [Google Scholar]
- 17. Miller JJ, Summers MK, Hansen DV, Nachury MV, Lehman NL, Loktev A, Jackson PK (2006) Emi1 stably binds and inhibits the anaphase‐promoting complex/cyclosome as a pseudosubstrate inhibitor. Genes Dev 20: 2410–2420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sivaprasad U, Machida YJ, Dutta A (2007) APC/C–the master controller of origin licensing? Cell Div 2: 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Garcia‐Higuera I, Manchado E, Dubus P, Canamero M, Mendez J, Moreno S, Malumbres M (2008) Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat Cell Biol 10: 802–811 [DOI] [PubMed] [Google Scholar]
- 20. Lafranchi L, de Boer HR, de Vries EG, Ong SE, Sartori AA, van Vugt MA (2014) APC/C(Cdh1) controls CtIP stability during the cell cycle and in response to DNA damage. EMBO J 33: 2860–2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Miller CW, Aslo A, Campbell MJ, Kawamata N, Lampkin BC, Koeffler HP (1996) Alterations of the p15, p16, and p18 genes in osteosarcoma. Cancer Genet Cytogenet 86: 136–142 [DOI] [PubMed] [Google Scholar]
- 22. Pfleger CM, Kirschner MW (2000) The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev 14: 655–665 [PMC free article] [PubMed] [Google Scholar]
- 23. Cohen M, Vecsler M, Liberzon A, Noach M, Zlotorynski E, Tzur A (2013) Unbiased transcriptome signature of in vivo cell proliferation reveals pro‐ and antiproliferative gene networks. Cell Cycle 12: 2992–3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sakaue‐Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H et al (2008) Visualizing spatiotemporal dynamics of multicellular cell‐cycle progression. Cell 132: 487–498 [DOI] [PubMed] [Google Scholar]
- 25. Clute P, Pines J (1999) Temporal and spatial control of cyclin B1 destruction in metaphase. Nat Cell Biol 1: 82–87 [DOI] [PubMed] [Google Scholar]
- 26. Peart MJ, Poyurovsky MV, Kass EM, Urist M, Verschuren EW, Summers MK, Jackson PK, Prives C (2010) APC/C(Cdc20) targets E2F1 for degradation in prometaphase. Cell Cycle 9: 3956–3964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Budhavarapu VN, White ED, Mahanic CS, Chen L, Lin FT, Lin WC (2012) Regulation of E2F1 by APC/C Cdh1 via K11 linkage‐specific ubiquitin chain formation. Cell Cycle 11: 2030–2038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Floyd S, Pines J, Lindon C (2008) APC/C Cdh1 targets aurora kinase to control reorganization of the mitotic spindle at anaphase. Curr Biol 18: 1649–1658 [DOI] [PubMed] [Google Scholar]
- 29. Clijsters L, Wolthuis R (2014) PIP‐box‐mediated degradation prohibits re‐accumulation of Cdc6 during S phase. J Cell Sci 127: 1336–1345 [DOI] [PubMed] [Google Scholar]
- 30. Kitamura E, Blow JJ, Tanaka TU (2006) Live‐cell imaging reveals replication of individual replicons in eukaryotic replication factories. Cell 125: 1297–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nakayama KI, Nakayama K (2006) Ubiquitin ligases: cell‐cycle control and cancer. Nat Rev Cancer 6: 369–381 [DOI] [PubMed] [Google Scholar]
- 32. van Leuken R, Clijsters L, Wolthuis R (2008) To cell cycle, swing the APC/C. Biochim Biophys Acta 1786: 49–59 [DOI] [PubMed] [Google Scholar]
- 33. Botz J, Zerfass‐Thome K, Spitkovsky D, Delius H, Vogt B, Eilers M, Hatzigeorgiou A, Jansen‐Durr P (1996) Cell cycle regulation of the murine cyclin E gene depends on an E2F binding site in the promoter. Mol Cell Biol 16: 3401–3409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sigl R, Wandke C, Rauch V, Kirk J, Hunt T, Geley S (2009) Loss of the mammalian APC/C activator FZR1 shortens G1 and lengthens S phase but has little effect on exit from mitosis. J Cell Sci 122: 4208–4217 [DOI] [PubMed] [Google Scholar]
- 35. Greil C, Krohs J, Schnerch D, Follo M, Felthaus J, Engelhardt M, Wäsch R (2015) The role of APC/C in replication stress and origin of genomic instability. Oncogene doi: 10.1038/onc.2015.367 [DOI] [PubMed] [Google Scholar]
- 36. Meyer HJ, Rape M (2011) Processive ubiquitin chain formation by the anaphase‐promoting complex. Semin Cell Dev Biol 22: 544–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Min M, Mayor U, Lindon C (2013) Ubiquitination site preferences in anaphase promoting complex/cyclosome (APC/C) substrates. Open Biol 3: 130097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ouseph MM, Li J, Chen HZ, Pecot T, Wenzel P, Thompson JC, Comstock G, Chokshi V, Byrne M, Forde B et al (2012) Atypical E2F repressors and activators coordinate placental development. Dev Cell 22: 849–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ping Z, Lim R, Bashir T, Pagano M, Guardavaccaro D (2012) APC/C (Cdh1) controls the proteasome‐mediated degradation of E2F3 during cell cycle exit. Cell Cycle 11: 1999–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Christensen J, Cloos P, Toftegaard U, Klinkenberg D, Bracken AP, Trinh E, Heeran M, Di Stefano L, Helin K (2005) Characterization of E2F8, a novel E2F‐like cell‐cycle regulated repressor of E2F‐activated transcription. Nucleic Acids Res 33: 5458–5470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang Z, Wang H, Li M, Rayburn ER, Agrawal S, Zhang R (2005) Stabilization of E2F1 protein by MDM2 through the E2F1 ubiquitination pathway. Oncogene 24: 7238–7247 [DOI] [PubMed] [Google Scholar]
- 42. Santos A, Wernersson R, Jensen LJ (2015) Cyclebase 3.0: a multi‐organism database on cell‐cycle regulation and phenotypes. Nucleic Acids Res 43: D1140–D1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zalmas LP, Zhao X, Graham AL, Fisher R, Reilly C, Coutts AS, La Thangue NB (2008) DNA‐damage response control of E2F7 and E2F8. EMBO Rep 9: 252–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Carvajal LA, Hamard PJ, Tonnessen C, Manfredi JJ (2012) E2F7, a novel target, is up‐regulated by p53 and mediates DNA damage‐dependent transcriptional repression. Genes Dev 26: 1533–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Aksoy O, Chicas A, Zeng T, Zhao Z, McCurrach M, Wang X, Lowe SW (2012) The atypical E2F family member E2F7 couples the p53 and RB pathways during cellular senescence. Genes Dev 26: 1546–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bassermann F, Frescas D, Guardavaccaro D, Busino L, Peschiaroli A, Pagano M (2008) The Cdc14B‐Cdh1‐Plk1 axis controls the G2 DNA‐damage‐response checkpoint. Cell 134: 256–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yamada M, Watanabe K, Mistrik M, Vesela E, Protivankova I, Mailand N, Lee M, Masai H, Lukas J, Bartek J (2013) ATR‐Chk1‐APC/CCdh1‐dependent stabilization of Cdc7‐ASK (Dbf4) kinase is required for DNA lesion bypass under replication stress. Genes Dev 27: 2459–2472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhu W, Giangrande PH, Nevins JR (2004) E2Fs link the control of G1/S and G2/M transcription. EMBO J 23: 4615–4626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, Dynlacht BD (2002) E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev 16: 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Robbins JA, Cross FR (2010) Requirements and reasons for effective inhibition of the anaphase promoting complex activator CDH1. Mol Biol Cell 21: 914–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kramer ER, Scheuringer N, Podtelejnikov AV, Mann M, Peters JM (2000) Mitotic regulation of the APC activator proteins CDC20 and CDH1. Mol Biol Cell 11: 1555–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002) Accurate normalization of real‐time quantitative RT–PCR data by geometric averaging of multiple internal control genes. Genome Biol 3: RESEARCH0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Movie EV1
Movie EV2
Review Process File