

Abstract
The common bean (Phaseolus vulgaris L.) is the world’s most important legume for human consumption. Anthracnose (ANT; Colletotrichum lindemuthianum) and angular leaf spot (ALS; Pseudocercospora griseola) are complex diseases that cause major yield losses in common bean. Depending on the cultivar and environmental conditions, anthracnose and angular leaf spot infections can reduce crop yield drastically. This study aimed to estimate linkage disequilibrium levels and identify quantitative resistance loci (QRL) controlling resistance to both ANT and ALS diseases of 180 accessions of common bean using genome-wide association analysis. A randomized complete block design with four replicates was performed for the ANT and ALS experiments, with four plants per genotype in each replicate. Association mapping analyses were performed for ANT and ALS using a mixed linear model approach implemented in TASSEL. A total of 17 and 11 significant statistically associations involving SSRs were detected for ANT and ALS resistance loci, respectively. Using SNPs, 21 and 17 significant statistically associations were obtained for ANT and angular ALS, respectively, providing more associations with this marker. The SSR-IAC167 and PvM95 markers, both located on chromosome Pv03, and the SNP scaffold00021_89379, were associated with both diseases. The other markers were distributed across the entire common bean genome, with chromosomes Pv03 and Pv08 showing the greatest number of loci associated with ANT resistance. The chromosome Pv04 was the most saturated one, with six markers associated with ALS resistance. The telomeric region of this chromosome showed four markers located between approximately 2.5 Mb and 4.4 Mb. Our results demonstrate the great potential of genome-wide association studies to identify QRLs related to ANT and ALS in common bean. The results indicate a quantitative and complex inheritance pattern for both diseases in common bean. Our findings will contribute to more effective screening of elite germplasm to find resistance alleles for marker-assisted selection in breeding programs.
Introduction
Common bean (Phaseolus vulgaris L.) is an annual legume crop with a relatively small genome of 473 Mb [1]. It is the most important grain legume for direct human consumption [2,3]. Dry beans provide a major source of quality protein, which is high in lysine and therefore complements most cereals. In addition, beans are high in carbohydrates, fiber, and minerals (calcium, potassium, phosphorus iron, zinc and magnesium) [4].
Anthracnose (ANT) and Angular Leaf Spot (ALS) are two important diseases that have the greatest impact on crop yield reduction. Yield losses caused by the ANT pathogen are extremely high in common bean, reaching up to 100% [5]. Anthracnose is caused by the specialized hemibiotrophic fungus Colletotrichum lindemuthianum (Sacc. and Magnus), which co-evolved with common bean into Andean and Mesoamerican races [6,7]. ALS leads to crop losses of up to 80% and is found in more than 60 countries worldwide [8,9]. This disease is caused by the hemibiotrophic fungus Pseudocercospora griseola (Sacc.) Crous & Braun (sin. Phaeoisariopsis griseola (Sacc.) Ferraris) [10], and can be identified by angular necrotic spots on plant leaves and pods [11].
Bi-parental populations used for QRL mapping accumulate a limited number of recombination events, typically leading to low mapping resolution or poor estimation of marker effects [12,13]. In this case, recombination has not had sufficient time to shuffle the genome into small fragments, and quantitative trait loci (QTL) are generally localized to large chromosomal regions of 10 to 20 centiMorgan [14].
Genome-wide association studies (GWAS) or association mapping (AM) and linkage disequilibrium (LD) offer high resolution through historical recombination accumulated in natural populations and collections of landraces, breeding materials, and varieties [15]. By exploiting broader genetic diversity, GWAS offers several advantages over linkage mapping, such as mapping resolution, allele number, time saved in establishing marker-trait associations, and application in breeding programs [16]. The strength of the correlation between two markers is a function of the distance between them: the closer two markers are, the stronger the LD. The resolution with which a QRL can be mapped is a function of how quickly LD decays over distance. Selfing reduces opportunities for recombination; thus, in self-pollinating species such as rice (Oryza Sativa), LD may extend to 100 Kb or more [17]. In general, high LD is expected between tightly linked loci, since the mutation should have eliminated LD between loci that are not in close proximity to one another [18,19]. In common bean, little information is available on the extent of LD [20,21,22].
The Common Bean Germplasm Bank of the Agronomic Institute (IAC, Campinas, S.P. Brazil) holds more than 1800 accessions representing the two principal centers of origin (Andean and Mesoamerican) of the species as well as ecotypes from different South American countries and a large number of lines from both Brazilian and international genetic breeding programs, most of which were obtained by germplasm interchange [23]. Some factors should be considered for the improvement association mapping, one of which is the selection of a group of individuals from the original germplasm collection with coverage of a high level of genetic diversity [24]. In a previous study [25], evaluated the diversity level and genetic organization present among 500 accessions from the Common Bean Germplasm Bank of the IAC and selected 180 accessions that represented the variability of the entire collection in order to use this panel for association mapping studies.
Some points in association mapping there are to consider: (1) selection of a germplasm collection with high level of genetic diversity; (2) recording or measuring the phenotypic characteristics of the selected population groups; (3) genotyping the individuals with available molecular markers; (4) quantification of the LD extent of the genome of a chosen population using molecular markers; (5) assessment of the level of genetic differentiation among groups within the sampled individuals and the coefficient of relatedness between pairs of individuals within a sample; and (6) taking the information gained through the quantification of LD and population structure into account for the correlation of phenotypic and genotypic/haplotypic data with the application of an appropriate statistical approach that reveals “marker tags” positioned within close proximity of the targeted trait of interest [26].
Several groups have performed AM studies for common bean [27,28,29], but none have focused on ANT and ALS resistance. Based on several linkage analysis studies, we have reported previously important resistance candidate genes in common bean [30,31,32,33]. However, to complement our initial findings and further understand the ANT and ALS plant disease interactions in common bean, the aims of this study were to (i) estimate the linkage disequilibrium (LD) in common bean; and (ii) identify QRLs associated with resistance to ANT and ALS using SSR and SNP data.
Materials and Methods
Plant material and DNA extraction
One hundred and eighty accessions previously identified and evaluated [25] from the common bean germplasm bank of the Agronomic Institute (IAC, Campinas, São Paulo, Brazil) were used in this work, with 24 genotypes are of Andean origin and 156 are of Mesoamerican origin. These genotypes displayed variance in many agronomic traits related to resistance to major common bean diseases (anthracnose, angular leaf spot, rust, fusarium wilt, bacterial blight, and gold mosaic virus), drought tolerance, as well as differences in grain size and tegument color. In summary, among the 180 accessions [25], 75 were chosen due to their great economic importance to the Brazilian common bean breeding program, as they include commercial varieties with carioca tegument, widely cultivated in at the state of São Paulo (Brazil), under the leadership of the Agronomic Institute (IAC). Total genomic DNA (gDNA) was extracted from lyophilized young leaf tissues using the CTAB method.
Simple sequence repeat (SSR) marker analysis
A total of 103 microsatellites (SSRs) were amplified. From these, 45 were EST-SSRs (EST—Expressed sequence tag) [34], while the others were genomic-SSRs [35]. Polymerase chain reaction (PCR) amplifications were performed in a 25 μL final volume containing 50 ng DNA template, 1× buffer, 0.2 μM forward primer, 0.2 μM reverse primer, 100 μM dNTP, 2.0 mM MgCl2, 10 mM Tris-HCl (pH 8,0), 50 mM KCl, and 0.5 U of Taq DNA polymerase. The following conditions were used for amplification: 1 min at 94°C, 30 cycles of 1 min at 94°C, 1 min at the specific annealing temperature for each SSR, and 1 min at 72°C, with a final extension of 5 min at 72°C. The PCR products were checked on a 3% agarose gel and separated using 6% silver-stained polyacrylamide.
Allele sizes were scored in base pairs (bp) by visual comparison with a 10-bp DNA ladder (Invitrogen), and the value was converted to gene and genotypic frequencies. After the binary allele scoring (1 or 0) was completed, genotyping was performed using the allele number in decreasing order; that is, alleles with greater size received the highest numbers. In the case of diploids such as common bean, homozygous bands with heterozygous genotypes were scored twice.
Single-nucleotide polymorphism (SNP) marker analysis
SNP genotyping was conducted using the technology Vera Code® BeadXpress platform (Illumina) at the Biotechnology Laboratory of Embrapa (Goiania, GO, Brazil). A set of 384 SNP markers, validated by a previously identified Prelim file (https://icom.illumina.com/Custom/UploadOpaPrelim/) for Phaseolus vulgaris [36], a derivative of polymorphisms between strains BAT477 and Pérola of Mesoamerican origins, was selected to compose the Oligo Pool Assay (OPA) SNP marker panel. Three oligonucleotides were used for each of the variants of the same SNP and the third specific-locus binding to the 3′ region of the DNA fragment containing the target SNP, generating a unique allele-specific fragment. Subsequently, this fragment was amplified using Taq DNA polymerase enzyme Titanium (Clontech®) and complementary primers labeled with Cy3 and Cy5 fluorophores. Genotyping was performed using Genome Studio software version 1.8.4 (Illumina, EUA) using Call Rate values ranging from 0.80 to 0.90 and GenTrain ≥ 0.26 for SNP grouping. Automated analyses were performed to cluster the SNP alleles of each line, based on signal intensities of Cy3 and Cy5 fluorophores. Groups were adjusted manually by determining the best clusters based on parental profiles.
Linkage disequilibrium (LD) analysis
Fisher’s exact tests [37] were performed for each possible pair of markers from the 103 SSRs. To avoid false positives due to multiple tests (i.e., markers in linkage equilibrium or non-causative associations), Bonferroni [38] and False Discovery Rate (FDR) [39] corrections were applied. FDR was applied in addition to Bonferroni because the latter is a very conservative method for type I error control. Furthermore, to determine the suitable marker density resolution for association mapping, we also analyzed the extent of LD against the genetic distance considering only Fisher’s exact test results between the mapped and linked SSRs, based on two previously published genetic linkage maps for common bean [35,40]. All the analyses were performed using R software (R Development Core Team 2014).
For 384 SNPs, pairwise tests for LD levels were performed using r2 [41], which was proposed in the context of biallelic loci. Initially, LD was estimated for each chromosome separately, and then a heat map for each one was generated to view LD patterns. Subsequently, the LD obtained for all associations involving linked SNPs from the different common bean chromosomes was plotted against the genetic distance. With these associations, it was possible to adjust a non-linear model for LD decay. All the analyses were performed using the package synbreed [42] available in R software (R Development Core Team 2014).
Phenotypic analyses of anthracnose and angular leaf spot
For the ANT phenotype the seeds of 180 common bean accessions were germinated on germination paper in a growth chamber at 25°C with a 12-hours photoperiod for 3 days. Four seedlings per accession were transplanted to boxes containing autoclaved vermiculite (Plantmax®) as substrate, constituting an experimental plot. Four different accessions were grown per box. The experimental design was carried out in randomized complete blocks with four replicates. Plants were inoculated 7 days after transplanting, where each experiment consisted in a single race of C. lindemuthianum inoculation (race 04). The experimental design was carried out in randomized complete blocks with four replicates.
Monosporic cultures of C. lindemuthianum were grown on PDA media (200 g L–1 potato, 30 g L–1 dextrose and 30 g L–1 agar), and conidia were collected in water suspension using a glass spreader. Plants were sprayed with spore suspension (106 spores mL–1) using a DeVilBiss apparatus (Fanem). Immediately after inoculation, plants were kept for 48 hours under 95–100% relative humidity at 23°C and a 12-hour photoperiod. Disease severity was evaluated 7 to 10 days post-inoculation using a diagrammatic scale proposed by Pastor-Corrales et al., in which 1–3 denotes resistant, 4–6 intermediate, and 7–9 susceptible [6].
For the ALS phenotype the seeds were sown in plastic boxes (29.5 cm × 46.5 cm × 12.5 cm) containing Dystrophic Red Latosol type soil, fertilized with NPK 04-14-08 (400 kg/ha), each with four accessions sown in rows at a distance of approximately 4 cm, containing four plants per row. The experimental design was carried out in randomized complete blocks with four replicates. Plants were inoculated when they reached the V3 development stage (first expanded trifoliate), 2 to 3 weeks after planting, by spraying both leaf surfaces with a 104 conidia/mL suspension prepared from P. griseola monosporic colonies grown in V8 medium [43]. The isolate used (14259–2) was classified into the 0–39 race based on the response of the differential cultivars according to Pastor-Corrales et al. [6].
After inoculation, the accessions were held for 48 hours at a temperature between 22°C to 24°C, relative humidity between 95–100%, and a photoperiod of 12 hours [43]. After this period, plants were transferred to the greenhouse. The evaluation of severity was made 17 days after inoculation.
The race 04 of C. lindemuthianum was used in this study because it previously detected ANT QTLs in greenhouse conditions [32], and the race 0–39 of P. griseola was also previously associated to ALS QTLs [30,31].
The phenotypic data obtained for each disease (ANT and ALS), based on the 9-point scale of severity, were analyzed according to the following statistical model:
where yijk corresponds to the level of ANT or ALS severity; μ μis the general average; bi is the fixed effect of the block i; gj is the random effect associated with accession j; rk(ij) is the random effect associated with the replicate k nested within genotype j and block i; and ɛij is the random residual term. The analyses were performed using the nlme package, available in R software (R Development Core Team 2014).
The presence of heterogeneous genetic variances and genetic covariances and correlations between the observations were investigated in the phenotypic analyses. We tested different mixed models for the genetic-effects matrix considering the interaction between the genotypes and the blocks or replicates, using the Akaike information criterion (AIC) [44] and Bayesian information criterion (BIC) [45] to compare and select the best models. The predicted means of the genotypes were obtained from the most likely models for ANT and ALS and used for association mapping analyses.
Genome mapping and functional annotation
Molecular markers associated with ANT and ALS resistance were aligned to the common bean genome [1] using the native nucleotide basic local alignment search tool (BLASTn) and default algorithm parameters (threshold E-value < 1 × 10−10) from Phytozome version 1.0 (http://www.phytozome.net/). Putative candidate genes involved in the resistance of common bean to these diseases were identified from the genome localization of the markers. GO annotations for putative functions of the genes were obtained by comparison to GeneOntology (GO, http://www.geneontology.org/) using Blast2GO program [46]. The annotations were separated for each marker and the corresponding disease. The KEGG pathway annotations were obtained by aligning sequences to the KEGG database comparing to the protein sequences of Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.genome.jp/) [47]. To predict gene structure and marker locations into the gene, these markers were alignedwith phytozome annotated genomic sequence (http://www.phytozome.net/).
Association mapping analyses
Marker’s sequences available on NCBI (http://www.ncbi.nlm.nih.gov/) or PhaseolusGenes (http://phaseolusgenes.bioinformatics.ucdavis.edu/) databases were used to localize the SSRs and SNPs in the P. vulgaris chromosomes using the native Phytozome’s BLAST and default algorithm parameters (http://www.phytozome.net/). The criteria used to assign putative chromosomes to the markers included E-values ≤ 1 × 10−15 and a minimum identity of 70% between query and database sequences.
Association mapping analyses were carried out using TASSEL version 2.01 software for SSRs and TASSEL version 5.0 for SNPs (http://www.maizegenetics.net/index.php?option=com_content&task=view&id=89&Itemid=119).
Mixed linear model (MLM) analyses were performed considering the estimated kinship matrix (K), calculated using SPAGeDi software [48], as random effects and the population structure matrix (Q), previously inferred by STRUCTURE software [49] in Perseguini et al. using SSRs, as part of the fixed effects [25]. The population structure consisted of a Q matrix that describes the percent subpopulation parentage for each line in the analysis. Cluster probabilities of K = 4 for SSRs and K = 3 for SNPs (results not shown) were used for the analyses.
Results
Analyses of linkage disequilibrium using SSRs and SNPs
The analysis of LD using SSRs is showed in Fig 1. Based on the Bonferroni threshold (p-value: 1.24 x 10−5; -LogP: 4.904) used to control type I error, it was observed that non-random associations extended up to 100 cM for the common bean genome and then declined rapidly from that distance. Using the FDR threshold (p-value: 2.48 x 10−2; -LogP: 1.606), which is a less conservative procedure to control multiple tests, LD extended up to more than 300 cM. Thus, despite the fact that few associations in LD were obtained and plotted above 100 cM with Bonferroni correction, a higher and more extensive LD was observed for the common bean genome using FDR, as can be expected for selfing species such as beans.
Fig 1. Linkage disequilibrium (r2) due to the genetic distance in bp, with SSRs markers.
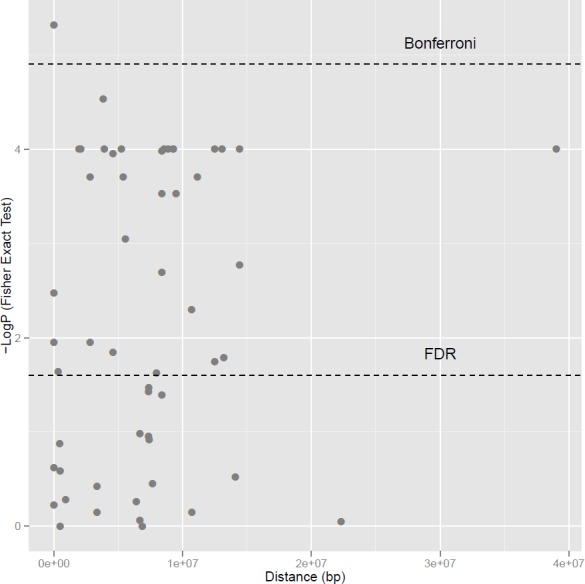
Thresholds corresponding to Bonferroni corrections and FDR are shown on a logarithmic scale with significant associations (in LD) and non-significant vectors above and below, respectively.
From the 369 available SNPs, 331 could be used to calculate pairwise LD along the common bean genome. Using SNP markers, it was possible to detect a rapid and unexpected decay in LD throughout the genome (Fig 2), despite having a high and expected LD (higher values of r2) in various chromosomal regions (S1 Fig). This pattern suggests extensive LD blocks, particularly on chromosomes 3, 8, and 11. In general, the common bean LD extended to several megabases, as can be observed in Fig 2.
Fig 2. Allele pair linkage disequilibrium (r2) across the entire common bean genome for the full group of genotypes, due to the genetic distance in bp, with SNP markers.
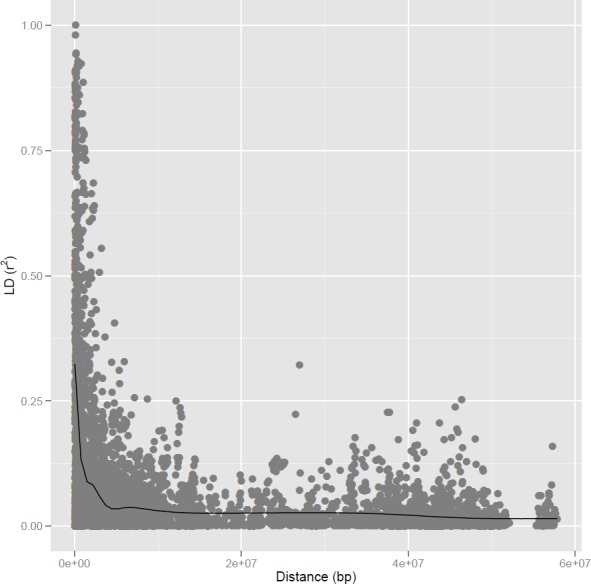
The extent of LD decay is usually affected by population structure. Assessing genome-wide LD patterns in beans is important not only for GWAS but also to shed light on historical effects of intensive directional selection due to domestication for vegetable use, compared with materials closer to the ancestral species. Examples have been reported between the indica and japonica groups of rice [50], wild and domesticated grape [51], landrace and modern varieties of wheat [52], and in asparagus bean (Vigna unguiculata ssp. sesquipedialis [53].
Phenotypic analyses of anthracnose and angular leaf spot
A mixed model approach was used to analyze phenotypic data from ANT and ALS experiments. To investigate different structures of variances and covariances for the genetic effects, we considered all the accessions from the core collection as a random effect into the model (Table 1). Grouping for block effects or replicates within the blocks, we found that our data fitted better to complex mixed models. For both ANT and ALS, the diagonal (DIAG) model explained satisfactorily the observed genetic variation, based on the lower values of AIC and BIC criteria. This model considers the presence of heterogeneous genetic variances and absence of genetic covariances and correlations between observations for each level of the accessions.
Table 1. Different structures of variances and covariances for the genetic-effects matrix related to the blocks (GBlock) and replicates within the genotypes and blocks (GRep).
Blocks and replicates are both used as grouping factor for each level of the random genotypes.
| Matrix | Model | Parameter Number | ALS | ANT | ||
|---|---|---|---|---|---|---|
| AIC | BIC | AIC | BIC | |||
| ID | 18 | 7,487.418 | 7,585.160 | 10,585.690 | 10,688.410 | |
| DIAG | 21 | 7,475.074 | 7,589.106 | 10,335.400 | 10,455.230 | |
| CS | 19 | 7,488.833 | 7,592.005 | 10,445.970 | 10,554.380 | |
| UNST | 27 | NC | NC | NC | NC | |
| ID | 18 | 7,474.923 | 7,572.665 | 10,331.630 | 10,434.340 | |
| DIAG | 34 | 7,318.306 | 7,502.930 | 10,313.830 | NC | |
| CS | 19 | 7,476.923 | 7,580.096 | 10,331.400 | 10,439.810 | |
| UNST | – | NC | NC | NC | NC | |
G: random genetic-effects matrix; ID: Identity; DIAG: Diagonal; CS: Compound Symmetry; UNST: Unstructured; NC: No Convergence. The selected model for both ALS and ANT diseases is indicated by the value in bold, which correspond to AIC and BIC criteria for the first and only AIC for the latter.
Association mapping using SSRs and SNPs
Of the 103 SSRs used in this study, 17 (16.5%) were associated with ANT (race 4) disease, of which three, PvM153 (Pv02), SSR-IAC254 (Pv08), and PvM98 (Pv11), were the most significant (P ≤ 0.001; Table 2). Statistically significant associations involving SSRs were observed for ANT in seven out of eleven chromosomes of the common bean (Table 2). For SNP markers, a total of 21 markers (6.3%) were associated with ANT disease (Table 2; S2 Fig). The SNP scaffold00024_916410 (Pv01), scaffold00060_874577 (Pv04), scaffold00021_89379 (Pv07), and sacaffold00034_860044 (Pv08) showed significant (P ≤ 0.001) associations with the trait. The SNP scaffold00021_89379 showed the most significant association to ANT, explaining 10.7% of the phenotypic variation (Table 2).
Table 2. Associations between SSR and SNP marker loci and Anthracnose (race 4) severity determined by unified mixed linear models (MLM).
| Marker | Chromosome (Pv) | p- valuea | R2_ markerb | R2_ marker (%) |
|---|---|---|---|---|
| PvM56 | 1 | 0.0101* | 0.0359 | 3.59 |
| PvM123 | 1 | 0.0235* | 0.0244 | 2.44 |
| PvM15 | 1 | 0.0042** | 0.0539 | 5.39 |
| BMc271 | 1 | 0.0367* | 0.0236 | 2.36 |
| scaffold00024_916410 | 1 | 0.00076488*** | 0.07697 | 7.697 |
| PvM93 | 2 | 0.0163* | 0.0435 | 4.35 |
| PvM153 | 2 | 0.000040712*** | 0.0877 | 8.77 |
| PvM126 | 3 | 0.002** | 0.0661 | 6.61 |
| SSR-IAC167 | 3 | 0.0084** | 0.047 | 4.7 |
| PvM95 | 3 | 0.0195* | 0.0283 | 2.83 |
| PvM124 | 3 | 0.0482* | 0.0209 | 2.09 |
| PVEST236 | 3 | 0.0366* | 0.0544 | 5.44 |
| scaffold00045_345513 | 3 | 0.00114** | 0.09041 | 9.041 |
| scaffold00060_874577 | 4 | 0.00052705*** | 0.08115 | 8.115 |
| scaffold00090_802505 | 4 | 0.00121** | 0.08733 | 8.733 |
| PvM07 | 5 | 0.031* | 0.0502 | 5.02 |
| scaffold00062_295319 | 5 | 0.04492* | 0.04494 | 4.494 |
| PvM14 | 6 | 0.0271* | 0.0446 | 4.46 |
| scaffold00001_1947432 | 6 | 0.04529* | 0.03401 | 3.401 |
| scaffold00128_112577 | 6 | 0.03906* | 0.03603 | 3.603 |
| scaffold00128_197955 | 6 | 0.0043** | 0.07229 | 7.229 |
| scaffold00001_2118513 | 6 | 0.04286* | 0.04503 | 4.503 |
| scaffold00021_89379 | 7 | 0.00058202*** | 0.10781 | 10.781 |
| scaffold00088_364454 | 7 | 0.01903* | 0.05802 | 5.802 |
| scaffold00021_767280 | 7 | 0.03442* | 0.03785 | 3.785 |
| scaffold00094_563857 | 7 | 0.01938* | 0.04311 | 4.311 |
| scaffold00098_217812 | 7 | 0.0138* | 0.03323 | 3.323 |
| SSR-IAC254 | 8 | 0.00097355*** | 0.0564 | 5.64 |
| PvM68 | 8 | 0.0247* | 0.0153 | 1.53 |
| scaffold00105_48480 | 8 | 0.04527* | 0.03448 | 3.448 |
| scaffold00034_860044 | 8 | 0.00043065*** | 0.09668 | 9.668 |
| scaffold00097_323110 | 8 | 0.03266* | 0.03752 | 3.752 |
| scaffold00097_164240 | 8 | 0.03657* | 0.03631 | 3.631 |
| PvM98 | 11 | 0.00078543*** | 0.066 | 6.6 |
| SSR-IAC127 | 11 | 0.019* | 0.0269 | 2.69 |
| scaffold00009_1366067 | 11 | 0.02796* | 0.04973 | 4.973 |
| scaffold00009_825782 | 11 | 0.01282* | 0.04752 | 4.752 |
| scaffold00096_204246 | 11 | 0.04505* | 0.04573 | 4.573 |
a *, p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001.
b R2_ marker = The fraction of the total variation explained by the marker after fitting the other model effects.
A total of 11 SSRs (10.6%; Table 3) were associated with ALS (race 0–39) disease, of which two, PvM97 (Pv01) and PvM62 (Pv05), were the most significant (P ≤ 0.01; Table 3). A total of 17 associations (5.1%) involving SNPs were associated with ALS disease (Table 3; S2 Fig), and the SNP scaffold00019_566327 (Pv11) showed the highest significance (P ≤ 0.01). The marker SSR-IAC66 (Pv04) was most significantly associated with ALS, explaining 16.66% of the phenotypic variation (Table 3).
Table 3. Associations between SSR and SNP marker loci and angular leaf spot (race 0–39) severity determined by unified mixed linear models (MLM).
| Marker | Chromosome (Pv) | p- valuea | R2_ markerb | R2_ marker (%) |
|---|---|---|---|---|
| PvM97 | 1 | 0.0041** | 0.0407 | 4.07 |
| SSR-IAC167 | 3 | 0.0296* | 0.041 | 4.10 |
| PvM95 | 3 | 0.0228* | 0.0276 | 2,76 |
| BMc215 | 3 | 0.0447* | 0.0218 | 2.18 |
| SSR-IAC66 | 4 | 0.0198* | 0.1666 | 16.66 |
| BMc300 | 4 | 0.0169* | 0.0327 | 3.27 |
| BMc225 | 4 | 0.0173* | 0.0661 | 6.61 |
| scaffold00060_115096 | 4 | 0.0383* | 0.03826 | 3.826 |
| scaffold00060_401853 | 4 | 0.04706* | 0.04779 | 4.779 |
| scaffold00076_331846 | 4 | 0.04686* | 0.02331 | 2.331 |
| PvM62 | 5 | 0.0086** | 0.0387 | 3.87 |
| scaffold00037_358238 | 6 | 0.02852* | 0.0536 | 5.36 |
| scaffold00001_2031371 | 6 | 0.04272* | 0.03811 | 3.811 |
| scaffold00021_89379 | 7 | 0.04195* | 0.05666 | 5.666 |
| scaffold00111_115892 | 7 | 0.01048* | 0.05333 | 5.333 |
| scaffold00126_28972 | 7 | 0.0329* | 0.04073 | 4.073 |
| scaffold00111_19536 | 7 | 0.03029* | 0.02764 | 2.764 |
| PvM01 | 8 | 0.0219* | 0.115 | 11.5 |
| scaffold00034_1236020 | 8 | 0.02152* | 0.04523 | 4.523 |
| scaffold00041_635678 | 8 | 0.04989* | 0.04717 | 4.717 |
| PvM61 | 9 | 0.0121* | 0.0356 | 3.56 |
| scaffold00043_193294 | 9 | 0.03672* | 0.05087 | 5.087 |
| scaffold00101_378095 | 9 | 0.04128* | 0.03813 | 3.813 |
| BMc273 | 10 | 0.0197* | 0.06 | 6.00 |
| scaffold00019_1159551 | 11 | 0.04345* | 0.03716 | 3.716 |
| scaffold00019_566327 | 11 | 0.00194** | 0.08497 | 8.497 |
| scaffold00019_960179 | 11 | 0.01919* | 0.05845 | 5.845 |
| scaffold00009_208616 | 11 | 0.02388* | 0.05398 | 5.398 |
a *, p ≤ 0.05
** p ≤ 0.01
***p ≤ 0.001.
b R2_ marker = The fraction of the total variation explained by the marker after fitting the other model effects.
The markers SSR-IAC167 and PvM95, both located on chromosome Pv03, and the SNP scaffold00021_89379, were associated with both diseases, suggesting a possible pleiotropic effect.
SSRs, SNPs, and genes associated with anthracnose and angular leaf spot resistance
The SSR and SNP markers found to be associated with both ANT (Table 2) and ALS (Table 3) resistance were physically located on the recently released common bean genome sequence [1]. All 64 markers were mapped to within a certain region on the common bean genome (E-value ≤4 x 10−50). In general, the markers were distributed across the entire common bean genome, with chromosomes Pv03 and Pv08 showing the greatest number of loci associated with ANT resistance. The chromosome Pv04 was the most saturated one, with six markers associated to ALS resistance (Tables 2 and 3). The telomeric region of this chromosome showed four markers located between approximately 2.5 Mb and 4.4 Mb (Fig 3).
Fig 3. Representative map of the genome positions of markers associated with ANT and ALS resistance.

SSR and SNP markers were located on the reference genome (Schmutz et al., 2014) using the Phytozome v1.0 BLAST tool (http://www.phytozome.net/). Markers in blue are associated with ANT resistance and those in red are associated with ALS resistance. The SSR-IAC167, PvM95, and sacaffold00021_89379 markers (green) were associated with resistance to both diseases. The map was drawn with MapChart (Voorrips 2002), and marker positions are represented in Mb.
Taking into account both ALS and ANT, chromosomes Pv03 and Pv11 portrayed the larger number of loci (nine) involved in the immune response to both diseases (Fig 3; S1 and S2 Tables). Telomeres also appeared to be enriched for disease-associated loci in chromosomes Pv06 (22.5 Mb to 26.4 Mb; seven markers), Pv07 (0.5 Mb to 3.4 Mb; four markers), and Pv11 (2.7 Mb to 5.5 Mb; six markers) (Fig 3).
The predicted candidate gene locations in the genome for each marker were identified using the reference common bean genome sequence [1]. A total of five markers were mapped on common bean genome regions with no expressed genes, based on the Phytozome RNA-seq and EST database [1]. In total, 57 genes were identified. Different functions could be assigned to the genes where the markers were located, including five involved with transcription factors, 12 of unknown functions, four kinases, and one with malectin-tyrosine kinase (S1 and S2 Tables).
Gene Ontology (GO) enrichment analysis was performed for all candidate genes to investigate whether the loci associated with ANT and ALS resistance corresponded to genes involved in known pathways. GO accessions, allowed them to be grouped into three functional categories: those related to Biological Processes, Molecular Functions and Cellular Components. Most of SSRs markers associated to Anthracnose were enriched into five biological processes: "Cellular metabolic process", "Biosynthetic process", "Primary metabolic process", "Organic substance metabolic process", "Regulation of biological process", followed by “Nitrogen compound metabolic process” and “Death”, whereas those associated to Angular Leaf Spot were distributed in equal proportions in similar process, except by “Establishment of localization”. In the molecular function and cellular component categories ANT-SSRs showed more categories than ALS-SSRs. Those related to ANT were mostly enriched in following molecular functions: “Sequence–specific DNA binding transcription factor activity”, “Ion binding”, “Heterocyclic compound binding” and “Organic cyclic compound binding”. “Protein binding”, “Oxidoreductase activity”, “Ion binding”, were the main functions assigned to SSR associated to ALS (S3 Fig). “Cell part” and “Membrane-bounded organelle” were the common cellular component for SSRs associated to both diseases.
Regarding to SNPs markers, similarly to SSRs, they were assigned to “Primary metabolic process”, “Organic substance metabolic process”, Cellular metabolic process”, “Establishment of localization”, “Single-organism cellular process” as main biological process for both diseases. In “Molecular Function category”, SNPs were assigned to eleven classes for ANT, with the most frequent corresponding to "Heterocyclic compound binding", "Ion binding", "Organic cyclic compound binding" and "Transferase activity". For ALS-SNPs, they were assigned to six classes, highlighting "Hydrolase activity”. It is interesting to note that only ANT-SNPs were assigned to “Cellular Component”, such as “Membrane part” and “Cell part” and no “Cell component” was found related to ALS-SNPs. In addition, markers linked to genes involved in biological process such as “methylation”, “biosynthetic process” and “programed cell death” are interesting to consider because they it may be involved in roles of stress response (S3 Fig).
Annotation using KEGG allowed the classification of the genes containing SSR and SNPs markers for ANT and ALS in agreement with their function in the context of specific metabolic pathways. Homologous for several enzymes revealed genes involved in the different pathways such as “Phenylpropanoid biosynthesis”, “Porphyrin and Chlorophyll metabolism”, “Carbapenem biosynthesis”, “Aminobenzoate degradation” and “Glycine, Serine and threonine metabolism” (S3 Table). The gene structure prediction allows identify where each marker is located into the gene, what is important to recognize it presence in putative coding regions. For example, specifically the PvM93 marker, which is a SSR associated to ANT, tags a exon of the gene that encode to a Glucosyltransferase involved in “Phenylpropanoid biosynthesis” and the SNP (Scaffold0024_916410) also associated to ANT, tags the gene that encode to a 5-Kinase dehydrogenase involved in “Carbapenem biosynthesis” pathways (Fig 4 and S4 Fig).
Fig 4. Example of two homologous gene structure predictions showing the marker location in the locus PvM93.

Discussion
The resolution of GWAS depends on the extent and structure of LD detected in the germplasm under consideration. As a predominantly selfing species, extended LD and low rates of effective recombination are expected for common bean [54].
Common bean allows the use of medium-sized populations as diversity panels for AM or GWAS [25,28]. Indeed, the size of the population depends on the relatedness of the individuals, the extent of LD, the type of study, and the polymorphism of the markers. The Brazilian core collection used in this study derived from the Agronomic Institute (IAC, Campinas, SP. Brazil) included domesticated genotypes from Andean and Mesoamerican genepools. A whole-genome-scan association study is feasible for bean domestic populations [21].
Population mating systems could have a strong influence on LD patterns in the common bean. Generally, there is a lower decline of LD in selfing species compared to out-breeding species [55], which could be partially explained by the reduced recombination events or selection effects in selfing species [16]. In this sense, LD tends to remain over tens to hundreds of kilobases in predominantly selfing species, such as rice, soybean, or common bean, showing that population mating could explain the high proportions of LD we detected. Therefore, the large extent of LD observed in this study with SSRs is in accordance with what has been previously reported for the common bean [20,21,22].
The rapid LD decay obtained with SNP markers was not expected. This could be attributed to the fact that these SNPs were developed from sequences that are related to water stress [36] and located within specific regions that are not randomly distributed across the bean genome.
Although LD is generally high in common bean breeding lines, there are some regions where the LD is much reduced, highlighting the importance of estimating LD in the core collection of distributed loci throughout the genome [26].
GWAS is an important tool for gene tagging in crops both for simple traits under additive genetic scenarios, as well as for the dissection of more complex genetic architectures. The advantage of GWAS for QRL discovery over traditional QRL-mapping in biparental crosses is primarily due to (1) the availability of broader genetic variations with wider background for marker-trait correlations (i.e., many alleles evaluated simultaneously), (2) the likelihood of higher resolution mapping because of the utilization of the majority of recombination events from a large number of meioses throughout the germplasm development history, (3) the possibility of exploiting historically measured trait data for associations, and (4) no need for the development of expensive and biparental populations that makes this approach time-saving and cost-effective [26].
According to Ferreira et al., in 2013 20 resistance genes (named Co) that condition specific isolates or races of ANT pathogenic on common bean were reported to date [56]. The authors related that the resistance of ANT has been mapped to seven of 11 common bean chromosomes (Pv01; Pv02; Pv03; Pv04; Pv07; Pv08 and Pv11). Recently, Campa et al. in 2014 found new locations for the ANT resistance gene in Pv09 [57].
Most identified ANT resistance genes have been located in the genetic map of common bean: genes Co-1, Co-x, and Co-w were mapped to chromosome Pv01 [58,59,60]; Co-u was located to Pv02 [58]; Co-13 to Pv03 [61]; Co-2 to Pv11 [62]; Co-3, Co-9, Co-y, Co-z, and Co-10 to Pv04 [59,63,64,65,66,67]; Co-4 to Pv08 [59]; and Co-5, Co-6, and Co-v to Pv07 [68,69]. Partial ANT resistance to races 23 and 1545 on Pv5 was previously reported by Gonzalez et al. in 2015 [70]. However, until now, no resistance locus to ANT has been found on chromosome Pv06. In fact, the ANT resistance system in common bean has been classically investigated by analyzing a limited number of isolates or races in different segregating populations [57]. Using GWAS, a broader range of genetic variation may be explored, not restricted to a bi parental source. Exploring the genetic architecture of the ANT response in common beans, Oblessuc et al. in 2014 reported a more quantitative response to ANT resistance (race 4), reinforcing the importance of unveiling minor effect alleles that may contribute to durable resistance [32]. Two markers were associated with resistance to ANT on chromosome Pv05 (PvM07 and scaffld00062_295319) and five markers on chromosome Pv06 (PvM14, scaffold00128_112577, scaffold00128_197955, scaffold00001_2118513 and scaffold0001_1947432), revealing new QRLs for ANT resistance.
The PvM07 marker was linked to putative genes related to disease resistance (Zinc finger proteins) (S1 Table). Zinc fingers proteins are members of a super family involved in resistance and regulatory mechanisms for various biotic stresses [71,72]. The presence of zinc finger DNA binding domains in nucleotide binding site-leucine rich repeats (NBS-LRR) determines the regulatory function of this protein in stress conditions [73]. Most plant disease resistance R proteins contain a series of NBS-LRRs. The LRRs of a wide variety of proteins from many organisms serve as protein interaction platforms, and as regulatory modules of protein activation. Genetically, the LRRs of plant R proteins are determinants of response specificity, and their action can lead to plant cell death in the form of the familiar hypersensitive response (HR) [74].
In chromosome Pv02, the PvM13 marker (Fig 3) was co-localized with QRL ANT02.4UC [32]. Three SNPs (scaffold00097_323110, scaffold00097_164240, and scaffold00034_860044; Fig 3) were co-localized with QRL ANT08.2UC [32]. The QRL ANT11.1UC observed in response to ANT race 4 was also positioned on the distal part of chromosome Pv11 [32]. The PvM98 marker was co-localized with this QRL.
Two markers in Pv01 (scaffold00024_916410 and PvM97; Fig 3) were localized in a position corresponding to the Co-1 resistance cluster. This resistance cluster was reported recently by Campa et al. in 2014, who found the genes Co-173-x and Co-165-x correspond to the resistance cluster against the ANT races 73 and 65, respectively [57].
Both scaffold00060_115096 and scaffold00060_401853 markers (Fig 3) were associated with ALS on chromosome Pv04. These two markers were localized in a position corresponding to Co-10, Phg-ON, and Ur-14 resistance clusters. These three genes appear very important for common bean breeding programs, as indicated by previous studies [63,75].
The ALS resistance was associated with nine of 11 common bean chromosomes (Pv01, Pv02, Pv03, Pv04, Pv05, Pv07, Pv08, Pv09, and Pv10) [76]. Until now, no ALS resistance QRL has been found on chromosome Pv06 (Fig 3). Two markers were associated with resistance to ALS on Pv06: scaffold00037_358238 and scaffold0001_2031371.
Oblessuc et al. in 2012 identified seven QRLs associated with ALS resistance to race 0–39 [30]. Comparing the former results with the associations detected herein, it is possible to verify similar regions for ALS resistance. On Pv03, the PvM124, scaffold00045_345513, and PvM95 markers (Fig 3) were co-localized near the QRL ALS3.1UC [30]. The PvM95 marker was associated with both diseases, while two other markers mapped on this genomic region were assigned specifically to ANT resistance.
On Pv04, BMc225 (Fig 3) was co-localized with QRL ALS4.2GS, UC [30], associated with ALS resistance to race 0–39. Keller et al. in 2015 reported a major QRL on Pv04, around 43.7 Mb, explaining 75.3% of the phenotypic variance to an Andean race (31–0) [76]. BMc225 was located at 42.28 Mb on Pv05, the same genomic region of the Pv-atgc002 marker, linked with ALS4.1GS,UC. The QTL5.2UC was co-localized to PvM07, PvM62, and scaffold00062_295319, as they were in the same genomic region of Pv05 that corresponded to this QRL region.
The discovery of gene clusters not only conditioning ANT resistance but linked to other gene clusters conditioning resistance to other pathogens such as ALS, bean rust, bean common mosaic virus, and halo blight has been reported [56]. In our study, new gene clusters were revealed for ANT and ALS resistance, preferentially localized on chromosome telomeres. The end of linkage group B4 was reported to have a high density of resistance genes and RGAs. A major-effect QRL against strain 45 (for leaf, stem, and petiole ANT resistance) and a reverse-effect QRL (i.e., coming from the susceptible Andean JaloEEP558 parent) for leaf ANT resistance against strain A7 were also located in this region [77].
Moreover, functional annotation analyses of markers from homologous sequence provided clues to the molecular bases of putatives responses to stress. In the present study, many GO terms and pathways relevant were identified, like phenylpropanoids, which are a group of plant secondary metabolites derived from phenylalanine, probably having a wide variety of functions as structural and signaling molecules [78]. Phenylalanine is first converted to cinnamic acid by deamination. It is followed by hydroxylation and frequent methylation to generate coumaric acid and other acids with a phenylpropane (C6-C3) unit. Reduction of the CoA-activated carboxyl groups of these acids results in the corresponding aldehydes and alcohols. The alcohols are called monolignols, the starting compounds for biosynthesis of lignin. Obviously, this is important to understanding biochemical bases of regulatory mechanisms induced by hormones in the processes of organogenesis or defense.
Although the genetic basis of durable resistance in plants is not fully understood [79], it is frequently presumed that quantitative resistance, conditioned by “minor” genes and supposed to act in a race-nonspecific manner, would provide durable resistance [80]. As pointed out by Oblessuc et al. positive QRL alleles for ANT and ALS resistance were identified using a recombinant inbred line (RIL) bi-parental population (IAC-UNA x CAL-143) [30,31,32,35]. New associations have been detected in this report using a GWAS approach. GWAS may provide a higher mapping resolution and the possibility to study various regions of the genome simultaneously, since it is based on germplasm collections with minimal genetic structure, preferentially a set of useful accessions for single or multiple traits of interest for breeding programs [54]. The putative genes identified in these regions may be potentially involved in the resistance response. Minor and major effect QRL revealed by this study may play a role in achieving a more comprehensive knowledge of the host-pathogen interactions of Colletotrichum lindemuthianum and Pseudocercospora griseola. Through cloning and characterizing candidate genes underlying resistance mechanisms in major QRL, the pathways involved in the race-specific and defense response to anthracnose and angular leaf spot infection of common beans can be further elucidated and understood.
Supporting Information
(TIF)
(A) Anthracnose–ANT, (B) Angular leaf spot–ALS. P values are shown on a log10 scale. Markers are considered significant when P ≤ 0.05. Axis x corresponds to the number of chromosomes in common bean.
(TIF)
In parenthesis, there are the numbers of genes of each category.
(TIF)
The blue boxes are for KEGG ECs that have homologies to Phaseolus vulgaris sequence target by PvM93.
(TIF)
(DOCX)
(DOCX)
(DOCX)
Acknowledgments
This research was supported by grants from São Paulo Research Foundation (FAPESP) (process numbers 2009/05284-1 and 2009/02502-8) and CNPq (process number 477239/2010-2).
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This research was supported by grants from São Paulo Research Foundation (FAPESP) (process numbers 2009/05284-1 LLB-R and 2009/02502-8 - JMKCP) and CNPq (process number 477239/2010-2 - LLB-R).
References
- 1.Schmutz J, McClean PE, Mamide S, Wu GA, Cannon SB, Grimwood J, et al. (2014) A reference genome for common bean and genome-wide analysis of dual domestications. Nat Genet 46: 707–713. 10.1038/ng.3008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Broughton WJ, Hernández G, Blair MW, Beebe S (2003) Beans (Phaseolus spp.)–model food legumes. Plant Soil 252: 55–128. 10.1023/A:1024146710611 [DOI] [Google Scholar]
- 3.Gepts P, Aragão FJL, de Barros E, Blair MW, Brondani R, Broughton W (2008) Phaseolus beans, a major source of dietary protein and micronutrients. In: Moore PH, Ming R (eds) Genomics of tropical crop molecular markers linked to angular leaf spot resistance genes in common beans accessions G10909. Mol Breed 28: 57–71. [Google Scholar]
- 4.Azarpazhooh E, Boye JI (2012) "Dry bean composition and effect of processing on protein, carbohydrates, fiber and minor constituents.",—Common Beans and Pulses: Production, Processing, and Nutrition, John Wiley & Sons, Ltd., 2012. [Google Scholar]
- 5.Pastor-Corrales MA, Tu JC (1989) Anthracnose. p. 77–104. In Pastor-Corrales MA, Schwartz HH (ed.) Bean production problems in the tropics. 2nd ed. CIAT, Cali, Colombia.
- 6.Pastor-Corrales MA, Jará CE (1995) La evoluciòn de Phaseolus vulgaris con el frijol comum en America Latina. Fitopatologia Colombiana 19: 15–24. [Google Scholar]
- 7.Melotto M, Balardin RS, Kelly JD (2000) Host-pathogen interaction and variability of Colletotrichum lindemuthianum In: Prusky D, Freeman S, Dickman MB (eds) Colletotrichum host specificity, pathology, and host-pathogen interaction. APS Press, St Paul, pp 346–361. [Google Scholar]
- 8.Stenglein S, Ploper LD, Vizgarra O, Balatti P (2003) Angular leaf spot: a disease caused by the fungus Phaeoisariopsis griseola (Sacc.) Ferraris o n Phaseolus vulgaris L. Adv Appl Microbiol 52: 209–243. 10.1016/S0065-2164(03)01009-8 [DOI] [PubMed] [Google Scholar]
- 9.Miklas PN, Kelly J, Beebe SE, Blair MW (2006) Common bean breeding for resistance against biotic and abiotic stresses: from classical to MAS breeding. Euphytica 147: 105–131. 10.1007/s10681-006-4600-5 [DOI] [Google Scholar]
- 10.Crous PW, Liebenberg MM, Braun U, Groenewald JZ (2006) Re-evaluating the taxonomic status of Phaeoisariopsis griseola, the causal agent of angular leaf spot of bean. Stud Mycol 55: 163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allorent D, Savary S (2005) Epidemiological characteristics of angular leaf spot of bean: a systems analysis. Eur J Plant Pathol 113: 329–341. [Google Scholar]
- 12.Zhu Q, Zheng X, Luo J, Gaut BS, Song E (2007) Multilocus analysis of nucleotide variation of Oryza sativa and its wild relatives: severe bottleneck during domestication of rice. Mol Biol Evol 24: 875–888. [DOI] [PubMed] [Google Scholar]
- 13.Huang X, Han B (2014) Natural Variations and Genome-Wide Association Studies in Crop Plants. Annual Rev of Plant Biol 65: 531–551. [DOI] [PubMed] [Google Scholar]
- 14.Myles S, Peiffer J, Brown PJ, Ersoz ES, Zhang Z, Costich DE, et al. (2009) Association mapping: Critical considerations shift from genotyping to experimental design. The Plant Cell 21: 2194–2202. 10.1105/tpc.109.068437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nordborg M, Tavaré S (2002) Linkage disequilibrium: what history has to tell us. Trends Genet 18: 83–90. [DOI] [PubMed] [Google Scholar]
- 16.Flint-Garcia SA, Thornsberry JM, Buckler ES (2003) Structure of linkage disequilibrium in plants. Annu Rev Plant Bio. 54: 357–374. [DOI] [PubMed] [Google Scholar]
- 17.Garris AJ, Tai TH, Coburn J, Kresovich S, McCouch S (2005) Genetic structure and diversity in Oryza sativa L. Genetics 169: 1631–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Breseghello F, Sorrells ME (2006a) Association analysis as a strategy for improvement of quantitative traits in plants. Crop Sci. 46: 1323–1330. [Google Scholar]
- 19.Breseghello F, Sorrells ME (2006b) Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 172: 1165–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwak M, Gepts P (2009) Structure of genetic diversity in the two major gene pools of common bean (Phaseolus vulgaris L, Fabaceae). Theor App Gen 118: 979–992. [DOI] [PubMed] [Google Scholar]
- 21.Rossi M, Bitocchi E, Bellucci E, Nanni L, Rau D, Attene G, et al. (2009) Linkage disequilibrium and population structure in wild and domesticated populations of Phaseolus vulgaris L. Evol Appl 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blair MW, Prieto S, Díaz LM, Buendía HF, Cardona C (2010) Linkage disequilibrium at the APA insecticidal seed protein locus of common bean (Phaseolus vulgaris L.). BMC Plant Biol 10: 79 10.1186/1471-2229-10-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chiorato AF, Carbonell SAM, Dias LAS, Moura RR, Chiavegato MB, Carlos Augusto Colombo CA (2006) Identification of common bean (Phaseolus vulgaris) duplicates using agromorphological and molecular data. Genetics and Molecular Biology 29:105–111. [Google Scholar]
- 24.Oliveira MF, Nelson RL, Geraldi IO, Cruz CD, De Toledo JFF (2010) Establishing a soybean germplasm core collection. Field Crops Research 119: 277–289. [Google Scholar]
- 25.Perseguini JMKC, Silva GMB, Rosa JRBF, Gazaffi R, Marçal JF, Carbonell SAM, et al. (2015) Developing a common bean core collection suitable for association mapping studies. Genetics Molec Biol 38: 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abdurakhmonov IY, Abdukarimov A (2008) Application of association mapping to understanding the genetic diversity of plant germplasm resources. Int. J. Plant Genomics 574927 10.1155/2008/574927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi C, Navabi A, Yu K (2011) Association mapping of common bacterial blight resistance QTL in Ontario bean breeding populations. BMC Plant Biol 11:52 10.1186/1471-2229-11-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galeano CH, Cortés JA, Fernández AC, Soler A, Franco-Herrera N, Makunde G, et al. (2012) Gene-based single nucleotide polymorphism markers for genetic and association mapping in common bean. BMC Genetics 13: 48 10.1186/1471-2156-13-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nemli S, Asciogul TK, Kaya HB, Kahtaman A, Esiyok D, Tanyolac B (2014) Association mapping of five agronomic traits in the common bean (Phaseolus vulgaris L.). Journal of the Sci of Food and Agric 94(15) 10.1002/jsfa.6664 [DOI] [PubMed] [Google Scholar]
- 30.Oblessuc PR, Baroni RM, Garcia AAF, Chioratto AF, Carbonell SAM, Camargo LEA, et al. (2012) Mapping of angular leaf spot resistance QTL in common bean (Phaseolus vulgaris L.) under different environments. BMC Genetics 13: 50 10.1186/1471-2156-13-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oblessuc PR, Perseguini JMKC, Baroni RM, Chiorato AF, Carbonell SAM, Mondego JMC, et al. (2013) Increasing the density of markers around a major QTL controlling resistance to angular leaf spot in common bean. Theor Appl Genet. 10.1007/s00122-013-2146-1 [DOI] [PubMed] [Google Scholar]
- 32.Oblessuc PR, Baroni RM, Pereira GS, Chioratto AF, Carbonell SAM, Briñez B, et al. (2014) Quantitative analysis of race-specific resistance to Colletotrichum lindemuthianum in common bean. Molecular Breeding 34: 1313–1329. [Google Scholar]
- 33.Oblessuc PR, Matiolli CC, Chiorato AF, Benchimol-Reis LL, Melotto M (2015) Common bean reaction to angular leaf spot comprises transcriptional modulation of genes in the ALS10.1 QTL. Front Plant Sci 6: 152 10.3389/fpls.2015.00152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hanai LL, Campos T, Camargo LEA, Benchimol LL, Souza AP, Melotto M, et al. (2007) Development, characterization, and comparative analysis of polymorphism at common bean-SSR loci isolated from genic and genomic sources. Genome 50: 266–277. [DOI] [PubMed] [Google Scholar]
- 35.Campos T, Oblessuc PR, Sforça DA, Cardoso JMK, Baroni RM, Sousa ACB, et al. (2011) Inheritance of growth habit detected by genetic linkage analysis using microsatellites in the common bean (Phaseolus vulgaris L.). Mol Breed 27: 549–560. [Google Scholar]
- 36.Vianello RP. QTL for drought tolerance in a Mesoamerican common bean (Phaseolus vulgaris L.) population Bat477 x Pérola. In press.
- 37.Fisher RA (1935) The logic of inductive inference. Journal of the Royal Statistical Society 98: 39–54. [Google Scholar]
- 38.Weir BS (1996) Genetic Data Analysis II. Sinauer Associates, Sunderland, MA.
- 39.Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society 57: 289–300. [Google Scholar]
- 40.Hanai LL, Santini L, Camargo LEA, Fungaro MHP, Gepts P, Tsai SM, et al. (2010) Extension of the core map of common bean with EST-SSR, RGA, AFLP, and putative functional markers. Mol Breeding 25: 25–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hill WG, Robertson A (1966) The effect of linkage on limits to artificial selection. Genet Res 8: 269–294. [PubMed] [Google Scholar]
- 42.Wimmer V, Albrecht T, Auinger H, Schön C (2011) Synbreed: a framework for the analysis of genomic prediction data using R. Bioinformatics 28: 2086–2087. [DOI] [PubMed] [Google Scholar]
- 43.Monda EO, Sanders FE, Hick A (2001) Infection and colonization of bean leaf by Phaeoisariopsis griseola. Plant Pathol 50: 103–110. [Google Scholar]
- 44.Akaike H (1974) A new look at the statistical model identification. IEEE Trans Aut Control 19: 716–723. [Google Scholar]
- 45.Schwarz G (1978) Estimating the dimension of a model. Annals of Statistics 6: 461–464. [Google Scholar]
- 46.Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21: 3674–3676. [DOI] [PubMed] [Google Scholar]
- 47.Altermann E, Klaenhammer TR (2005) PathwayVoyager: pathway mapping using Kyoto Encyclopedia of Genes and Genomes (KEGG) database. BMC Genomics 6:60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 618–620. [Google Scholar]
- 49.Pritchard JK, Stephens P, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mather KA, Caicedo AL, Polato NR, Olsen KM, McCouch S, Purugganan MD (2007) The extent of linkage disequilibrium in rice (Oryza sativa L.). Genetics 177: 2223–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barnaud A, Laucou V, This P, Lacombe T, Doligez A (2010) Linkage disequilibrium in wild French grapevine, Vitis vinifera L. subsp. Silvestris. Heredity 104: 431–437. 10.1038/hdy.2009.143 [DOI] [PubMed] [Google Scholar]
- 52.Hao C, Wang L, Ge H, Dong Y, Zhang X (2011) Genetic diversity and linkage disequilibrium in Chinese bread wheat (Triticum aestivum L.) revealed by SSR markers. PLoS ONE 6(2): e17279 10.1371/journal.pone.0017279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu P, Wang B, Luo J, Liu Y, Ehlers JD, Close TJ, et al. (2012) Genome wide linkage disequilibrium in Chinese asparagus bean (Vigna unguiculata ssp. sesquipedialis) germplasm: implications for domestication history and genome wide association studies. Heredity 109: 34–40. 10.1038/hdy.2012.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Korte A, Ashley F (2013) The advantages and limitations of trait analysis with GWAS: a review. Plant Methods 9:29 10.1186/1746-4811-9-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nordborg M (2000) Linkage disequilibrium, gene trees and selfing: an ancestral recombination graph with partial self-fertilization. Genetics 154: 923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ferreira JJ, Campa A, Kelly JD (2013) “Organization of genes conferring resistance to anthracnose in common bean,” in Translational Genomic for Crop Breeding: Biotic Stress, eds Varshney R. K. and Tuberosa R. (Chichester: John Wiley & Sons; ), 151–182. 10.1002/9781118728475.ch9 [DOI] [Google Scholar]
- 57.Campa A, Rodríguez-Suárez C, Giraldez R, Ferreira JJ (2014) Genetic analysis of the response to eleven Colletotrichum lindemuthianum races in a RIL population of common bean (Phaseolus vulgaris L.). BMC Plant Biol 14: 115 10.1186/1471-2229-14-115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Geffroy V, Sevignac M, Billant P, Dron M, Langin T (2008) Resistance to Colletotrichum lindemuthianum in Phaseolus vulgaris: a case study for mapping two independent genes. Theor Appl Genet 116: 407–415. [DOI] [PubMed] [Google Scholar]
- 59.Rodríguez-Suárez C, Méndez-Vigo B, Pañeda A, Ferreira JJ, Giraldez R (2007) A genetic linkage map of Phaseolus vulgaris L. and localization of genes for specific resistance to six races of anthracnose (Colletotrichum lindemuthianum). Theor Appl Genet 114: 713–722. [DOI] [PubMed] [Google Scholar]
- 60.Richard MMS, Pflieger S, Sévignac M, Thareau V, Blanchet S, Li Y, et al. (2014) Fine mapping of Co-x, an anthracnose resistance gene to a highly virulent strain of Colletotrichum lindemuthianum in common bean. Theor Appl Genet 127: 1653–1666. 10.1007/s00122-014-2328-5 [DOI] [PubMed] [Google Scholar]
- 61.Lacanallo GF, Gonçalves-Vidigal MC, Vidigal Filho PS, Kami J, Gonela A (2010) Mapping of an Andean gene for resistance to anthracnose in the landrace Jalo Listras Pretas. Annu Rept Bean Improv Coop 53: 95–96. [Google Scholar]
- 62.Adam-Blondon AF, Sévignac M, Dron M, Bannerot H (1994) A genetic map of common bean to localize specific resistance genes against anthracnose. Genome 37: 915–924. [DOI] [PubMed] [Google Scholar]
- 63.Alzate-Marin AL, Costa MR, Arruda KM, Gonçalves de Barros E, Alves Moreira M (2003) Characterization of the anthracnose resistance gene present in Ouro Negro (Honduras 35) common bean cultivar. Euphytica 133: 165–169. [Google Scholar]
- 64.Geffroy V, Sicard D, de Oliveira JCF, Sévignac M, Cohen S, Gepts P, et al. (1999) Identification of an ancestral resistance gene cluster involved in the coevolution process between Phaseolus vulgaris and its fungal pathogen Colletotrichum lindemuthianum. Mol Plant-Microbe Interact 12: 774–784. [DOI] [PubMed] [Google Scholar]
- 65.Gonçalves-Vidigal MC, Cruz AS, Lacanallo GF, Vidigal Filho PS, Sousa LL, Pacheco CMNA, et al. (2013) Co-segregation analysis and mapping of the anthracnose Co-10 and angular leaf spot Phg-ON disease-resistance genes in the common bean cultivar Ouro Negro. Theor Appl Genet 126: 2245–255.http://www.biomedcentral.com/sfx_links?ui=1471-2229-14-115&bibl=B24 10.1007/s00122-013-2131-8 [DOI] [PubMed] [Google Scholar]
- 66.Méndez-Vigo B, Rodríguez-Suárez C, Pañeda A, Ferreira JJ, Giraldez R (2005) Molecular markers and allelic relationships of anthracnose resistance gene cluster B4 in common bean. Euphytica 141: 237–245. [Google Scholar]
- 67.Rodríguez-Suárez C, Ferreira JJ, Campa A, Pañeda A, Giraldez R (2008) Molecular mapping and intra-cluster recombination between anthracnose race-specific resistance genes in the common bean differential cultivars Mexico 222 and Widusa. Theor Appl Genet 116: 807–814. 10.1007/s00122-008-0714-6 [DOI] [PubMed] [Google Scholar]
- 68.Campa A, Giraldez R, Ferreira JJ (2009) Genetic dissection of the resistance to nine anthracnose races in the common bean differential cultivars MDRK and TU. Theor Appl Genet 119: 1–11. 10.1007/s00122-009-1011-8 [DOI] [PubMed] [Google Scholar]
- 69.Geffroy V, Creusot F, Falquet J, Sévignac M, Adam-Blondon A- F, Gepts P, et al. (1998) A family of LRR sequences at the Co-2 locus for anthracnose resistance in Phaseolus vulgaris and its potential use in markerassisted selection. Theor Appl Genet 96: 494–502. 10.1007/s001220050766 [DOI] [PubMed] [Google Scholar]
- 70.González AM, Yuste-Lisbona FJ, Rodiño AP, De Ron AM, Capel C, García-Alcázar M, et al. (2015) Uncovering the genetic architecture of Colletotrichum lindemuthianum resistance through QTL mapping and epistatic interaction analysis in common bean. Front. Plant Sci. 6:141 10.3389/fpls.2015.00141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Feurtado JA, Huang D, Wicki-Stordeur L, Hemstock LE, Potentier MS, et al. (2011) The Arabidopsis C2H2 zinc finger Indeterminate Domain1/Enhydrous promotes the transition to germination by regulating light and hormonal signaling during seed maturation. The Plant Cell 23: 1772–1794. 10.1105/tpc.111.085134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Giri J, Vij S, Dansana PK, Tyagi AK (2011) Rice A20/AN1 zinc-finger containing stress-associated proteins (SAP1/11) and a receptor-like cytoplasmic kinase (OsRLCK253) interact via A20 zinc-finger and confer abiotic stress tolerance in transgenic Arabidopsis plants. New Phytologist 191: 721–732. 10.1111/j.1469-8137.2011.03740.x [DOI] [PubMed] [Google Scholar]
- 73.Zhu H, Cannon AB, Young ND, Cook DR (2002) Phylogeny and Genomic Organization of the TIR and Non-TIR NBS-LRR Resistance Gene family in Medicago truncatula. The American Phyttop Soc 15: 529–539. [DOI] [PubMed] [Google Scholar]
- 74.Belkhadir Y, Subramaniam R, Dangl JL (2004) Plant disease resistance protein signaling: NBS-LRR proteins and their partners. Current Opinion in Plant Biology 4: 391–399. [DOI] [PubMed] [Google Scholar]
- 75.Gonçalves-Vidigal MC, Cruz AS, Lacanallo GF, Vidigal-Filho PS, Sousa LL, Pacheco CMNA, et al. (2013) Co-segregation analysis and mapping of the anthracnose Co-10 and angular leaf spot Phg-ON disease-resistance genes in the common bean cultivar Ouro Negro. Theor Appl Genet 126: 2245–2255. 10.1007/s00122-013-2131-8 [DOI] [PubMed] [Google Scholar]
- 76.Keller B, Manzanares C, Jara C, Lobaton JD, Studer B, Raatz B (2015) Fine-mapping of a major QTL controlling angular leaf spot resistance in common bean (Phaseolus vulgaris L.). Theor and Appl Genetics 128: 813–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Geffroy V, Sevignac M, de Oliveira JCF, Fouilloux G, Skroch P, Thoquet P, et al. (2000) Inheritance of partial resistance against Colletotrichum lindemuthianum in Phaseolus vulgaris and co-localization of quantitative trait loci with genes involved in specific resistance. Mol Plant Microbe Interact 13: 287–296. [DOI] [PubMed] [Google Scholar]
- 78.Nair RB, Bastress KL, Ruegger MO, Denault JW, Chapple C (2004) The Arabidopsis thaliana REDUCED EPIDERMAL FLUORESCENCE1 gene encodes an aldehyde dehydrogenase involved in ferulic acid and sinapic acid biosynthesis.Plant Cell 16: 544–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Johnson R (1981) Durable resistance: Definition of genetic control and attainment in plant breeding. Phytopathology 71: 567–568. [Google Scholar]
- 80.Vanderplank JE (1968) Disease Resistance in Plants. Academic Press, New York. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
(A) Anthracnose–ANT, (B) Angular leaf spot–ALS. P values are shown on a log10 scale. Markers are considered significant when P ≤ 0.05. Axis x corresponds to the number of chromosomes in common bean.
(TIF)
In parenthesis, there are the numbers of genes of each category.
(TIF)
The blue boxes are for KEGG ECs that have homologies to Phaseolus vulgaris sequence target by PvM93.
(TIF)
(DOCX)
(DOCX)
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.