

Abstract
Background
Cisplatin‐based chemotherapy is the standard first‐line treatment for non‐small‐cell lung cancers (NSCLCs); however, the long‐term therapeutic effect is reduced by chemoresistance. Brain and reproductive organ‐expressed (BRE) proteins are overexpressed in several cancers and have an anti‐apoptotic function. However, their biological role in the development of the chemoresistant phenotype of human NSCLC remains unknown. We investigate the differential expression of the BRE gene in human lung adenocarcinoma cell lines A549 and the cisplatin‐resistant variant A549/cisplatin (DDP), and the mechanisms of cisplatin‐resistance induced by the BRE gene.
Methods
Cell counting kit‐8 assay was employed to determine the sensitivity of A549 and A549/DDP cell lines to cisplatin. BRE expression was measured using quantitative real time‐polymerase chain reaction and western blot analysis. The apoptosis rate of lung adenocarcinoma cells was determined by flow cytometry.
Results
BRE expression in A549 cells, derived from human lung cells, was markedly decreased compared with parental cisplatin‐resistant A549/DDP cells at messenger ribonucleic acid and protein levels. BRE overexpression in A549 significantly decreased sensitivity to DDP by inhibiting cell apoptosis. Conversely, BRE knockdown in A549/DDP cells increased their chemosensitivity. Importantly, we demonstrate that BRE overexpression induces the expression of phosphoprotein kinase B (p‐Akt) in lung cancer cells, while BRE silencing inhibits p‐Akt expression. Furthermore, downregulation of p‐Akt by LY294002 reversed the DDP resistance induced by BRE by increasing apoptosis. BRE enhances the DDP resistance of lung cancer cells through the Akt signaling pathway.
Conclusion
Our findings provide new insight into the mechanism of DDP resistance in NSCLC cells and suggest BRE as an attractive new target for NSCLC treatment.
Keywords: BRE, cisplatin, NSCLC, resistant
Introduction
Cisplatin (DDP) is the most frequently used chemotherapeutic agent for lung cancer. Initiation of DDP is mediated by its interaction with DNA to form DNA adducts, primarily intrastrand crosslink adducts, which activate several signal transduction pathways, including those involving ATM Rad3‐related protein kinase (ATR), p53, p73, and mitogen‐activated protein kinases (MAPK), followed by culmination in the activation of apoptosis.1 DDP exerts anticancer effects via multiple mechanisms, yet its most prominent mode of action involves the generation of DNA lesions, followed by the activation of the DNA damage response and the induction of mitochondrial apoptosis.2 DDP resistance, however, can attenuate DNA damage‐mediated apoptotic signals. The resistance ensues, which is a major limitation of DDP‐based chemotherapy. DDP resistance represents a major obstacle in effective clinical treatment. There is an urgent need to elucidate the mechanisms that regulate DDP resistance in lung cancer. The molecular mechanism of DDP resistance in lung cancer is complicated, including chromosome abnormality, apoptosis‐related cellular signal transduction, apoptosis inhibitory factor gene expression, and DNA damage repair gene expression.3, 4, 5, 6 Although there are many factors inducing chemoresistance in lung cancer therapy, the exact mechanisms of DDP resistance remain unclear.
Brain and reproductive organ‐expressed (BRE) proteins, known as BRCC45, encode a protein with a molecular weight of 44 kD.7 BRE is distributed in the nucleus and cytoplasm of cells and is a member of the breast cancer 1 (BRCA1) complex, which is involved in DNA repair.8, 9 In cytosol, the overexpression of BRE in transfected cells results in the attenuation of death receptor and stress stimuli‐activated apoptosis, through inhibition of the mitochondria‐dependent apoptotic pathway. Moreover, BRE also downregulates tumor necrosis factor alpha (TNFα) signaling by binding death receptors and an overexpression of BRE inhibits apoptosis in hepatocelluar, breast, esophageal, and mouse Lewis lung cancers.10, 11, 12, 13 However, the role of BRE in apoptosis and DDP resistance in human lung cancer is still unknown.
Apoptosis influences cell survival and proliferation, and defends against abnormal cellular changes and infection.14, 15, 16 Recent research shows that apoptosis also plays an important role in DDP resistance in Ewing's sarcoma.17 Apoptotic inhibitor molecules, such as survivin and X‐linked inhibitor of apoptosis protein (XIAP), both of which belong to the IAP superfamily, can exacerbate DDP resistance when overexpressed.18, 19 These inhibitors directly or indirectly impact the activities of caspases (cysteinyl aspartate specific proteinase), the direct effectors of apoptosis, irrespective of the DNA damage pathway, and mediate the apoptotic signal. For DDP, caspases 3, 8, and 9 are critical, and their activation is attenuated in DDP‐resistant cells.20 Overexpression of Bcl‐2 is also associated with DDP resistance, and this is probably facilitated by an increase of glutathione levels and compounded by the presence of mutant p53.21 Similarly, upregulation of Bcl‐xL is also observed in DDP‐resistant tumor cells, possibly as a result of repression of the negative regulator BAD.22 Among these inhibitory mechanisms of DDP‐induced apoptosis, the phosphatidylinositol‐4,5‐bisphosphate 3‐kinase (PI3K)/protein kinase B (Akt) pathway plays an important role.23
Protein kinase B was originally identified as the likely transforming gene component (v‐Akt) of the Akt8 provirus.24 We now know that the Akt family comprises three closely related isoforms: Akt1, Akt2, and Akt3. We have a very good understanding of the mechanisms by which Akt isoforms are activated by growth factors and other extracellular stimuli, as well as by oncogenic mutations in key upstream regulatory proteins, including Ras, PI3‐kinase subunits, and phosphatase and tensin homolog.25 Activation of the PI3K/Akt signal pathway is involved in resistance to DDP‐induced apoptosis.26
In the present study, we sought to investigate the putative role of BRE in DDP‐resistant non‐small cell lung cancer (NSCLC). We found that BRE was decreased in A549 cells compared with parental DDP‐resistant A549/DDP cells, suggesting that BRE might act as a DDP‐resistant gene in NSCLC. We identified that BRE overexpression in A549 cells decreased chemosensitivity to DDP by inhibiting cell apoptosis. By contrast, BRE knockdown in A549/DDP cells increased their chemosensitivity. Furthermore, we found that the Akt signaling pathway was involved in BRE‐induced apoptosis and DDP resistance in lung cancer.
Methods and materials
Cell culture and transfection
The human lung adenocarcinoma cell line A549 and the DDP‐resistant variant A549/DDP (obtained from the Tianjin Lung Cancer Institute, Tianjin, China) were cultured in RMPI 1640 medium containing 10% fetal bovine serum (Gibco, Carlsbad, CA, USA) at 37°C with 5% CO2 incubator, and for A549/DDP, 2 μg/mL DDP was added (Sigma‐Aldrich, St. Louis, MO, USA). Cells were cultured to 80% confluence and transfected with recombinant eukaryotic vector and empty vector using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's recommendation.
Small interfering ribonucleic acids
The small interfering ribonucleic acids (siRNA) targeting human BRE messenger (m)RNA, negative control siRNA (siControl) were purchased from GenePharma (Shanghai, China). BRE siRNA sense: 5′‐GCC CGU AGA UUU CAG CAA UTT ‐3′; anti‐sense: 5′‐AUU GCU GAA AUC UAC GGG CTT ‐3′. Control siRNA sense: 5′‐UUC UCC GAA CGU GUC ACG UTT ‐3′; anti‐sense: 5′‐ACG UGA CAC GUU CGG AGA ATT ‐3′.
Plasmid constructs
Ectopic expression of BRE sequences were obtained from GenBank (Gene ID: 9577). Human BRE was constructed by polymerase chain reaction (PCR) and cloned into pcDNA3.1 vector (Invitrogen, USA), named pcDNA3.1‐ BRE. The primers used in this study were as follows: sense 5′‐ AAG CTT AAA ATG TCC CCA GAA GTG GCC TTG AAC CG ‐3′ and antisense 5′‐ TCT AGA AAT AAG CCC AAA GTG ATC AAA ATT ACT G ‐3′.
Quantitative real‐time polymerase chain reaction
Quantitative real‐time PCR (qRT‐PCR) was performed to validate the micro (mi)RNA expression level. qRT‐PCR was carried out using SYBR Premix Ex TaqTM (Takara, Japan). PCR were carried out in triplicate and analyzed using the ABI Prism 7900HT fast real‐time PCR system (Applied Biosystems, Life Technologies, Carlsbad, CA, USA). The relative quantification values for each gene were calculated by the 2‐ΔΔCt method using glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) as an internal reference. The primers used in this study were as follows: BRE forward, 5′‐ GAT TCA AGG GTA TCA CAA A ‐3′ and reverse, 5′‐CCA CAT CAG CAG CAG AGT ‐3′; GAPDH forward, 5′‐TGC ACC ACC AAC TGC TTA GC‐3′ and reverse, 5′‐GGC ATG GAC TGT GGT CAT GAG ‐3′.
Western blot analysis
Cells were treated with pcDNA3.1‐BRE, pcDNA3.1, si‐BRE, or a blocking agent to target the PI3K/Akt pathway (LY294002). Total cell extracts prepared from cells using radio immunoprecipitation assay buffer (RIPA) buffer (Beyotime, Guangzhou, China), were resolved on 10% gradient sodium dodecyl sulfate‐polacrylamide gel and transferred nitrocellulose (NC) membranes. Membranes were blocked for one hour in 5% skim milk in tris‐buffered saline plus tween 20 (TBST) and incubated with primary antibody overnight at 4°C, followed by the incubation with appropriate horseradish peroxidase‐conjugated secondary antibody at optimized concentration. The primary antibodies anti‐phosphoprotein (p)‐Akt, anti‐Akt, anti‐p‐signal transducer and activator of transcription 3 (Stat3), anti‐Stat3, and anti‐β‐actin antibody were purchased from Cell Signaling Technology (Danvers, MA, USA) The densitometry of western blot results was measured using ImageJ software.
Cell cycle analysis
Cell cycle analysis was performed with flow cytometry assay. A549 and A549/DDP cells were harvested, fixed overnight at 4°C, and stained with a propidium iodide solution for 30 minutes, and were then analyzed using flow cytometry (Beckman Coulter, Brea, CA, USA) and WinCycle (Windsor, VT, USA).
Apoptosis analysis
Cells were transfected with pcDNA3.1/BRE, pcDNA3.1, siBRE, or siNC, 48 hours after transfection. Cells were then harvested and washed with ice cold phosphate‐buffered saline (PBS), and then subjected to Annexin V‐FITC Apoptosis Detection kit I (BD Pharmingen, Franklin, NJ, USA) for staining, which was followed by flow cytometric analysis using a FACScan instrument (Beckman Coulter, USA). The test was repeated three times per experiment.
Statistical analysis
The data were presented as mean ± standard deviation (SD). A t‐test was used to determine significant differences between the control and treatment groups. Statistical analysis was performed using SPSS version 15.0 (SPSS Inc., Chicago, IL, USA), and P < 0.05 was considered a statistically significant difference.
Results
Parental A549 cells and cisplatin (DDP)‐resistant A549/DDP cells differed in biology
To better understand the biological theories of chemoresistance in lung cancer cells, we established a DDP‐resistant human lung adenocarcinoma cell line by subjecting A549 cells to drug pressure. The resistant line was termed A549/DDP. The cell counting kit‐8 (CCK‐8) assay was performed on A549 and A549/DDP cells, which produced IC50 values for DDP of 2.24 ± 0.62 ug/mL and 12.78 ± 0.66 ug/mL (P < 0.01), respectively (Fig 1a). A proliferation assay indicated that A549 grew at a faster rate than A549/DDP (Fig 1b). Using flow cytometric analysis, we found that A549/DDP cells displayed predominant accumulation in the S phase and a reduction in the G2 phase compared with A549 cells (P < 0.05; Fig 1c). The parental line also demonstrated a greater rate of apoptosis (17.59 ± 2.19%) than in the resistant cells (5.91 ± 0.20%; P < 0.05; Fig 1d).
Figure 1.
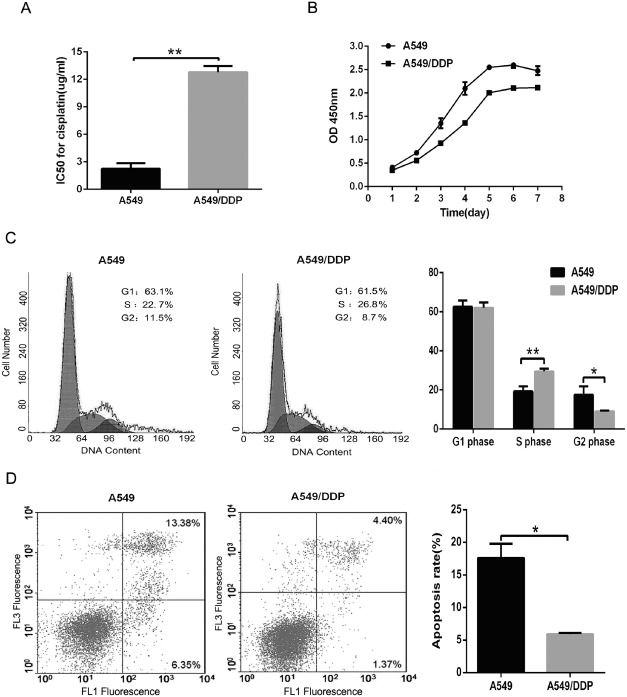
Characteristics of A549/cisplatin (DDP) and parental A549 cells. (a) Cell counting kit‐8 assay was used to measure cells inhibitory concentration (IC)50 for DDP. (b) Cell growth was detected by a cell viability assay.  , A549;
, A549;  , A549/DDP. (c) Cell cycle was investigated by flow cytometry.
, A549/DDP. (c) Cell cycle was investigated by flow cytometry.  , A549;
, A549;  ,A549/DDP. (d) Cell apoptosis was measured by flow cytometry. Both A549 and A549/DDP cells were cultured with 2.0 μg/mL DDP. The data are expressed as mean ± standard deviation of three independent experiments. *P < 0.05 and **P < 0.01 compared with A549 cells.
,A549/DDP. (d) Cell apoptosis was measured by flow cytometry. Both A549 and A549/DDP cells were cultured with 2.0 μg/mL DDP. The data are expressed as mean ± standard deviation of three independent experiments. *P < 0.05 and **P < 0.01 compared with A549 cells.
Brain and reproductive organ (BRE) enhanced resistance to DDP in lung cancer cells
Brain and reproductive organ expression was measured in A549 and DDP‐resistant A549/DDP cells using qRT‐PCR and western blot analysis. The mRNA and protein expression of BRE in A549 cells was markedly lower than in A549/DDP cells. The data indicate that BRE may be involved in DDP resistance in human lung cancer cells.
We therefore investigated the role of BRE in DDP resistance. We performed a cell viability assay (CCK‐8) to validate the inhibitory concentration (IC)50 values for A549 and A549/DDP cells exposed to DDP with and without BRE expression. High BRE expression in A549 cells, achieved via transfection, significantly increased the IC50 values for DDP; silencing BRE by siRNA in A549/DDP cells reduced the IC50 values. The transfecting or silencing efficiency of BRE in the cells was established using western blot analysis (Fig 2d and f, lower). We concluded that BRE expression conferred DDP resistance to A549 cells.
Figure 2.
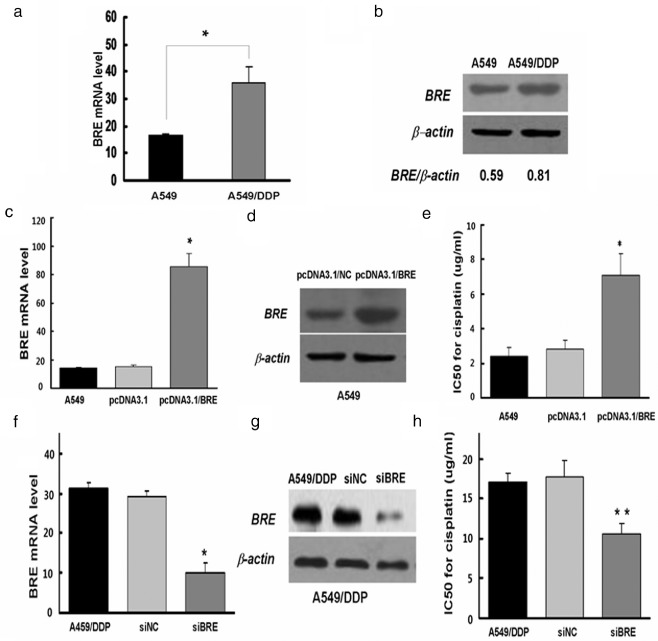
Effect of brain and reproductive organ‐expressed (BRE) protein on cisplatin (DDP) resistance in lung cancer cells. (a,b) BRE expression was measured by real‐time polymerase chain reaction (PCR) and western blot in A549 and A549/DDP cells. (c‐e) Ectopic expression of BRE in A549 cells. The messenger ribonucleic acid (mRNA) and protein levels of BRE were examined by real‐time PCR and western blot. Additionally, inhibitory concentration (IC)50 for DDP was measured using cell counting kit‐8 (CCK‐8) assay. (f‐h) Silencing of BRE by small interfering (RNA) in A549/DDP cells. The mRNA and protein levels of BRE were examined by real‐time PCR and western blot. Additionally, IC50 for cisplatin was measured using CCK‐8 assay. Data are reported as mean ± standard deviation for three independent experiments (*P < 0.05, t‐test).
BRE affected resistance to DDP through regulation of apoptosis in lung cancer cells
To investigate the effect of BRE on cell viability, flow cytometry was used to measure apoptosis. BRE upregulation in A549 cells inhibited apoptosis, reducing the apoptotic rate from 18.49 ± 2.19% to 12.84 ± 1.47%, compared with the control group (Fig 3a). However, the apoptotic rate in A549/DDP cells increased from 7.91 ± 0.95% to 14.9 ± 1.34% when BRE was silenced by siRNA (Fig 3b). This result suggests that BRE may be involved in the suppression of DDP‐induced apoptosis.
Figure 3.
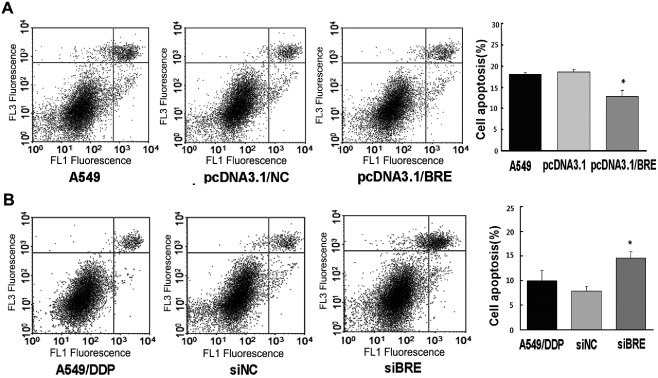
Effect of brain and reproductive organ‐expressed (BRE) protein on apoptosis in lung cancer cells. (a) Ectopic expression of BRE in A549 cells. Flow cytometry assay was used to measure the apoptosis rate of cells. (b) Silencing BRE by small interfering ribonucleic acid (siRNA) in A549/cisplatin (DDP) cells. The apoptosis rate of cells was measured by flow cytometry assay.
The protein kinase B pathway contributes to BRE‐induced DDP resistance
To investigate the antiapoptotic signal transduction pathway affected by BRE upregulation, we measured apoptosis‐related proteins Akt and Stat3 using western blot analysis. Compared with the control cells, Akt phosphorylation in A549 cells significantly increased when BRE was overexpressed (Fig 4a), and significantly decreased in A549/DDP cells when BRE was knocked down (Fig 4b). In contrast, phosphorylated and total Stat3 showed no significant change in either scenario (Fig 4a). These results demonstrated that BRE‐inhibited apoptosis may involve, at least in part, the PI3K/Akt signaling pathway.
Figure 4.
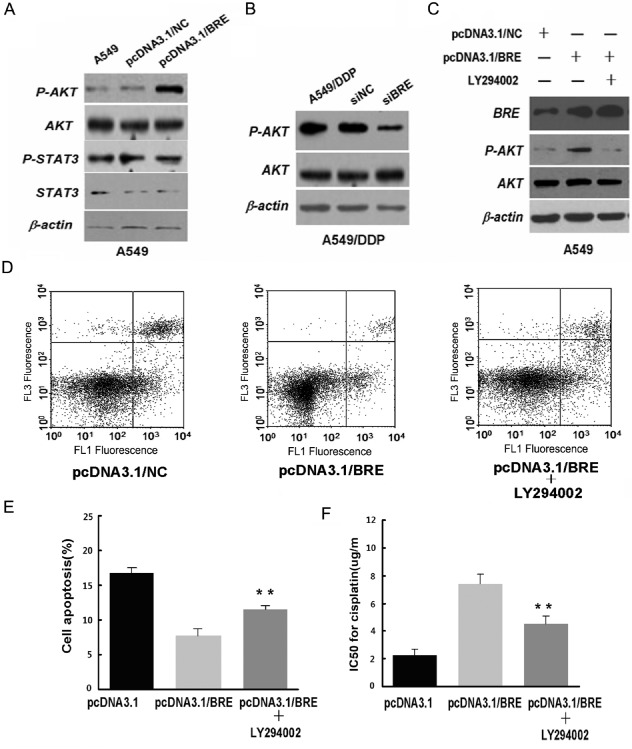
Phosphatidylinositol‐4,5‐bisphosphate 3‐kinase/protein kinase B (Akt) pathway involvement in brain and reproductive organ‐expressed (BRE)‐induced cisplatin (DDP) resistance. (a) Ectopic expression of BRE in A549 cells. The protein levels of phosphor (p)‐Akt, Akt, p‐signal transducer and activator of transcription 3 (Stat3), and Stat3 were examined by western blot. (b) Silencing BRE by small interfering ribonucleic acid (siRNA) in A549/DDP cells. The protein levels of p‐Akt and Akt were examined by western blot. (c–f) A549 cells after transfection of pcDNA3.1/BRE with or without LY294002, followed by the addition of 2 g/mL cisplatin. (c) Western blot was used to detect the protein levels of p‐Akt and Akt. (d,e) Cell apoptosis rate was measured by flow cytometry assay. (f) Inhibitory concentration (IC)50 for cisplatin was measured by cell counting kit‐8 assay.
Protein kinase B is a major downstream target of PI3K. To validate the relationship between BRE and the PI3K/Akt signal pathway, we used LY294002 (PI3 kinase inhibitor) to restrict the level of phosphorylated Akt. We transfected A549 cells with pcDNA3.1/BRE in the presence or absence of LY294002 (20 μM). In the presence of LY294002, p‐Akt levels were markedly reduced in BRE‐overexpressing A549 cells (Fig 4c). Apoptosis also increased significantly in these cells following treatment with 2.5 μg/mL DDP for 24 hours in the presence of LY294002 (Fig 4d). Moreover, MTT analysis showed that cell viability was significantly decreased in the presence of LY294002 (Fig 4e).
Discussion
Chemotherapy is, at present, one of the most effective treatments for lung cancer. Among the many chemotherapeutic agents, DDP is the most commonly prescribed for treating NSCLC. Unfortunately, the majority of tumors acquire drug resistance. Most patients with advanced NSCLC will eventually relapse and die because of acquired drug resistance. Therefore, DDP resistance is a major clinical challenge. The mechanisms involved in DDP resistance remain largely unexplored, and, thus, are not yet fully understood.
In this study, we found that the mRNA and protein expression levels of BRE in parental A549cells were markedly decreased compared with DDP‐resistant A549/DDP cells. The reintroduction of BRE in A549 cells significantly increased the DDP IC50 values. Meanwhile, silencing BRE by siRNA in A549/DDP cells decreased the IC50 values. These findings strongly suggest that BRE potentiates DDP resistance in lung cancer. BRE, also known as BRCC45, localizes to different subcellular compartments. Recently, BRE overexpression has been reported in several types of tumors, such as hepatocellular and esophageal cancers and mouse Lewis lung cancer, and experimental evidence suggests that BRE could inhibit cancer cell apoptosis.10, 11, 12, 13, 27 These findings raise the possibility that BRE may induce DDP resistance in lung cancer through an apoptosis‐related pathway.
To explore this hypothesis, we first investigated the function of BRE in apoptosis. Li et al. found that the anti‐apoptotic role of cytosolic BRE is independent of its nuclear counterpart and is not dependent on direct targeting of the mitochondria.28 BRE overexpression could inhibit apoptosis by repressing TNF‐induced NF‐κB activation. The TNF‐related apoptosis‐inducing ligand is correlated with DDP‐induced apoptosis in esophageal squamous cell carcinoma.29, 30 These studies support our hypothesis that BRE may inhibit DDP‐induced apoptosis through one or more apoptosis‐related pathways. Our data showed that the reintroduction of BRE dramatically reduced apoptosis in NSCLC cells in vitro. In contrast, silencing of BRE by siRNA in A549/DDP cells increased apoptosis. Because decreased apoptosis is a key feature of drug resistance, our data suggest that increased BRE expression may facilitate the DDP resistance of human cancers like NSCLC.
Protein kinase B is a major downstream target of PI3K. Research has shown that the PI3K/Akt signaling pathway is frequently overactivated in human lung, breast, endometrial, and ovarian cancers, glioblastomas, and medulloblastomas. Current research has also demonstrated that Akt activation confers resistance to DDP‐induced apoptosis in ovarian cancer. In this study, we found that p‐Akt levels were significantly increased in BRE overexpressed A549 cells and decreased in BRE silenced A549/DDP cells. After inhibiting PI3K/Akt signaling by LY294002, the apoptotic ratio of BRE overexpressed A549 cells was significantly increased.
Conclusion
This study has shown that BRE plays an important role in promoting DDP resistance in human lung adenocarcinoma. Thus, clarifying the mechanism of BRE‐induced DDP resistance and identification of specific agents able to inhibit the expression of BRE is a potential future strategy for the treatment of NSCLC.
Disclosure
No authors report any conflict of interest.
Acknowledgments
This study was partly supported by the grants from the National Natural Science Foundation of China (No. 81201852), the National 863 Program (No. 2012AA02A201, No. 2012AA02A502), and the National 973 Program (No. 2010CB529405).
References
- 1. Siddik ZH. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003; 22: 7265–7279. [DOI] [PubMed] [Google Scholar]
- 2. Galluzzi LL, Senovilla I, Vitale J et al Molecular mechanisms of cisplatin resistance. Oncogene 2012; 31: 1869–1883. [DOI] [PubMed] [Google Scholar]
- 3. Branch PM, Masson G, Aquilina M, Bignami M, Karran P. Spontaneous development of drug resistance: Mismatch repair and p53 defects in resistance to cisplatin in human tumor cells. Oncogene 2000; 19: 3138–3145. [DOI] [PubMed] [Google Scholar]
- 4. Takano MK, Kudo K, Goto T, Yamamoto K, Kita T, Kikuchi Y. Analyses by comparative genomic hybridization of genes relating with cisplatin‐resistance in ovarian cancer. Hum Cell 2001; 14: 267–271. [PubMed] [Google Scholar]
- 5. Chen HY, Yu SL, Chen CH et al A five‐gene signature and clinical outcome in non‐small‐cell lung cancer. NEJM 2007; 356: 11–20. [DOI] [PubMed] [Google Scholar]
- 6. Pabla NS, Huang QS, Mi R, Daniel R, Dong Z. ATR‐Chk2 signaling in p53 activation and DNA damage response during cisplatin‐induced apoptosis. J Biol Chem 2008; 283: 6572–6583. [DOI] [PubMed] [Google Scholar]
- 7. Gu C, Castellino A, Chan JY, Chao MV. BRE: A modulator of TNF‐alpha action. FASEB J 1998; 12: 1101–1108. [DOI] [PubMed] [Google Scholar]
- 8. Dong Y, Hakimi MA, Chen X et al Regulation of BRCC, a holoenzyme complex containing BRCA1 and BRCA2, by a signalosome‐like subunit and its role in DNA repair. Mol Cell 2003; 12: 1087–1099. [DOI] [PubMed] [Google Scholar]
- 9. Kim H, Chen J, Yu X. Ubiquitin‐binding protein RAP80 mediates BRCA1‐dependent DNA damage response. Science 2007; 316: 1202–1205. [DOI] [PubMed] [Google Scholar]
- 10. Chui YL, Ma CH, Li W et al Anti‐apoptotic protein BRE/BRCC45 attenuates apoptosis through maintaining the expression of caspase inhibitor XIAP in mouse Lewis lung carcinoma D122 cells. Apoptosis 2014; 19: 829–840. [DOI] [PubMed] [Google Scholar]
- 11. Noordermeer SM, Wennemers M, Bergevoet SM et al Expression of the BRCA1 complex member BRE predicts disease free survival in breast cancer. Breast Cancer Res Treat 2012; 135: 125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen HB, Pan K, Tang MK et al Comparative proteomic analysis reveals differentially expressed proteins regulated by a potential tumor promoter, BRE, in human esophageal carcinoma cells. Biochem Cell Biol 2008; 86: 302–311. [DOI] [PubMed] [Google Scholar]
- 13. Chui YL, Ching AK, Chen S et al BRE over‐expression promotes growth of hepatocellular carcinoma. Biochem Biophys Res Commun 2010; 391: 1522–1525. [DOI] [PubMed] [Google Scholar]
- 14. Vaux DL, Korsmeyer SJ. Cell death in development. Cell 1999; 96: 245–254. [DOI] [PubMed] [Google Scholar]
- 15. Strasser A, O'Connor L, Dixit VM. Apoptosis signaling. Annu Rev Biochem 2000; 69: 217–245. [DOI] [PubMed] [Google Scholar]
- 16. Barber GN. Host defense, viruses and apoptosis. Cell Death Differ 2001; 8: 113–126. [DOI] [PubMed] [Google Scholar]
- 17. Mitsiades N, Yu WH, Poulaki V, Tsokos M, Stamenkovic I. Matrix metalloproteinase‐7‐mediated cleavage of Fas ligand protects tumor cells from chemotherapeutic drug cytotoxicity. Cancer Res 2001; 61: 577–581. [PubMed] [Google Scholar]
- 18. Asselin E, Mills GB, Tsang BK. XIAP regulates Akt activity and caspase‐3‐dependent cleavage during cisplatin‐induced apoptosis in human ovarian epithelial cancer cells. Cancer Res 2001; 61: 1862–1868. [PubMed] [Google Scholar]
- 19. Ikeguchi M, Liu J, Kaibara N. Expression of survivin mRNA and protein in gastric cancer cell line (MKN‐45) during cisplatin treatment. Apoptosis 2002; 7: 23–29. [DOI] [PubMed] [Google Scholar]
- 20. Henkels KM, Turchi JJ. Cisplatin‐induced apoptosis proceeds by caspase‐3‐dependent and ‐independent pathways in cisplatin‐resistant and ‐sensitive human ovarian cancer cell lines. Cancer Res 1999; 59: 3077–3083. [PubMed] [Google Scholar]
- 21. Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ. Bcl‐2 functions in an antioxidant pathway to prevent apoptosis. Cell 1993; 75: 241–251. [DOI] [PubMed] [Google Scholar]
- 22. Hayakawa J, Ohmichi M, Kurachi H et al Inhibition of BAD phosphorylation either at serine 112 via extracellullar signal‐regulated protein kinase cascade or at serine 136 via Akt cascade sensitizes human ovarian cancer cells to cisplatin. Cancer Res 2000; 60: 5988–5994. [PubMed] [Google Scholar]
- 23. Yang X, Fraser M, Abedini MR, Bai T, Tsang BK. Regulation of apoptosis‐inducing factor‐mediated, cisplatin‐induced apoptosis by Akt. Br J Cancer 2008; 98: 803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Staal SP. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: Amplification of AKT1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci U S A 1987; 84: 5034–5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hers I, Vincent EE, Tavaré JM. Akt signalling in health and disease. Cell Signal 2011; 23: 1515–1527. [DOI] [PubMed] [Google Scholar]
- 26. Yang X, Fraser M, Moll UM, Basak A, Tsang BK. Akt‐mediated cisplatin resistance in ovarian cancer: Modulation of p53 action on caspase‐dependent mitochondrial death pathway. Cancer Res 2006; 66: 3126–3136. [DOI] [PubMed] [Google Scholar]
- 27. Chan BC, Ching AK, To KF et al BRE is an antiapoptotic protein in vivo and overexpressed in human hepatocellular carcinoma. Oncogene 2008; 27: 1208–1217. [DOI] [PubMed] [Google Scholar]
- 28. Li Q1, Ching AK, Chan BC et al A death receptor‐associated anti‐apoptotic protein, BRE, inhibits mitochondrial apoptotic pathway. J Biol Chem 2004; 279: 52106–52116. [DOI] [PubMed] [Google Scholar]
- 29. Kondo KS, Yamasaki T, Sugie N et al Cisplatin‐dependent upregulation of death receptors 4 and 5 augments induction of apoptosis by TNF‐related apoptosis‐inducing ligand against esophageal squamous cell carcinoma. Int J Cancer 2006; 118: 230–242. [DOI] [PubMed] [Google Scholar]
- 30. Lu H, Yang X, Duggal P et al TNF‐alpha promotes c‐REL/DeltaNp63alpha interaction and TAp73 dissociation from key genes that mediate growth arrest and apoptosis in head and neck cancer. Cancer Res 2011; 71: 6867–6877. [DOI] [PMC free article] [PubMed] [Google Scholar]