

Abstract
The POU4F2/Brn-3b transcription factor has been identified as a potentially novel regulator of key metabolic processes. Loss of this protein in Brn-3b knockout (KO) mice causes profound hyperglycemia and insulin resistance (IR), normally associated with type 2 diabetes (T2D), whereas Brn-3b is reduced in tissues taken from obese mice fed on high-fat diets (HFD), which also develop hyperglycemia and IR. Furthermore, studies in C2C12 myocytes show that Brn-3b mRNA and proteins are induced by glucose but inhibited by insulin, suggesting that this protein is itself highly regulated in responsive cells. Analysis of differential gene expression in skeletal muscle from Brn-3b KO mice showed changes in genes that are implicated in T2D such as increased glycogen synthase kinase-3β and reduced GLUT4 glucose transporter. The GLUT4 gene promoter contains multiple Brn-3b binding sites and is directly transactivated by this transcription factor in cotransfection assays, whereas chromatin immunoprecipitation assays confirm that Brn-3b binds to this promoter in vivo. In addition, correlation between GLUT4 and Brn-3b in KO tissues or in C2C12 cells strongly supports a close association between Brn-3b levels and GLUT4 expression. Since Brn-3b is regulated by metabolites and insulin, this may provide a mechanism for controlling key genes that are required for normal metabolic processes in insulin-responsive tissues and its loss may contribute to abnormal glucose uptake.
Keywords: POU4F2/Brn-3b, transcription factor, glucose intolerance, GLUT4
metabolic homoeostasis is achieved by the coordination of complex and highly regulated processes that involve cross talk between different tissues (36). Many of these changes are mediated by circulating metabolites such as glucose, which drive genetic changes in different tissues, thereby maintaining blood glucose levels within narrow functional ranges. Under normal conditions, elevated blood glucose causes increased insulin secretion by the pancreas, which facilitates glucose uptake, in responsive tissues, but inhibits gluconeogenesis and lipolysis in the liver and adipose tissue (9). However, deregulation of one or more of these processes can cause elevated blood glucose (hyperglycemia), which is characteristic of type 2 diabetes mellitus (T2D). Uncontrolled hyperglycemia can drive pathophysiological changes such as inflammation and atherosclerotic plaque formation in the vasculature but also contributes to the development of cardiovascular, renal, and neuropathic diseases (2, 14, 15, 21, 22). However, the molecular mechanisms that drive early pathological changes are not fully elucidated.
In the postprandial state, skeletal muscles and adipose tissues are responsible for the majority of glucose utilization, and this is facilitated by insulin-responsive glucose transporters such as GLUT4, which are translocated from intracellular vesicles to the plasma membrane where they facilitate glucose uptake from the bloodstream (27). Hyperglycemia and T2D are often associated with insulin resistance, whereby normally insulin-sensitive tissues fail to respond to increased circulating insulin (19, 36). Although these complex effects may be attributable to multiple factors, reduced GLUT4 expression has been implicated in impaired glucose responses and insulin resistance (13, 23, 27). In this regard, disruption of one allele in heterozygote (GLUT4+/−) mutants has been sufficient to cause peripheral insulin resistance (32). Furthermore, GLUT4 gene transcription is dynamically regulated by feeding in rodent models of streptozocin-induced diabetes since fasted animals showed significant reduction of GLUT4 mRNA in adipose tissues, which was rapidly reversed upon refeeding (20, 34). Therefore, transcription factors that regulate GLUT4 expression in a tissue-specific manner will be important for controlling metabolic processes in insulin-dependent tissues and may contribute to initiation or progression of dysfunction/disease (18, 24).
The POU4F2/Brn-3b transcription factor (Brn-3b), which belongs to subclass IV of POU homeodomain proteins, is characterised by the highly conserved DNA binding POU (Pit-Oct-Unc) domain (7), which binds to regulatory regions of target genes, thereby controlling the rate of transcription by the RNA pol II enzyme. The gene encoding Brn-3b consists of two exons separated by an intron, which can give rise to distinct protein isoforms that are completely conserved in the DNA binding COOH terminus but differ in NH2 terminal domain (26, 28). Thus, the shorter Brn-3b(s) protein is encoded by exon 2 only, whereas Brn-3b(l) protein is encoded by exons 1 and 2 and therefore contains an additional NH2 terminal domain, which is not found in Brn-3b(s) (3, 8). However, the functions of distinct isoforms are still to be elucidated.
Brn-3b mediates diverse effects on cell fate by regulating the expression of multiple target genes, but these are highly dependent on the cell types and/or growth conditions. For instance, Brn-3b can transactivate cell cycle proteins cyclin D1/CDK4 (3) in epithelial cells and, as such, has been implicated in some cancers, where elevated Brn-3b enhances cell proliferation and tumor growth (5, 7, 17). However, Brn-3b also confers drug resistance and migratory potential after chemotherapeutic treatment by driving expression of other target genes such as the small heat shock protein HSP27, which protects cells under these conditions (6, 33). On the other hand, Brn-3b is essential for survival and specification of retinal ganglion cells (RGCs), since KO mutant mice are blind due to loss of RGC after birth (12, 26). However, if coexpressed with growth inhibitory p53 protein, Brn-3b can promote apoptosis by interacting and cooperating with p53 to increase transcription of proapoptotic target genes, e.g., Bax and Noxa. As such, it is crucial to analyze the effects of this transcriptional regulator in the context of cell/tissue-specific effects in order to determine its mechanism of action on cell growth, behaviour or fate.
More recently, Brn-3b was identified as the only transcription factor to show significant changes in skeletal muscle taken from subjects after 2 mo of endurance training, when comparing gene expression in subjects with high or low posttraining insulin sensitivity (37). The results of these microarray analyses suggested that Brn-3b may be important for controlling gene expression and function in metabolically active tissues and led us to hypothesize that this transcription factor may regulate genes that control important metabolic processes.
These studies report on the expression of Brn-3b in insulin-responsive tissues and investigate how loss of this transcription factor affects metabolic function in Brn-3b KO mutant mice. Furthermore, the regulation of Brn-3b expression in response to glucose and insulin have been characterized using the skeletal myoblast-derived cell line C2C12. Finally, analysis of gene expression changes upon loss of Brn-3b has identified potential target genes such as GLUT4, which may be regulated by this transcription factor in skeletal muscle and adipose tissues. The results herein suggest that Brn-3b represents a novel regulator of target genes that are important for controlling metabolic function and glucose homeostasis in insulin-responsive tissues.
MATERIALS AND METHODS
General laboratory reagents.
Reagents were from Merck (Nottingham, UK) or Sigma (Dorset, UK) unless otherwise stated. Tissue culture reagents/plastics were from the following: Gibco/Life Technologies (Paisley, UK); Nunc (Paisley, UK); Greiner (Stonehouse, Gloucester, UK), or Corning (Scientific Laboratory Supplies, Nottingham, UK). Primary antibodies were sourced as follows: rabbit-Brn-3b pAb (Abcam, Cambridge, UK); goat-Brn-3b pAB and goat-actin pAb (Santa Cruz Biotechnology, Santa Cruz, CA); β-tubulin mAb, (Merck Millipore, Darmstad, Germany); Glut4 (1F8) Mouse mAb, Cell Signaling Technology (Danvers, MA); and all secondary Ab were from Dako (Cambridgeshire, UK). GLUT4 promoter, cloned in the pGL3 luciferase vector, was a generous gift from Prof M. Quon (University of Maryland, Baltimore, MD) and Prof E. Karnieli (Rambam Medical Center, Haifa, Israel).
Methods.
For in vivo models, studies using rodent models were undertaken in accordance with Home Office guidelines (Animals Scientific Procedures Act 1986) and approved by local Ethics Review Board. C57BL/6J strain was used for studies, with outbred C57BL6 mice obtained from Harlan UK, and Brn-3b heterozygote mice were used to generate Brn-3bKO mutant mice and aged-matched wild-type (WT) littermates. For longitudinal studies (>1 yr), experimental animals were weighed monthly, whereas mice fed a high-fat diet (HFD) or normal chow diet (ND) were weighed weekly. Dissected tissues (e.g., skeletal muscle, liver, adipose tissue, pancreas) were either snap-frozen (for subsequent RNA/protein extraction) or fixed in 4% paraformaldehyde (PFA) and embedded for subsequent sectioning.
Glucose tolerance tests and serum insulin measurements.
For glucose tolerance tests (GTT), experimental animals were fasted for 12 h, and baseline blood glucose was evaluated using a glucometer. Glucose bolus (2 mg glucose/g BW) was administered by intraperitoneal injection, and blood glucose was recorded after 15, 30, 60, and 120 min. For serum insulin measurements, whole blood was collected from the saphenous vein at the start and end of experiments into serum separator tubes (BD Microtainer; Becton-Dickinson, Franklin Lakes, NJ). Serum insulin levels were measured using a Mercodia Mouse Insulin ELISA kit (Upsalla, Sweden).
Calorie consumption.
Calorie consumption was calculated by monitoring dietary intake and then using the known caloric value of the feed to calculate intake. Briefly, mice were housed in separate cages, and the chow was weighed at the start of experiments, and this was repeated at the end of each week, before topping up or replacing of the feed. This was also measured if it was deemed necessary to top up on at other times. The differences in weight (grams) of the feed at the start and end of the week were multiplied by the known caloric value (5.1 kcal/g) to calculate calorie consumption per week for each mouse. The mean and standard error of each group (n ≥ 5) were used to determine differences in calorie consumption over time.
Cell culture and treatments.
Skeletal muscle satellite cell-derived C2C12 myoblasts were maintained in full growth medium (FGM) [Dulbecco's modified Eagle's medium (DMEM), 10% fetal bovine serum (FBS), 1% penicillin-streptomycin] grown in 5% CO2 at 37°C. Cells plated onto 6-well (5 × 105/well) or 12-well (105/well) culture dishes were transfected or treated as specified. For treatment of cells with free fatty acids (FFA), unsaturated long-chain fatty acids (oleic acid) or saturated fatty acids (palmitic acid) were dissolved in ethanol and then added to cells at appropriate concentrations. Transfections were carried out using Fugene (Promega, Hampshire, UK) as previously described (6, 33), and reporter assays were done using the Dual Luciferase Reporter Assay System (Promega, Hampshire, UK).
RNA extraction, cDNA synthesis, and quantitative RT-PCR.
Tissues homogenized in liquid nitrogen were resuspended in TRIzol (Invitrogen, Paisley, UK); C2C12 cells were harvested in TRIzol and then processed according to the manufacturer's protocol. DNAse1-treated RNA was used for cDNA synthesis (RNA Superscript II RT) (Invitrogen). qRT-PCR was performed on an Opticon 2 DNA engine thermal cycler (Bio-Rad, UK), using SYBR Green master mix (Qiagen, Manchester, UK) and Brn-3b primers (forward-ATCGCCGAAAAGCTGGAT; reverse-TTCTCTTCTGTTTCTGCCTCTG) or QuantiTect Assay primers (Qiagen) for selected target genes. Variability between samples was adjusted using GAPDH and fold changes were calculated using the 2−ΔΔCT method (25).
PCR array analysis.
cDNA from Brn-3b KO skeletal muscle and WT controls (see above) were used to screen the Mouse Diabetes RT2 (SAB BioSciences, Qiagen, West Sussex, UK), which facilitates the screening of 84 genes associated with onset, development, and progression of diabetes. Quantitative PCR was undertaken according to the manufacturers' protocol using the Opticon 2 DNA thermal cycler and analysis done using PCR Array Data Analysis Software (https://www.qiagen.com/gb/products/genesandpathways/data-analysis).
Protein extraction and immunoblotting.
Cells were harvested in Laemmli buffer; mouse tissues were pulverized in liquid nitrogen and then resuspended in Laemmli buffer and homogenized. Total protein extraction and polyacrylamide gel electrophoresis (SDS-PAGE) were carried out as described (4). Proteins were quantified using densitometry (Quantity One Software, Bio-Rad Laboratories) or Image-J, and the invariant β-tubulin protein was used to adjust for differences in protein loading.
Chromatin immunoprecipitation assay (ChIP) was carried out as described by Lee et. al. (22), using anti-goat Brn-3b Ab (Santa Cruz Biotechnology) to immunoprecipitate Brn-3b on chromatin in intact cells. Anti-GAPDH (Abcam) was used as negative control. Sonicated ChIP DNA was amplified with PCR primers 1+2 or 3+4, designed to amplify different regions of GLUT4 promoter, as follows: primer 1, CAGGTACACATGTAGTACACA; primer 2, TGGCTGTTCTGGAACTCACT; primer 3, CTTGAGTATACATGTGGCACAC; primer 4, TGTCAGCTTCTTGATGGCATC.
Transient transfections and reporter assays.
To analyze effects of Brn-3b on GLUT4 promoter activity in C2C12 cells, 5 × 104 cells/well (in 6-well plates) were cotransfected with GLUT4 reporter construct and Brn-3b expression vector or control LTR, using Fugene reagent (Roche, UK). To analyze effects of glucose ± insulin on Brn-3b promoter activity, the Brn-3b reporter construct was transfected into C2C12 cells grown in FGM, NG, or NG + glucose with or without insulin. Cells were harvested after 24 h, and promoter activity was measured using a Dual-Luciferase reporter kit (Promega, UK) and a TD-20/20 luminometer (Turner Designs).
Statistics.
Statistical analysis was performed using Microsoft Excel or GraphPad Prism 6 (San Diego, CA). Parametric tests (paired t-test) or nonparametric tests (Mann-Whitney and Kruskal-Wallis), followed by post hoc analysis (Dunn's multiple comparisons test) were used to determine differences between independent samples.
RESULTS
Brn-3bKO mutant mice develop increased body weight and glucose intolerance.
Casual observation of differences in the size of Brn-3b KO mice compared with WT littermates led us to undertake longitudinal studies to compare body weights (BW) over time (>12 mo). Results showed that mutant mice had consistently higher BW compared with WT controls [Fig. 1A(i)], although there were no significant changes in body length (e.g., Brn-3b KO ∼9.3 ± 0.05 cm vs. WT females ∼9.1 ± 0.1 cm). Closer inspection showed increased visceral fat in the abdomens of Brn-3b KO mutants compared with littermate controls [Fig. 1A(ii)], suggesting increased fat deposition in KO mice. To test whether changes in BW were caused by differences in calorie consumption, food intake was closely monitored for WT and Brn-3b KO mutants fed a HFD (see materials and methods) and used to determine caloric intake. Figure 1A(iii) showed that there were no statistically significant differences in food consumption by KO mutants and WT controls, suggesting that increased BW in Brn-3b KO mutants may result from potential changes in metabolic processes. Since Brn-3b was shown to be differentially expressed in skeletal muscle of insulin-responsive subjects (37), we next tested for changes in glucose handling in Brn-3b KO mutants by undertaking glucose tolerance tests (GTT). Therefore, Brn-3b KO and WT control mice were fasted for 12 h, and blood glucose levels were measured at baseline, after which a glucose bolus was administered intraperitoneally. This was followed by half-hourly blood glucose measurements for up to 120 min. The results (Fig. 1B) showed no significant differences in blood glucose between KO and WT mice at baseline (t = 0), but marked differences were evident after administration of the intraperitoneal glucose bolus. As expected, WT mice showed increased blood glucose at 30 min, which decreased by 60 min and returned to baseline by 120 min. However, Brn-3b KO mice had significantly higher blood glucose levels at 30 min, which continued to rise at 60 min and remained significantly elevated after 120 min.
Fig. 1.

Metabolic dysfunction in Brn-3b KO mutant mice. A(i): change in body weight in Brn-3b KO mutants (red line; n = 5) compared with age-matched WT control littermates (blue; n = 5). Weights were measured at monthly intervals for up to 14 mo. (ii): representative images of abdominal viscera of 3-mo-old Brn-3b KO and WT mice. *Increased fatty deposits around internal organs such as the kidney in Brn-3b KO mutants, which is not seen in WT controls. (iii): graph showing calorie consumption by WT mice (dark gray) or Brn-3b KO mutant (light gray). Values represent means ± SE of calorie consumption of 6 individual animals in each group measured at weekly intervals. B: comparison of blood glucose levels in Brn-3b KO mutant mice (red line) compared with WT controls (black line). After overnight fasting, baseline blood glucose was measured before administration of peritoneal injection of glucose bolus, after which blood glucose levels were measured half-hourly for 120 min. Values represent means ± SD of 6 animals. C(i): table showing serum insulin levels in WT and Brn-3b KO mutant mice at baseline (t0) and at the end of experiments (120 min). (ii): graph showing means ± SD of serum insulin levels in WT (dark gray) or mutants (light gray) at 2 different time points. *Statistically significant differences (P < 0.05) between WT and mutant levels at 120 min; ns, no significant differences at time 0. D: representative Western blot analysis showing Brn-3b proteins. Brn-3b(s) protein (30–35 kDa) was the main protein isoform in skeletal muscle, whereas liver expressed high levels of Brn-3b(l) (∼40–43 kDa) and lower levels of Brn-3b(s). Brn-3b protein was not detected in pancreas taken from WT mice.
ELISA assays were used to examine insulin levels in serum collected at baseline and after 120 min. Although baseline serum insulin levels in the mutant animals were slightly higher than in WT, this was not statistically significant. However, at the end of experiments (120 min), serum insulin was significantly higher in Brn-3b KO mutants than in controls (Fig. 1C). The observed hyperglycemia, accompanied by increased insulin levels, in mutant mice suggested changes in metabolic processes such as insulin production or glucose uptake by insulin-responsive tissues e.g., skeletal muscle, adipose tissue, and liver. However, since Brn-3b expression was not previously investigated in such tissues, Western blot analyses were carried out using protein extracts from pancreas, skeletal muscle, liver, and adipose tissue. Figure 1D shows that Brn-3b was not detectable in pancreatic tissue, but significant levels were observed in insulin-responsive tissues such as skeletal muscle and liver. The shorter Brn-3b(s) isoform was more abundant in skeletal muscle, whereas the longer Brn-3b(l) isoform was predominant in the liver (Fig. 1D) and adipose tissues (Fig. 2B). Although the potential roles for these distinct isoforms are unclear at present, high Brn-3b expression in insulin-responsive tissues such as skeletal muscle may suggest that persistent hyperglycemia and insulin resistance in Brn-3b KO mutants are likely to be associated with defects in metabolically active tissues rather than the pancreas, in which Brn-3b is not readily detected.
Fig. 2.

Reduced Brn-3b expression in insulin-responsive tissues in animals fed a high-fat diet (HFD) correlates with obesity, hyperglycemia, and insulin resistance. A(i): blood glucose levels in WT mice fed normal diet (ND; dotted lines) or HFD (solid lines) for >3 mo. Baseline measurement was undertaken after 12-h fasting and before administration of glucose bolus, after which analyses were done at intervals shown, for up to 120 min. (ii): table showing blood glucose and insulin at baseline (t0) and at the end of experiments (120 min). (iii): graph showing means ± SD of serum insulin levels in animals fed ND (dark gray) or HFD (light gray) at 2 different time points. **Statistically significant differences between ND control and HFD (n = 6). B: representative Western blot analysis showing Brn-3b expression in skeletal muscle (i) and adipose tissue (iii) taken from groups of animals fed ND or HFD. Arrows indicate position of distinct isoforms Brn-3b(l) or -(s); β-tubulin blots show differences in protein loading. (ii); (iv) shows quantification of changes in Brn-3b(s) protein in skeletal muscle or adipose tissue taken from mice fed ND or HFD. Values were normalized to β-tubulin in corresponding samples and represent means ± SE from 4 independent animals. *Statistically significant reduction of Brn-3b protein in tissues taken from animals fed HFD vs. ND as determined by t-test (n ≥ 3).
Brn-3b is reduced in insulin-responsive tissues of WT mice fed a high-saturated fat diet.
Since the loss of Brn-3b was associated with increased BW and hyperglycemia, we considered whether reduction of this protein might be linked to diet-induced obesity, which can also cause hyperglycemia and insulin resistance in rodent models (38). To test this hypothesis, we generated and analyzed a diet-induced obese mouse model in which WT mice, fed a diet containing 60% saturated fat (HFD) for >12 wk, were compared with control mice fed a ND. Most of the mice fed the HFD developed asignificant increase in BW compared with age-matched controls fed ND. However, one animal fed the ND developed obesity, whereas one animal fed the HFD did not show increased BW. Since these studies aimed to analyze the link between BW and Brn-3b, these two mice were excluded. At the end of these studies, a GTT was undertaken on fasted mice, during which blood glucose and serum insulin levels were analyzed at baseline (t = 0) and then at 30, 60, and 120 min after administration of a glucose bolus. Although there were no significant differences in blood glucose or serum insulin levels at baseline between these groups (Fig. 2A), differences were detected after 120 min when animals fed the ND showed an expected drop in blood glucose and serum insulin similar to baseline levels. However, animals fed the HFD demonstrated significantly elevated blood glucose, and this was associated with increased serum insulin levels [Fig. 2A(ii and iii)], similar to the effect seen in Brn-3bKO mutants.
To examine potential links between changes in Brn-3b expression and development of pathophysiological effects in these mice, we analyzed for protein expression in insulin-responsive tissues taken from mice fed a HFD or ND. Analyses were carried out using skeletal muscle, which contributes to postprandial glucose uptake and adipose tissue, which was considerably increased in animals fed on HFD. Figure 2B(i) shows protein levels in skeletal muscle, and as expected, Brn-3b(s) was highly expressed in tissues taken from ND-fed mice but was significantly reduced in those fed the HFD [Fig. 2B(ii)]. Adipose tissues expressed both Brn-3b isoforms, but higher levels of Brn-3b(l) were detected in mice fed the ND [Fig. 2B(iii)]. However, both isoforms were significantly reduced in tissues from HFD-fed mice. These results suggest that Brn-3b proteins are highly regulated in such metabolically active tissues and that its reduction may contribute to the hyperglycemia observed in obese WT animals or in KO mutants.
Brn-3b expression is regulated by glucose and insulin in C2C12 myoblasts.
Since loss or reduction of Brn-3b was associated with high serum insulin and hyperglycemia, we next tested whether Brn-3b expression was affected by glucose and/or insulin in C2C12 myoblasts. Therefore, cells were grown either in NG medium lacking glucose or NG medium supplemented with 25 mmol glucose in the absence of insulin (NG-GLU) or with added insulin (NG-GLU-INS). Control cells were grown in full growth medium (FGM). Figure 3A shows that Brn-3b mRNA was reduced in cells grown in glucose-free medium, and this was restored upon addition of 25 mmol glucose (Fig. 4A), whereas chronic insulin treatment (10 nmol for 24 h) downregulated Brn-3b mRNA, although insulin had no effect at early time points (1 or 4 h; data not shown). These results were confirmed by immunoblotting to analyze changes in protein levels. Although both Brn-3b isoforms were detectable in these cells, Brn-3b(l) was more abundant than Brn-3b(s) (Fig. 4B). Cells grown in NG medium had reduced levels of both proteins, with Brn-3b(s) being undetectable, but levels were restored and sustained by adding glucose to the NS medium. In contrast, addition of insulin caused reduction of both Brn-3b isoforms.
Fig. 3.

Brn-3b expression is regulated by glucose and insulin in myoblast-derived C2C12 cells: A: results of qRT-PCR showing relative changes in Brn-3b mRNA in C2C12 cells grown in full growth medium (FGM), no-glucose medium (NG), NG + 25 mmol glucose without insulin (NG-GLU), or with insulin (NG-GLU-INS). Values represent means ± SD from 3 independent experiments after Brn-3b levels were adjusted with house keeping gene GAPDH. B: Western blot analysis showing changes in Brn-3b protein in C2C12 cells grown in different media including FGM, NG, or with glucose added at different concentrations (as shown). Left: protein levels in the absence of insulin; right: similar treatments, but in the presence of insulin. The 2 different isoforms of Brn-3b are indicated (< >), and levels of tubulin protein were used to adjust for variation in protein loading. C: results of luciferase assays showing changes in activity of the Brn-3b promoter, transfected into C2C12 cells, which were treated as indicated. Values were equalized with Renilla luciferase activity (internal control), and data represent means ± SD from at least 3 independent experiments. * and **Statistically significant (P < 0.05, or < 0.01, respectively).
Fig. 4.
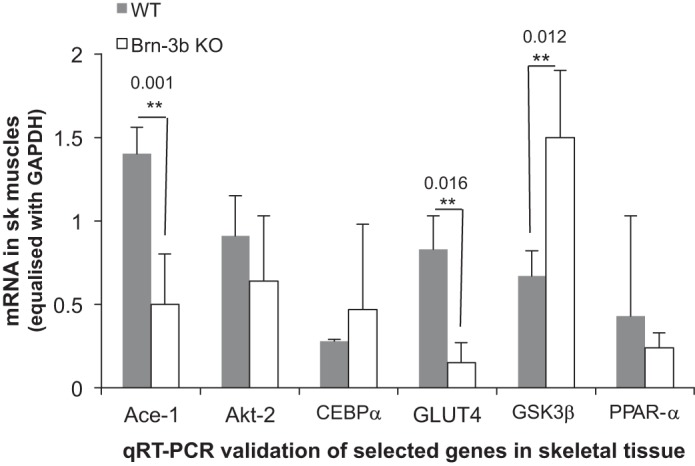
Brn-3b KO tissue displays significant changes in key metabolic genes. Results of qRT-PCR validate changes in selected genes in skeletal muscles taken from groups of >6 independent Brn-3bKO or WT mice. Means ± SE of gene expression are shown.
Since Brn-3b expression was stimulated by glucose but reduced by insulin treatment, we next tested whether these effects were mediated at the level of transcription of the Brn-3b gene by analyzing for changes in promoter activity. Therefore, C2C12 myoblasts were transfected with Brn-3b reporter construct and then transferred into different growth conditions. Cells were harvested after 24 h, and dual luciferase assays were used to measure promoter activity. Figure 3C shows that Brn-3b promoter activity was significantly repressed when cells were grown in medium lacking glucose (NG) compared with FGM, whereas addition of 25 mM glucose was sufficient to increase promoter activity (>60-fold) compared with NG; however, the promoter was repressed by insulin. These results suggest that Brn-3b expression is highly regulated by glucose and insulin in skeletal muscle.
Gene expression changes associated with loss of Brn-3b in skeletal muscle.
To identify gene expression changes in Brn-3bKO mice that might contribute to glucose intolerance, the disease-focused RT2 Profiler PCR 337021 Array was screened with cDNA synthesized using RNA extracted from skeletal muscle taken from mutant mice or age-matched WT littermates (see materials and methods). Table 1 shows a selection of genes that were either overexpressed or underexpressed in Brn-3bKO mutants compared with WT controls. To validate changes in selected genes, qRT-PCR was carried out using RNA from skeletal muscle taken from independent Brn-3b KO or WT mice. Figure 4 shows that statistically significant changes were reproduced for glycogen synthase kinase-3β (GSK3β) mRNA, which was increased in Brn-3b KO mutants, whereas angiotensin-1-converting enzyme (ACE) and the solute carrier family 4 glucose transporter (GLUT4) were both reduced in mutant tissue. However, the expected changes in other genes were not significant or reproducible. Therefore, these results suggest that changes in GSK3β, ACE, and GLUT4 may be closely linked to altered Brn-3b expression.
Table 1.
Selection of genes that show differential expression in RNA prepared from skeletal muscle taken from Brn-3b KO mutants compared with WT control littermates and used to screen the disease-focused RT2 Profiler PCR 337021 Array
| Gene | Definition | Fold Change |
|---|---|---|
| Overexpressed in Brn-3b KO vs. WT | ||
| Akt-2 | Thymoma viral protooncogene | (+) 321 |
| CEBPα | CCAAT/enhancer binding protein-α | (+) 35 |
| Ide | Insulin-degrading enzyme | (+) 24.9 |
| Pparα | Peroxisome proliferator-activated receptor-α | (+) 17.9 |
| Gsk3β | Glycogen synthase kinase-3β | (+) 12 |
| Irs1 | Insulin receptor substrate 1 | (+) 7 |
| Underexpressed in Brn-3b KO vs. WT | ||
| Ace | Angiotensin-1-converting enzyme | (−) 12 |
| Slc2a4 | Solute carrier family 4 (GLUT4) | (−) 5 |
KO, knockout; WT, wild type. Gene symbols and details of the genes that were significantly altered are shown alongside fold changes in expression.
Brn-3b binds to and activates GLUT4.
The profound dysfunction in glucose handling observed in Brn-3b KO mutants was similar to the impaired glucose response and insulin resistance reported for GLUT4+/− or GLUT4−/− mutants (13, 23). We therefore considered whether Brn-3b might affect glucose uptake by regulating the expression of this transporter. Genomatix Transfac analysis identified multiple V$BRNF sites in the mouse GLUT4 promoter sequences that could act as potential binding sites for Brn-3b transcription factor (Fig. 5A). These were clustered in two regions that were either proximal to the coding sequence (indicated by p1 and p2) or distal promoter sites (p3 and p4). Therefore, to test whether Brn-3b regulated transcription of this promoter, cotransfection studies were carried out in which the GLUT4 luciferase reporter was cotransfected with different concentrations of Brn-3b expression vectors in C2C12 cells. Results of the luciferase assays showed that at 1:1 ratio, Brn-3b transactivated the promoter by more than fourfold but that promoter activity was significantly enhanced (>12-fold) when Brn-3b was increased to 2:1 (Fig. 5B), confirming that Brn-3b does indeed regulate GLUT4 promoter activity.
Fig. 5.

Brn-3b can activate GLUT4 expression. A: schematic diagram to show positions of potential Brn-3b binding sites (V$BRNF) in the mouse GLUT4 promoter sequence as determined by Genomatix Transfac analysis for transcription factor binding sites. Primers were designed to span each of 2 clusters of binding sites, with p1 and p2 flanking the proximal sites while p3 and p4 flanked the sites at the distal end of the promoter. B: results of luciferase assays showing effect of cotransfecting different ratios of Brn-3b expression vector with GLUT4 promoter reporter construct (2:1 or 4:1) into C2C12 cells. Values were equalized with internal control, Renilla luciferase activity and expressed relative to the promoter activity with empty expression vector (LTR; set at 1). Data shown represent means ± SD from at least 3 independent experiments. C: PCR products obtained when using ChIP DNA immunoprecipitated with Brn-3b or GAPDH antibody (negative control). Primers 1 and 2 amplified ∼500-bp PCR product from the proximal promoter, whereas primers 3 and 4 were designed to amplify the distal region of the promoter. D(i): results of qRT-PCR showing changes in GLUT4 mRNA levels in skeletal muscle prepared from Brn-3b KO mutant mice (n = 5) compared with WT littermate controls (n = 4). Variability in total RNA from different tissues was equalized using GAPDH. (ii): representative immunoblots showing changes in protein expression in Brn-3b KO adipose tissue compared with WT controls. Quantification of GLUT4 protein expression in Brn-3b KO tissue and age-matched WT control was adjusted with invariant protein β-tubulin. *Statistical significance P < 0.01 (t-test analysis). E(i): representative immunoblot showing GLUT4 protein levels in untransfected control C2C12 (C) in cells transfected with 1 or 2 μg of Brn-3b expression with empty expression or empty vector (V). (ii): regression analysis showing relationship between GLUT4 and Brn-3b mRNA expression in C2C12 cells grown under different conditions (n > 6 independent points).
Since there were multiple potential binding sites spanning relatively large regions of the GLUT4 promoter, it was not feasible to undertake site-directed mutagenesis to identify specific site(s) required for Brn-3b-mediated promoter activity. Therefore, to test whether Brn-3b binds directly to the GLUT4 promoter in vivo in intact cells, we carried out chromatin immunoprecipitation (ChIP) assays (26) in which Brn-3b Abs were used to immunoprecipitate the protein bound to chromatin in intact C2C12 cells, and GAPDH antibody was used as a negative control for specificity. ChIP DNA isolated using Brn-3b Ab or GAPDH Ab were used for PCR, with primers designed to amplify the two regions of GLUT4 promoter that were enriched for Brn-3b binding sites; i.e., the proximal sites (primers 1 and 2) or distal sites (primers 3 and 4), and the PCR products were resolved with 2% agarose gel. As shown in Fig. 5C, amplification with primers 1 and 2 produced ∼500 bp products, which was observed in the positive control (input) sample and ChIP DNA immunoprecipitated with Brn-3b Ab but not control Ab. However, although primers 3 and 4 gave rise to ∼350 bp products with the input DNA, no significant bands were produced when the Brn-3b ChIP DNA was used. These results suggest that Brn-3b binds directly to one or more of the sites in the proximal GLUT4 promoter to activate gene expression.
To determine whether Brn-3b was necessary for maximal transcription of this gene, we next analyzed for changes in GLUT4 mRNA in insulin-responsive tissues such as skeletal muscle and adipose tissue taken from Brn-3b KO mice or age-matched WT controls. Therefore, RNA from KO mutants or WT tissues was used for cDNA synthesis and quantification of GLUT4 transcripts using qRT-PCR. Figure 5D(i) shows that GLUT4 mRNA was significantly lower in skeletal muscle taken from Brn-3b KO mutants compared with levels in WT tissues, confirming that Brn-3b is necessary for transcription of this gene. To test whether reduced GLUT4 transcripts in Brn-3b KO mutants also affected protein levels, immunoblotting was carried out using total cellular protein extracted from insulin-responsive tissues from Brn-3b KO and WT controls. The representative immunoblots in Fig. 5D(ii) show that the GLUT4 protein was reduced in Brn-3b KO adipose tissues, with ∼50% less protein compared with controls [Fig. 5D(iii)].
We next tested whether overexpressing Brn-3b affected basal GLUT4 protein levels in myoblasts by transfecting C2C12 cells with an expression vector containing Brn-3b cDNA (at 1 or 2 μg). Untransfected control cells (C) or cells transfected with the empty expression vector (V) were used as controls, and after 48 h protein extracts from transfected cells were used for immunoblotting to analyze for changes in GLUT4 and Brn-3b expression. Figure 5E shows a representative immunoblot from these experiments and demonstrates increased Brn-3b protein in cells transfected with the Brn-3b expression vector, which correlated with significant upregulation of GLUT4 protein compared with control cells. Furthermore, regression analysis showed a statistically significant correlation between Brn-3b expression and GLUT4 mRNA expression in C2C12 cells grown under different conditions e.g., FGM, NG, or NG + glucose [Fig. 5E(ii)]. These results strongly support Brn-3b as a key regulator of GLUT4 in insulin-responsive cells/tissues.
DISCUSSION
Hyperglycemia, which is commonly associated with insulin resistance, T2D, and obesity, can drive pathological processes that contribute to comorbidities such as cardiovascular diseases, renal dysfunction, and neurological damage (1, 14, 29). These effects result from complex molecular changes that are triggered in different tissues and which lead to the development of pathological changes and disease progression, but the factors that control such changes are not fully understood (36). Hyperglycemia, insulin resistance, and T2D are set to rise, in line with a predicted global obesity epidemic that is linked to dietary and lifestyle changes (11, 25, 36). Therefore, it is important to understand the molecular mechanisms that drive pathological changes, since this may help to identify strategies that could be used to block or reverse tissue damage, thereby minimizing morbidity and mortality.
In this regard, we have identified Brn-3b as a novel regulator of glucose homeostasis. The first evidence suggesting that this transcription factor may be involved in regulating metabolic processes was inferred from microarray analysis, which showed that Brn-3b was significantly altered in skeletal muscle taken from individuals with high or low insulin sensitivity following endurance training (37). Results from our studies have shown that Brn-3b protein is expressed in insulin-responsive tissues such as skeletal muscle and adipose tissue as well as in liver but has not been detected in the pancreas. Furthermore, higher body weights in constitutive Brn-3b KO mice are associated with increased fat deposition in the abdominal cavity despite no statistically significant changes in body length or differences in calorie consumption compared with control mice. Brn-3b KO mutants also developed persistent hyperglycemia in the presence of high serum insulin levels, suggesting that the pancreas continues to produce insulin and that high blood glucose may be linked to defects in insulin responsiveness and/or glucose uptake in tissues such as skeletal muscle and adipose tissues. In line with this, Brn-3b expression is also reduced in skeletal muscle and adipose tissues taken from WT mice that develop obesity and hyperglycemia/insulin resistance after being fed a high-saturated-fat diet. These results suggest that Brn-3b is dynamically regulated in such metabolically active tissues and that the metabolic dysfunction in constitutive KO mutants may result from direct loss of this transcription factor in insulin-responsive tissues.
Two distinct Brn-3b isoforms have previously been described (3, 8), and we have shown that Brn-3b(s) is the predominant isoform in skeletal muscle whereas Brn-3b(l) is more abundant in liver and adipose tissues. Moreover, both isoforms are regulated in response to high-fat diet. Although the functions of these distinct isoforms are still to be elucidated, both proteins can be regulated by similar signals but can also regulate each other's expressions (8, 17). Therefore, it will be interesting to investigate potential tissue-specific effects for distinct Brn-3b isoforms in insulin-responsive tissues in later studies.
These results have provided evidence to support potential roles for the Brn-3b transcription factor in controlling metabolic function and have been reinforced by findings in C2C12 myoblast cells, which show that Brn-3b can, itself, be regulated by insulin and metabolites such as glucose. In this regard, Brn-3b mRNA and proteins are reduced in cells grown in low glucose media or in the presence of insulin but can be restored by addition of physiological levels of glucose (25 mM). These effects are likely to be mediated at the level of transcription, since low glucose or insulin can repress the Brn-3b promoter, whereas addition of glucose strongly stimulated promoter activity. Such tight regulation of Brn-3b by insulin and glucose indicate that this transcription factor will have important role(s) for regulating genes associated with glucose uptake.
In line with this, analysis of differential gene expression in skeletal muscle taken from Brn-3b KO and WT mice have identified changes in key genes that are associated with metabolic processes. For example, increased GSK3β in Brn-3b KO tissues may contribute to insulin resistance by phosphorylating and inhibiting key molecules such as glycogen synthase and insulin receptor substrate proteins, and importantly, elevated GSK3β has been detected in skeletal muscle of patients with T2D (10, 16, 30). On the other hand, reduced GLUT4 in tissues taken from Brn-3b KO mice may underlie the glucose intolerance detected in these mutants, since Brn-3b directly regulates transcription of GLUT4 transporter. Thus, identification of potential Brn-3b binding sites by bioinformatic analysis and cotransfection studies combined with reporter assays have confirmed that increasing exogenous Brn-3b can strongly transactivate the GLUT4 promoter in C2C12 cells. The presence of multiple sites, clustered in two regions of the GLUT4 promoter, means that it has not been possible to use site-directed mutagenesis to identify the specific sequence(s) required for Brn-3b binding and promoter activation. However, ChIP analysis has conclusively demonstrated that Brn-3b binds directly to the proximal sites in the GLUT4 promoter in vivo in intact myoblast cells. There is also strong evidence for a significant correlation between Brn-3b and GLUT4 expression from in vivo and in vitro studies. Thus, GLUT4 mRNA and protein are reduced in insulin-responsive tissues (skeletal muscle and adipose) taken from Brn-3b KO mutant mice, and altered Brn-3b correlates well with GLUT4 expression in C2C12 myoblasts grown under different conditions. In addition, overexpressing Brn-3b in C2C12 cells is sufficient to increase basal GLUT4 levels.
These findings strongly suggest that Brn-3b may be a key regulator of GLUT4 expression in insulin-responsive tissues and that loss of this transporter may contribute, in part, to the hyperglycemia and insulin resistance observed in Brn-3b KO mutants or in obese mice with reduced Brn-3b expression. The GLUT4 transporter is important for insulin-mediated glucose uptake in adipose tissues and skeletal muscle, which accounts for the majority of postprandial glucose utilization (27). Although GLUT4 translocation from intracellular vesicles to the cell membrane is an important mechanism for regulating glucose uptake, changes in expression levels have also been implicated in inefficient glucose utilization. Thus, reduced GLUT4 expression in heterozygote mice is sufficient to cause defective glucose handling, since mutant mice develop severe peripheral insulin resistance, which can be reversed by recapitulating expression in skeletal muscle (32, 35). Although the relevance of reduced GLUT4 in humans with non-insulin-dependent T2D has been brought into question more recently (31), the clear correlation between Brn-3b and GLUT4 expression in insulin-responsive tissues, in these studies strongly suggests that this transcription factor will be important for regulating the expression of this glucose transporter in different tissues. However, it is likely that GLUT4 represents one of multiple target genes that are regulated by the Brn-3b transcription factor in insulin-responsive tissue, which may contribute to the complex phenotypes seen in the constitutive KO mutants.
Thus, Brn-3b represents a novel and important regulator of metabolic processes in insulin-responsive tissues. However, since its expression is also controlled by circulating levels of metabolites and insulin, this transcription factor may act as part of a feedback loop that maintains glucose homoeostasis under normal conditions.
GRANTS
This research was funded by the Dunhill Medical Trust (SA24/0712), the British Heart Foundation (PG/08/074/25533 and FS/14/32/30729), and the Rosetrees Trust.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: S.B., H.A., L.M., and S.O. performed experiments; S.B., H.A., and S.O. analyzed data; S.B., H.A., and V.S.B.-M. interpreted results of experiments; S.B., H.A., and V.S.B.-M. prepared figures; S.B., H.A., V.M.-A., and V.S.B.-M. edited and revised manuscript; V.M.-A. and V.S.B.-M. conception and design of research; V.S.B.-M. drafted manuscript; V.S.B.-M. approved final version of manuscript.
ACKNOWLEDGMENTS
We are grateful to Prof. M. Quon, University of Maryland, Baltimore, and Prof. E. Karnieli, Rambam Medical Center, Haifa, Israel, for providing the GLUT4 promoter constructs and to Dr. Gautam Mehta and Katayoon Gardner for technical assistance.
REFERENCES
- 1.Belkina AC, Denis GV. Obesity genes and insulin resistance. Curr Opin Endocrinol Diabetes Obes 17: 47477, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boyle PJ. Diabetes mellitus and macrovascular disease: mechanisms and mediators. Am J Med 120: S12–S17, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Budhram-Mahadeo V, Moore A, Morris PJ, Ward T, Weber B, Sassone-Corsi P, Latchman DS. The closely related POU family transcription factors Brn-3a and Brn-3b are expressed in distinct cell types in the testis. Int J Biochem Cell Biol 33: 1027–1039, 2001. [DOI] [PubMed] [Google Scholar]
- 4.Budhram-Mahadeo V, Morris PJ, Latchman DS. The Brn-3a transcription factor inhibits the pro-apoptotic effect of p53 and enhances cell cycle arrest by differentially regulating the activity of the p53 target genes encoding Bax and p21(CIP1/Waf1). Oncogene 21: 6123–6131, 2002. [DOI] [PubMed] [Google Scholar]
- 5.Budhram-Mahadeo V, Ndisang D, Ward T, Weber BL, Latchman DS. The Brn-3b POU family transcription factor represses expression of the BRCA-1 anti-oncogene in breast cancer cells. Oncogene 18: 6684–6691, 1999. [DOI] [PubMed] [Google Scholar]
- 6.Budhram-Mahadeo VS, Irshad S, Bowen S, Lee SA, Samady L, Tonini GP, Latchman DS. Proliferation-associated Brn-3b transcription factor can activate cyclin D1 expression in neuroblastoma and breast cancer cells. Oncogene 27: 145–154, 2008. [DOI] [PubMed] [Google Scholar]
- 7.Budhram-Mahadeo VS, Latchman DS. Targeting Brn-3b in breast cancer therapy. Expert Opin Ther Targets 10: 15–25, 2006. [DOI] [PubMed] [Google Scholar]
- 8.Dennis JH, Budhram-Mahadeo V, Latchman DS. The Brn-3b POU family transcription factor regulates the cellular growth, proliferation, and anchorage dependence of MCF7 human breast cancer cells. Oncogene 20: 4961–4971, 2001. [DOI] [PubMed] [Google Scholar]
- 9.Desvergne B, Michalik L, Wahli W. Transcriptional regulation of metabolism. Physiol Rev 86: 465–514, 2006. [DOI] [PubMed] [Google Scholar]
- 10.Eldar-Finkelman H, Krebs EG. Phosphorylation of insulin receptor substrate 1 by glycogen synthase kinase 3 impairs insulin action. Proc Natl Acad Sci USA 94: 9660–9664, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Freeman H, Cox RD. Type-2 diabetes: a cocktail of genetic discovery. Hum Mol Genet 15, Spec No 2: R202–R209, 2006. [DOI] [PubMed] [Google Scholar]
- 12.Fujita R, Ounzain S, Wang AC, Heads RJ, Budhram-Mahadeo VS. Hsp-27 induction requires POU4F2/Brn-3b TF in doxorubicin-treated breast cancer cells, whereas phosphorylation alters its cellular localisation following drug treatment. Cell Stress Chaperones 2011. [DOI] [PMC free article] [PubMed]
- 13.Giacchetti G, Faloia E, Taccaliti A, Morosini PP, Arnaldi G, Soletti F, Mantero F, Accili D, De PR. Decreased expression of insulin-sensitive glucose transporter mRNA (GLUT-4) in adipose tissue of non-insulin-dependent diabetic and obese patients: evaluation by a simplified quantitative PCR assay. J Endocrinol Invest 17: 709–715, 1994. [DOI] [PubMed] [Google Scholar]
- 14.Goran MI, Ball GD, Cruz ML. Obesity and risk of type 2 diabetes and cardiovascular disease in children and adolescents. J Clin Endocrinol Metab 88: 1417–1427, 2003. [DOI] [PubMed] [Google Scholar]
- 15.Grundy SM, Benjamin IJ, Burke GL, Chait A, Eckel RH, Howard BV, Mitch W, Smith SC Jr Sowers JR. Diabetes and cardiovascular disease: a statement for healthcare professionals from the American Heart Association. Circulation 100: 1134–1146, 1999. [DOI] [PubMed] [Google Scholar]
- 16.Henry RR, Ciaraldi TP, Mudaliar S, Abrams L, Nikoulina SE. Acquired defects of glycogen synthase activity in cultured human skeletal muscle cells: influence of high glucose and insulin levels. Diabetes 45: 400–407, 1996. [DOI] [PubMed] [Google Scholar]
- 17.Irshad S, Pedley RB, Anderson J, Latchman DS, Budhram-Mahadeo V. The Brn-3b transcription factor regulates the growth, behavior, and invasiveness of human neuroblastoma cells in vitro and in vivo. J Biol Chem 279: 21617–21627, 2004. [DOI] [PubMed] [Google Scholar]
- 18.Kadonaga JT. Regulation of RNA polymerase II transcription by sequence-specific DNA binding factors. Cell 116: 247–257, 2004. [DOI] [PubMed] [Google Scholar]
- 19.Kadowaki T. Insights into insulin resistance and type 2 diabetes from knockout mouse models. J Clin Invest 106: 459–465, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kahn BB, Charron MJ, Lodish HF, Cushman SW, Flier JS. Differential regulation of two glucose transporters in adipose cells from diabetic and insulin-treated diabetic rats. J Clin Invest 84: 404–411, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kahn CR, Vicent D, Doria A. Genetics of non-insulin-dependent (type-II) diabetes mellitus. Annu Rev Med 47: 509–531, 1996. [DOI] [PubMed] [Google Scholar]
- 22.Kamide K, Nagano M, Nakano N, Yo Y, Kobayashi R, Rakugi H, Higaki J, Ogihara T. Insulin resistance and cardiovascular complications in patients with essential hypertension. Am J Hypertens 9: 1165–1171, 1996. [DOI] [PubMed] [Google Scholar]
- 23.Kouidhi S, Berrhouma R, Rouissi K, Jarboui S, Clerget-Froidevaux MS, Seugnet I, Bchir F, Demeneix B, Guissouma H, Elgaaied AB. Human subcutaneous adipose tissue Glut 4 mRNA expression in obesity and type 2 diabetes. Acta Diabetol 2011. [DOI] [PubMed]
- 24.Latchman DS. Gene Regulation: A Eukaryotic Perspective. 2002.
- 25.Lazar MA. How obesity causes diabetes: not a tall tale. Science 307: 373–375, 2005. [DOI] [PubMed] [Google Scholar]
- 26.Lee SA, Ndisang D, Patel C, Dennis JH, Faulkes DJ, D'Arrigo C, Samady L, Farooqui-Kabir S, Heads RJ, Latchman DS, Budhram-Mahadeo VS. Expression of the Brn-3b transcription factor correlates with expression of HSP-27 in breast cancer biopsies and is required for maximal activation of the HSP-27 promoter. Cancer Res 65: 3072–3080, 2005. [DOI] [PubMed] [Google Scholar]
- 27.Leto D, Saltiel AR. Regulation of glucose transport by insulin: traffic control of GLUT4. Nat Rev Mol Cell Biol 13: 383–396, 2012. [DOI] [PubMed] [Google Scholar]
- 28.Liu YZ, Dawson SJ, Latchman DS. Alternative splicing of the Brn-3a and Brn-3b transcription factor RNAs is regulated in neuronal cells. J Mol Neurosci 7: 77–85, 1996. [DOI] [PubMed] [Google Scholar]
- 29.Matheus AS, Tannus LR, Cobas RA, Palma CC, Negrato CA, Gomes MB. Impact of diabetes on cardiovascular disease: an update. Int J Hypertens 2013: 653789, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nikoulina SE, Ciaraldi TP, Mudaliar S, Carter L, Johnson K, Henry RR. Inhibition of glycogen synthase kinase 3 improves insulin action and glucose metabolism in human skeletal muscle. Diabetes 51: 2190–2198, 2002. [DOI] [PubMed] [Google Scholar]
- 31.Pedersen O, Bak JF, Andersen PH, Lund S, Moller DE, Flier JS, Kahn BB. Evidence against altered expression of GLUT1 or GLUT4 in skeletal muscle of patients with obesity or NIDDM. Diabetes 39: 865–870, 1990. [DOI] [PubMed] [Google Scholar]
- 32.Rossetti L, Stenbit AE, Chen W, Hu M, Barzilai N, Katz EB, Charron MJ. Peripheral but not hepatic insulin resistance in mice with one disrupted allele of the glucose transporter type 4 (GLUT4) gene. J Clin Invest 100: 1831–1839, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Samady L, Dennis J, Budhram-Mahadeo V, Latchman DS. Activation of CDK4 Gene Expression in Human Breast Cancer Cells by the Brn-3b POU Family Transcription Factor. Cancer Biol Ther 3: 317–323, 2004. [PubMed] [Google Scholar]
- 34.Sivitz WI, DeSautel SL, Kayano T, Bell GI, Pessin JE. Regulation of glucose transporter messenger RNA in insulin-deficient states. Nature 340: 72–74, 1989. [DOI] [PubMed] [Google Scholar]
- 35.Stenbit AE, Tsao TS, Li J, Burcelin R, Geenen DL, Factor SM, Houseknecht K, Katz EB, Charron MJ. GLUT4 heterozygous knockout mice develop muscle insulin resistance and diabetes. Nat Med 3: 1096–1101, 1997. [DOI] [PubMed] [Google Scholar]
- 36.Taubes Insulin resistance G. Prosperity's plague. Science 325: 256–260, 2009. [DOI] [PubMed] [Google Scholar]
- 37.Teran-Garcia M, Rankinen T, Koza RA, Rao DC, Bouchard C. Endurance training-induced changes in insulin sensitivity and gene expression. Am J Physiol Endocrinol Metab 288: E1168–E1178, 2005. [DOI] [PubMed] [Google Scholar]
- 38.Winzell MS, Ahren B. The high-fat diet-fed mouse: a model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes 53, Suppl 3: S215–S219, 2004. [DOI] [PubMed] [Google Scholar]