

Abstract
17β-Estradiol (estradiol) inhibits microglia proliferation. 2-Methoxyestradiol (2-ME) is an endogenous metabolite of estradiol with little affinity for estrogen receptors (ERs). We hypothesize that 2-ME inhibits microglial proliferation and activation and contributes to estradiol's inhibitory effects on microglia. We compared the effects of estradiol, 2-hydroxyestradiol [2-OE; estradiol metabolite produced by cytochrome P450 (CYP450)], and 2-ME [formed by catechol-O-methyltransferase (COMT) acting upon 2-OE] on microglial (BV2 cells) DNA synthesis, cell proliferation, activation, and phagocytosis. 2-ME and 2-OE were approximately three- and 10-fold, respectively, more potent than estradiol in inhibiting microglia DNA synthesis. The antimitogenic effects of estradiol were reduced by pharmacological inhibitors of CYP450 and COMT. Inhibition of COMT blocked the conversion of 2-OE to 2-ME and the antimitogenic effects of 2-OE but not 2-ME. Microglia expressed ERβ and GPR30 but not ERα. 2,3-Bis(4-hydroxyphenyl)-propionitrile (ERβ agonist), but not 4,4′,4′′-(4-propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol (ERα agonist) or G1 (GPR30 agonist), inhibited microglial proliferation. The antiproliferative effects of estradiol, but not 2-OE or 2-ME, were partially reversed by ICI-182,780 (ERα/β antagonist) but not by 1,3-bis(4-hydroxyphenyl)-4-methyl-5-[4-(2-piperidinylethoxy)phenol]-1H-pyrazole (ERα antagonist) or G15 (GPR30 antagonist). Lipopolysaccharide increased microglia iNOS and COX-2 expression and phagocytosing activity of microglia; these effects were inhibited by 2-ME. We conclude that in microglia, 2-ME inhibits proliferation, proinflammatory responses, and phagocytosis. 2-ME partially mediates the effects of estradiol via ER-independent mechanisms involving sequential metabolism of estradiol to 2-OE and 2-ME. 2-ME could be of potential therapeutic use in postischemic stroke injuries. Interindividual differences in estradiol metabolism might affect the individual's ability to recover from stroke.
Keywords: stroke, microglia, 17β-estradiol, 2-hydroxyestradiol, 2-methoxyestradiol
evidence suggests that ovarian hormones like 17β-estradiol (estradiol) protect against stroke (20). For example, compared with age-matched men, the incidence of stroke is rare in premenopausal women; however, following the onset of menopause the incidence of stroke increases. The protective role of estradiol is also supported by the fact that the incidence of stroke is low during pregnancy and higher following delivery when estradiol levels fall dramatically (31). Similarly, stroke risk is increased in premenopausal women who have undergone bilateral oophorectomy but are not treated with estradiol (31). Animal experiments also provide strong evidence that estradiol is protective against ischemic stroke (31). However, the timing of estradiol delivery pre- or poststroke seems to be critical in defining its protective actions (25), and some studies suggest that estradiol treatment actually worsens poststroke outcomes (23). Since multiple factors and cells contribute to the pathophysiology of ischemic stroke, understanding the mechanisms of estradiol's actions in the brain may provide leads for developing therapeutic agents against stroke.
Estradiol is known to exert anti-inflammatory effects, and animal studies suggest that this may contribute to the neuroprotective effects of estradiol (7, 11, 24, 31, 32). Irreversible activation of microglia from the quiescent state results in increased release of proinflammatory cytotoxic molecules and phagocytosing activity and is associated with neurodegeneration (4, 29). Increases in microglia number and activation are known to occur following stroke (35). Interestingly, via estrogen receptor-α (ERα) and ERβ, estradiol abrogates the release of proinflammatory molecules from lipopolysaccharide (LPS)-activated microglia (2, 5), suggesting that estradiol may induce poststroke protection via this mechanism.
Recent studies provide evidence that 2-methoxyestradiol (2-ME), an endogenous metabolite of estradiol with little affinity for ERs, is biologically active and of pathophysiological relevance (13). Our previous studies show that 2-ME inhibits vascular smooth muscle cell proliferation and migration and is protective against multiple proliferative disorders (12). Indeed, 2-ME is a potential therapeutic agent that is protective against cardiovascular disease and multiple proliferative and immune disorders such as cancer and rheumatoid arthritis (12, 28). Importantly, 2-ME inhibits experimental autoimmune encephalomyelitis via suppression of lymphocyte activation (16), induces neuroprotection following traumatic brain injury by inhibiting maladaptive HIF-1α responses (33), and reduces brain infarct size in a middle cerebral artery occlusion-induced focal ischemia rat model (8). Decreased levels of 2-ME are observed in patients with Parkinson's disease (18), and significant increases in 2-ME levels during the last months of pregnancy seem to correlate temporally with remission of clinical symptoms in some pregnant multiple sclerosis and rheumatoid arthritis patients (27, 28). Together these findings suggest that 2-ME may attenuate poststroke damaging cellular processes.
We hypothesize that 2-ME may induce its neuroprotective actions by inhibiting microglia proliferation and activation. The rationale for this hypothesis is as follows: 1) abnormal proliferation and activation of microglia are thought to contribute to neuronal damage following ischemic injury/stroke (29); 2) the microglial reaction parallels the severity of neuronal damage in an animal model of stroke (40); and 3) suppression of microglial activation after ischemic injury is beneficial for the healing process (9, 38). Based on the fact that estradiol is synthesized in the brain (1), and the brain expresses the enzymes cytochrome P4501B1 (CYP1B1) and catechol-O-methyltransferase (COMT) (17, 30), the enzymes responsible for the sequential metabolism of estradiol to 2-hydroxyestradiol (2-OE) and 2-ME, we also hypothesize that 2-ME mediates in part the actions of estradiol on microglia (Fig. 1).
Fig. 1.
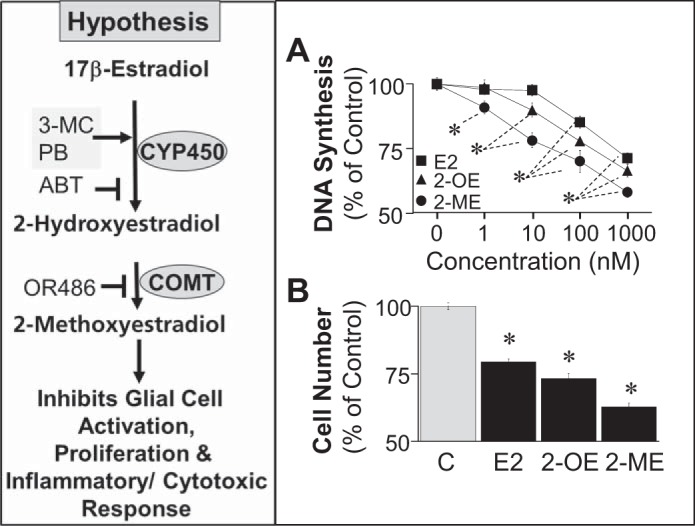
Schematic at left illustrates our hypothesis that the inhibitory effects of 17β-estradiol (E2) on BV2 cell proliferation and activity may be mediated in part via the sequential metabolism of E2 to 2-hydroxyestradiol (2-OE) and 2-methoxyestradiol (2-ME) by cytochrome P450 (CYP450) and catechol-O-methyltransferase (COMT), respectively. ABT, 1-aminobenzotriazole; PB, phenobarbital; 3-MC, 3-methylcholantherene; OR486, COMT inhibitor. →, Inducer; ⊣, inhibitor. A: effects of increasing concentrations (1–1,000 nmol/l) of E2, 2-OE, and 2-ME on 2.5% FBS-induced DNA synthesis ([3H]thymidine incorporation) by microglia. B: effects of E2, 2-OE, and 2-ME (100 nmol/l) on microglia proliferation (cell no.). Results are presented as means ± SE (n = 4). *P < .05 vs. control (C).
To test our hypothesis, in the present study we investigated the effects of estradiol, 2-OE, and 2-ME on the proliferation and activation of microglia. Moreover, using selective inhibitors and inducers, we investigated the role of estradiol metabolism in mediating estradiol's effects on microglia. Furthermore, we assessed the role of ER-dependent and ER-independent pathways in mediating the biological actions of estradiol on the proliferation and activation of microglia. Additionally, using LPS stimulation, we investigated whether 2-ME attenuates microglia-mediated inflammation, phagocytosis, and neuronal cytotoxicity.
MATERIALS AND METHODS
Growth studies.
The BV2 cell line was established to study microglial cells in vitro (19), and BV2 cells retain most of their phenotypical as well as morphological and functional properties (e.g., proliferation upon stimulation, phagocytosing ability, and constitutive as well as inducible and secretory functions). BV2 cells were cultured under standard tissue culture conditions in DMEM (Gibco, Paisley, UK) supplemented with 10% fetal bovine serum (FBS; Gibco), glutamine (Sigma-Aldrich, St. Louis, MO), and 1× antibiotic-antimycotic (Gibco). Subconfluent cells were growth arrested in phenol red-free medium containing 0.1% charcoal-stripped FBS (Hyclone, MA) for 24 h. Cell growth was induced by 2.5% FBS in the presence or absence of ICI-182,780 (ERα/β antagonist, 5 μmol/l; Tocris, Ballwin, MO), 1,3-bis(4-hydroxyphenyl)-4-methyl-5-[4-(2-piperidinylethoxy)phenol]-1H-pyrazole (MPP; ERα antagonist, 5 nmol/l; OBITER Research, Champaign, IL), G1 (GPR30 agonist; Cayman Chemical, Ann Arbor, MI), G15 (GPR30 antagonist, 100 nmol/l; Cayman Chemical), 1H-benzotriazol-1-amine (ABT; cytochrome P450 inhibitor, 5 μmol/l; Sigma-Aldrich), OR486 (COMT inhibitor, 5 μmol/l, Tocris), phenobarbital (cytochrome P450 inducer, 5 μmol/l; Sigma-Aldrich), 3-methylcholanthrene (3-MC; cytochrome P450 inducer, 5 μmol/l; Sigma-Aldrich), ellipticine (CYP1A1 inhibitor, 10 μmol/l; Sigma-Aldrich), ketoconazole (CYP3A4; 10 μmol/l; Sigma-Aldrich), pyrene (CYP1B1 inhibitor, 5 nmol/l; Sigma-Aldrich), 17β-estradiol (Steraloids, Newport, RI), 2-OE (Steraloids), 2-methoxyestradiol (Steraloids), 2,3-bis(4-hydroxyphenyl)-propionitrile (DPN; ERβ agonist; Tocris), or 4,4′,4′′-(4-propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol (PPT; ERα agonist; Tocris). 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT; Sigma-Aldrich) was used for cell viability assay. DNA synthesis ([3H]thymidine incorporation) and cell proliferation (cell no.) experiments were determined as described previously by us (15).
Western blotting.
To investigate the expression of ERα/β and to analyze cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) expression, BV2 cells were grown to subconfluence, treated as indicated, lysed, and analyzed by Western blotting using specific antibodies against ERα (Alexis, Lausen, CH 210-201-C050, no cross-reactivity with ERβ), ERβ (Alexis, 210-180-C050, no cross-reactivity with ERα), COX-2 (Cayman Chemical), iNOS (2977; Cell Signaling Technology, Danvers, MA), GPR30 (39742; Abcam, Cambridge, MA), or CYP1B1 (GTX104424; GeneTex, Irvine, CA). The antibody against β-actin (A5441; Sigma-Aldrich) was used as a loading control.
High-performance liquid chromatography analysis.
Formation of estradiol metabolites in cells was measured by high-performance liquid chromatography (HPLC) analysis after extraction from the culture medium, as described previously (39), with minor modifications. Confluent cells were treated with 5 μmol/l 2-OE in the presence or absence of 10 μmol/l OR486 in DMEM supplemented with 10 mmol/l HEPES buffer, 0.1% sodium bicarbonate, and 1 mmol/l ascorbic acid (to avoid oxidation of estradiol metabolites). The medium was harvested after 2 h of treatment, and 16-α-hydroxyestradiol (100 μl of 2.5 μmol/l) was added as an internal standard. Steroids were extracted with chloroform, dried, resuspended in H2O-MeOH (80:20) and analyzed by HPLC [C-8 reversed-phase column, mobile phase gradient H2O-MeOH (80:20 to 20:80 in 30 min), UV detection at 280 nm].
Radioassay for conversion of estradiol to 2-OE.
To demonstrate that BV2 cells can metabolize estradiol to 2-OE, we used 17β-[2-3H(N)]estradiol, which upon hydroxylation to 2-OE releases [3H]H2O in a stoichiometric fashion (6,14). Measuring radiolabeled [3H]H2O formation is well established to provide a reliable estimate for the conversion of estradiol to 2-OE (6, 14). Briefly, BV2 cells grown to 80% confluence in 33-mm culture dishes were pretreated for 24 h with 5 μmol/l of 3-MC and subsequently fed 2 ml of medium supplemented with 1 μCi/ml of 17β-[2-3H(N)]estradiol (specific activity, 20 Ci/mmol; ANAWA/American Radiolabeled Chemicals, St. Louis, MO) in the presence of ABT or pyrene. After 36 h the supernatants were collected, and the protein content was precipitated by adding an equal volume (1:1) of 60% perchloric acid. The samples were neutralized with 6 N NaOH. Activated charcoal was added to 2-ml aliquots of each supernatant (final concentration: 1 mg/ml), and the samples were incubated overnight with gentle shaking at 4°C. Subsequently, 100 μl of protamine sulfate (10 mg/ml) was added to the supernatants, and the samples were centrifuged at 3,000 g at 4°C. Next, 2 ml of supernatant was passed through activated C18-Bond Elut Columns (Varian, Lake Forest, CA) and collected, and the amount of [3H]H2O formed was assayed by counting in 500-μl aliquots on a β-scintillation counter. 17β-[2-3H(N)]estradiol incubated in the absence of cells and processed in parallel served as the control for subtracting the background counts after extraction. To investigate whether BV2 expresses CYP1B1, cell lysates from cultured BV2 cells were analyzed by Western blotting and probed with antibodies to CYP1B1.
Phagocytosis assay.
Phagocytosis was measured using the pH-Rodo BioParticle Conjugates assay (Invitrogen) according to the manufacturer's protocols. Briefly, BV2 cells were plated in 96-well plates (50,000 cells/well). After overnight incubation, 10 μl of pH-Rodo Bioparticle suspension was added to each well. The plate was incubated at 37°C under standard tissue culture conditions, and the fluorescence was determined by employing the Tecan Spectrofluor Plus reader (Tecan Group, Männedorf, Switzerland) using 540 nm excitation and 595 nm emission. Fluorescence microscopy was also employed to monitor and confirm the uptake of Bioparticles under various treatment conditions.
Nitrite assay.
Nitrite secretion into the supernatant was measured using the Griess reagent [5 g of sulfanilamide, 0.5 g of ethylenediamine dihydrochloride, 29.41 ml of phosphoric acid (H3PO4), and 570.59 ml of H2O]. Fifty microliters of the Griess reagent was added to 50 μl of medium. The mixture was incubated at room temperature for 20 min, and the absorbance at 540 nm was analyzed using the Spectrofluormeter Plus reader.
Cell viability assay.
Cells were plated overnight in a 96-well plate (10,000 cells/well) and allowed to attach. The attached cells were treated with test compounds for different times. Subsequently, the medium was replaced with fresh medium containing 0.5 mg/ml of MTT and the test compounds. After additional incubation for 3–4 h, the medium was aspirated, and the cells were lysed by the adding 200 μl of DMSO. The absorbance was quantitated at 540 nm using a Tecan Spectrofluorometer Plus reader.
BV2 activation and neuroblastoma cell toxicity.
To assess whether factors released from LPS-activated microglia induce neuronal toxicity and whether these actions are abrogated by 2-ME, we conducted “no-contact” coculture experiments. Briefly, conditioned medium from BV2 cells activated with LPS (1 μg/ml) in the presence or absence of different concentrations of 2-ME (1-1,000 nmol/l) for 24 h was collected and layered on top of cultured SH-SY5Y neuroblastoma cells (kindly provided by Prof. Michael Weller, ETH Zurich, Switzerland). After 24 h the viability of SH-SY5Y cells was measured by MTT assay. To confirm that the conditioned medium was devoid of 2-ME, we analyzed 2-ME levels in the medium of BV2 cells treated under identical conditions, and they were not detectable. Moreover, direct treatment of SH-SY5Y cells with 2-ME in presence of conditioned medium from LPS-stimulated BV2 cells failed to prevent neuronal toxicity.
Statistics.
Experiments were performed in quadruplicate. Data were analyzed using analysis of variance, and post hoc analyses were calculated using the Fisher's least significant difference test. The criterion of significance was P < 0.05. All data are expressed as means ± SE.
RESULTS
In growth-arrested BV2 cells, treatment with estradiol and its metabolites 2-OE and 2-ME inhibited FBS-induced DNA synthesis (Fig. 1A) and cell number (Fig. 1B). The relative potencies for inhibition of FBS-induced growth were 2-ME > 2-OE > estradiol. In this regard, the lowest concentrations of 2-ME, 2-OE, and estradiol that significantly inhibited DNA synthesis were 1, 10, and 100 nmol/l, respectively. At a concentration of 100 nmol/l, DNA synthesis was significantly inhibited by estradiol, 2-OE, and 2-ME by 14.9, 22.2, and 30.1%, respectively, and proliferation was inhibited by 20.6, 26.8, and 37.3%, respectively.
The effects of estradiol (100 nmol/l) on DNA synthesis (Fig. 2A) and cell proliferation (Fig. 2B) were significantly decreased by the broad-spectrum CYP450 inhibitor ABT (5 μmol/l) and by the COMT inhibitor OR486 (5 μmol/l) yet significantly augmented by the CYP450 inducers phenobarbital (5 μmol/l) and 3-MC (5 μmol/l). In this regard, ABT and OR486 significantly decreased the ability of estradiol to inhibit FBS-induced DNA synthesis from 16.5 to 8.5 and 9.4%, respectively, and FBS-induced proliferation from 21 to 7.2 and 10.3%, respectively. In contrast, phenobarbital and 3-MC significantly augmented the inhibition of FBS-induced DNA synthesis from 16.5 to 32.8 and 24.3%, respectively, and the inhibition of FBS-induced proliferation from 21.0 to 36.7 and 28.1%, respectively. Treatment with ICI-182,780 (non-selective ER antagonist) and MPP (ERα antagonist) did not modulate the inhibitory effects of 2-OE (100 nmol/l) or 2-ME (100 nmol/l) on DNA synthesis (Fig. 2C) or proliferation (Fig. 2D).
Fig. 2.

A and B: bar graphs show the effects of E2 (100 nmol/l) on DNA synthesis ([3H]thymidine incorporation; A) by microglia and proliferation (cell no.; B) of microglia in the absence and presence of ABT (5 μmol/l, CYP450 inhibitor), OR486 (OR; COMT inhibitor, 5 μmol/l), PB (5 μmol/l, cytochrome P450 inducer), and 3-MC (5 μmol/l, cytochrome P450 inducer). C and D: bar graphs show the effects of 2-OE (100 nmol/l) and 2-ME (100 nmol/l) on 2.5% FBS-induced DNA synthesis by microglia (C) and cell proliferation of microglia (D) in the absence and presence of ICI-182,780 [ICI; 5 μmol/l, estrogen receptor (ER)α/β antagonist] and 1,3-bis(4-hydroxyphenyl)-4-methyl-5-[4-(2-piperidinylethoxy)phenol]-1H-pyrazole (MPP; 5 nmol/l, ERα antagonist). Results are presented as means ± SE (n = 4). *P < .05 vs. control; §P < .05 vs. E2 alone.
As shown in the representative chromatogram in Fig. 3A and the bar graph in Fig. 3B, treatment of BV2 cells with 2-OE (5 μmol/l) resulted in the formation of 2-ME, and this conversion was blocked by the COMT inhibitor OR486 (5 μmol/l) (Fig. 3C). Moreover, BV2 cells expressed CYP1B1 protein, and treatment of BV2 cells with the CYP450 inducer 3-MC (5 μmol/l) induced CYP1B1 expression (Fig. 4A). Additionally, incubation of BV2 cells for 48 h with 17β-[2-3H(N)]estradiol resulted in 2-OE formation, as assayed by the release of [3H]H2O. The formation of [3H]H2O, a stoichiometric indicator for 2-OE formation, was inhibited by the CYP450 inhibitor ABT (5 μmol/l) as well as by pyrene, a CYP1B1 inhibitor (5 nmol/l) (Fig. 4B). Furthermore, inhibition of 2-OE metabolism to 2-ME by OR486 abrogated the inhibitory effect of 2-OE on cell number (Fig. 5A) and DNA synthesis (Fig. 5B) in BV2 cells. In this regard, inhibition of DNA synthesis was attenuated from 23.1 to 4.4%, and the inhibition of proliferation was decreased from 30.3 to 3.5%. Additionally, as shown in Fig. 5C, the inhibitory effects of estradiol were significantly decreased by pyrene (selective CYP1B1 inhibitor, 5 nmol/l) from 15.1 to 7.3%, but not by ellipticine (selective CYP1A1 inhibitor, 10 μmol/l) or by ketoconazole (selective CYP3A4 inhibitor, 10 μmol/l).
Fig. 3.
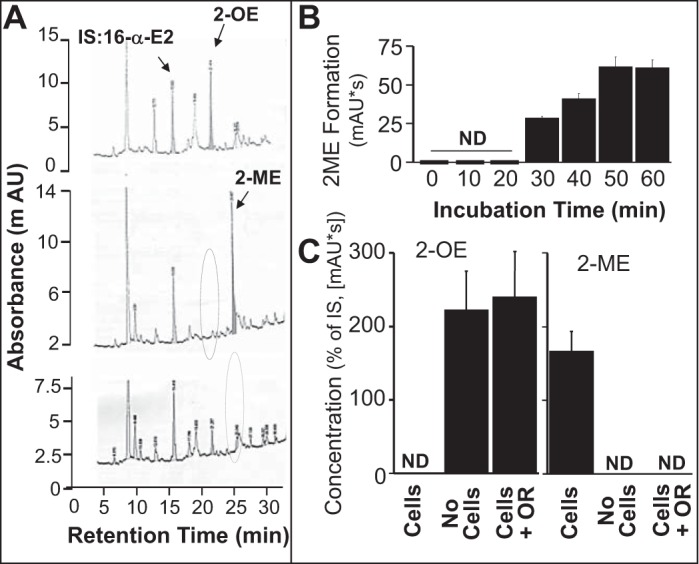
A: when microglia were incubated with 2-OE (5 μmol/l) in the presence of OR (COMT inhibitor; top), a peak was observed corresponding to 2-OE, but no peak occurred corresponding to 2-ME. However, in the absence of OR (middle), the same experiment resulted in a peak corresponding to 2-ME but the absence of a peak corresponding to 2-OE. In the absence of 2-OE (bottom), neither 2-OE nor 2-ME was detected. The internal standard used was 16α-E2. B: time-dependent conversion of 2-OE to 2-ME by microglia. C: effect of OR (10 μmol/l, COMT inhibitor) on the metabolism of 2-OE (5 μmol/l) to 2-ME as calculated as a percentage of the internal standard (IS) 16α-E2. Results are presented as means ± SE (n = 4). AU, arbitrary units; ND, below detection limit.
Fig. 4.

A: representative Western blot showing the expression of CYP1B1 in BV2 cells and its upregulation in cells treated for 36 h with 3-MC (5 μmol/l). Bar graph depicts the optical density (OD) ratio of CYP1B1/β-actin. *P < 0.05 vs. control (Con) BV2 cells. B: graph showing the metabolic capability of BV2 cells to convert estradiol to 2-OE using the radiolabeled 17β-[2-3H(N)]estradiol assay for determining the stoichiometric conversion of estradiol to 2-OE by analyzing [3H]H2O release. Bar graph depicts the formation of [3H]H2O assessed in BV2 treated for 48 h with medium (Con), 3-MC (5 μmol/l), 3-MC + ABT (5 μmol/l), or 3-MC + pyrene (5 nmol/l). Values for each data point represent the mean ± SE from 4 samples. *P < 0.05 vs. Con (BV2 in medium alone); §P < 0.05, significant inhibition of [3H]H2O formation.
Fig. 5.
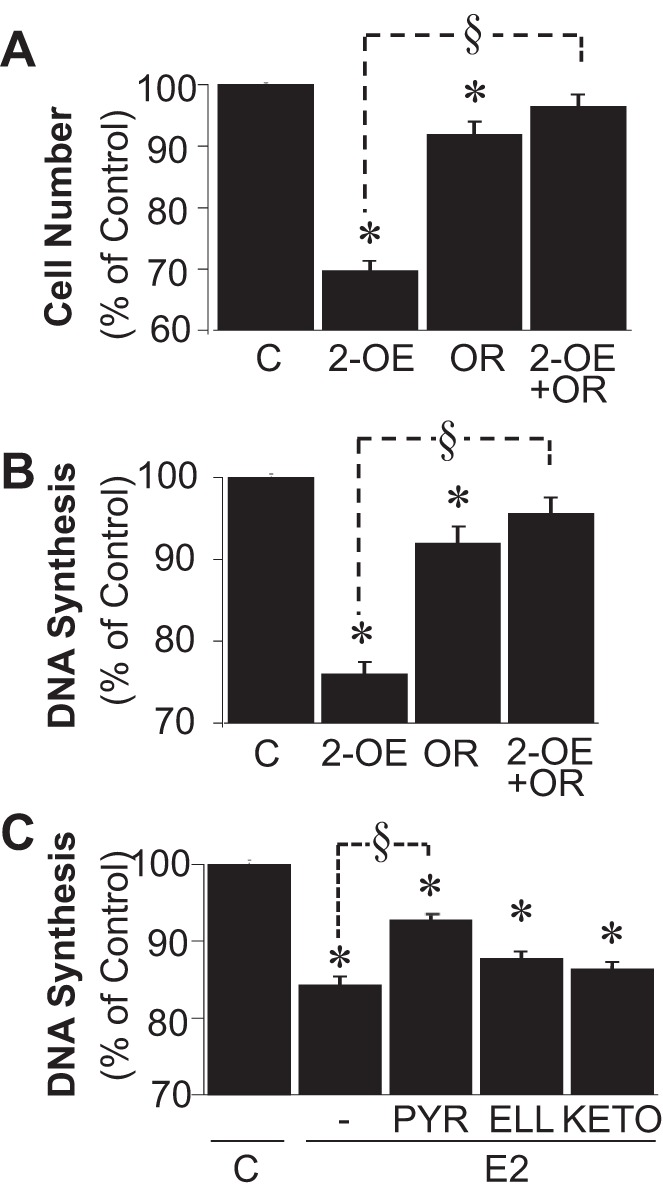
A and B: bar graphs show the effects of 2-OE (100 nmol/l) on cell no. (A) and DNA synthesis ([3H]thymidine incorporation; B) in the absence and presence of OR (COMT inhibitor, 5 μmol/l). C: modulatory effects of pyrene (PYR; selective CYP1B1 inhibitor, 5 nmol/l), ellipticine (ELL; selective CYP1A1 inhibitor, 10 μmol/l), and ketoconazole (KETO; selective CYP3A4 inhibitor, 10 μmol/l) on the ability of E2 (100 nmol/l) to inhibit DNA synthesis. Results are presented as means ± SE (n = 4). *P < .05 vs. C; §P < 0.05 between indicated treatments.
In BV2 cells treated with LPS, iNOS and COX-2 expression were upregulated, and this upregulation was significantly abrogated by preincubation with 2-ME (Fig. 6, A–C). Additionally, LPS-induced nitrite secretion was decreased by 2-ME treatment (Fig. 6D). The lowest concentration (1 nmol/l) of 2-ME significantly inhibited nitrite secretion by 14.2%; moreover, >50% inhibition was observed at 100 nmol/l. Treatment of BV2 cells with LPS induced phagocytosis, and cotreatment with 2-ME inhibited phagocytosing activity in a concentration-dependent manner (Fig. 7). As shown in the representative photomicrographs and bar graph in Fig. 7, treatment with 100 nmol/l 2-ME inhibited phagocytosing activity by 39.7%. Inhibition of phagocytosing activity by 15% was also observed at 10 nmol/l of 2-ME; however, this response did not achieve statistical significance. In contrast to 2-ME, in LPS-treated BV2 cells, estradiol (1-1,000 nmol/l) and DPN (10 and 100 nmol/l) did not inhibit phagocytosing activity (Fig. 7).
Fig. 6.

A and B: Western blots show the concentration-dependent (1–100 nmol/l) inhibitory effects of 2-ME on lipopolysaccharide (LPS; 1 μg/ml)-induced inducible nitric oxide synthase (iNOS; A) and cyclooxygenase (COX-2; B) expression. C: densitometry results for these experiments are shown in the bar graphs. D: concentration-dependent inhibitory effects of 2-ME on nitrite production from LPS-stimulated microglia. Nitrite secretion was quantified using the Griess reagent. Results are presented as means ± SE (n = 3). §P < 0.05 vs. no LPS; *P < 0.05 vs. LPS-treated cells.
Fig. 7.

Effects of increasing concentrations of 2-ME and 17β-estradiol on LPS-induced (1 μg/ml for 90 min) phagocytosing activity in microglia. Top: representative photomicrographs depicting the concentration-dependent (1-1,000 nmol/l) inhibition of phagocytosis by 2-ME. Bar graphs depict the comparative effects between 2-ME, 2-OE, 17β-estradiol, and 2,3-bis(4-hydroxyphenyl)-propionitrile (DPN) on phagocytosis. In contrast to 2-ME, 17β-estradiol and DPN were unable to inhibit phagocytosis by activated microglia under identical conditions. Results are presented as means ± SE (n = 3). *P < .05 vs. LPS-treated cells.
To assess neurotoxic effects of factors generated by LPS-activated BV2 cells, the supernatant of LPS-stimulated BV2 cells was placed on SH-SY5Y cells, and this treatment decreased the viability of the latter (Fig. 8). Pretreatment of BV2 cells with 2-ME prior to LPS stimulation partially abrogated the decrease of SH-SY5Y viability (Fig. 8). Even pretreatment of BV2 with 0.1 nmol/l of 2-ME slightly but significantly decreased the inhibition of SH-SY5Y viability. This effect was not due to the presence of 2-ME, since 2-ME was washed out after treatment of the BV2 cells prior to LPS stimulation. Indeed, 2-ME levels were not detectable by HPLC in the conditioned medium; moreover, direct treatment of SH-SY5Y cells with 2-ME (0.1–1,000 nmol/l) in presence of conditioned medium solely from LPS-stimulated BV2 cells was ineffective and failed to prevent neuronal toxicity.
Fig. 8.

Analysis of viability of SH-SY5Y cells after treatment with conditioned microglia supernatant. Microglia (BV2 cells) were pretreated with 2-ME for 24 h, washed twice with PBS, and treated in the presence or absence of LPS. After 24 h, the medium was transferred onto SH-SY5Y cells. The viability of SH-SY5Y cells after 24 h of incubation with the conditioned medium was assessed using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay. Results are presented as means ± SE (n = 3). *Significant reversal of cell toxicity by 2-ME.
We assessed the contribution of ER-dependent mechanisms in mediating the antiproliferative effects of estradiol. As shown in Fig. 9A, BV2 cells expressed ERβ, but not ERα, and treatment with estradiol upregulated ERβ expression by ∼200%. We also assessed whether ERβ is involved in mediating the inhibitory effects of estradiol. Treatment with estradiol (100 nmol/l) inhibited FBS-induced DNA synthesis (Fig. 9B) and cell number (Fig. 9C). The nonselective ER antagonist ICI-182,780 (5 μmol/l) did not alter the effects of FBS but attenuated the ability of 100 nmol/l estradiol to inhibit DNA synthesis from 17.7 to 8.3% (Fig. 9B) and cell number from 20.6 to 5.5% (Fig. 9C). The inhibitory effects of estradiol on DNA synthesis (Fig. 9D) and cell number (Fig. 9E) were mimicked by the ERβ agonist DPN, but not by ERα agonist PPT. At a concentration of 1 nmol/l, DPN inhibited DNA synthesis by 9% and cell proliferation by 9.8%; at 10 nmol/l, DPN inhibited DNA synthesis by 18.1% and cell proliferation by 20.9%, and at 100 nmol/l, DPN inhibited DNA synthesis by 20.6% and proliferation by 23.5%. The inhibitory effects of estradiol on DNA synthesis (Fig. 9F) and cell proliferation (Fig. 9G) were not blocked by the ERα antagonist MPP, a finding consistent with our observation that BV2 cells did not express ERα.
Fig. 9.

A: representative Western blot demonstrating the presence of ERβ, but not ERα, in microglia and the modulatory effects of 10, 100, and 1,000 nmol/l of E2 on the expression of ERβ. B–G: modulatory effects of ER agonists and antagonist on FBS-induced DNA synthesis (B, D, and F) and cell number (C, E, and G). Antiproliferative effects of 100 nmol/l E2 were attenauted by ICI-182,780 (5 μmol/l, ERα/β antagonist; B and C). Moreover, the antiproliferative effects of E2 were mimicked by DPN (ERβ agonist; D and E) but not by 4,4′,4′′-(4-propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol (PPT; ERα agonist; D and E). The effects of 100 nmol/l estradiol were not blocked by 1,3-bis(4-hydroxyphenyl)-4-methyl-5-[4-(2-piperidinylethoxy)phenol]-1H-pyrazole (MPP; ERα antagonist, 5 nmol/l; F and G). Results are presented as means ± SE (n = 4). *P < 0.05 vs. C; §P < 0.05 between indicated treatments. NS, not significant.
Previously, we have argued that the effects of 2-ME are ER independent; however, based on a recent publication by Kognati et al. (22), it has been suggested that 2-ME may be a GPR30 ligand. Hence, we assessed the role of GPR30 in mediating the effects of 2-ME in BV2 cells. As shown in Fig. 10A, BV2 cells expressed GPR30. However, in contrast to 2-ME, treatment of BV2 cells with G1, a GPR30 agonist, did not inhibit BV2 cell growth (Fig. 10B) or phagocytosis (Fig. 10C). Furthermore, the inhibitory effects of 2-ME were not blocked by the GPR30 antagonist G15. Taken together, our observations provide evidence that the inhibitory effects of 2-ME are not GPR30 mediated.
Fig. 10.

A: representative Western blot showing the expression of GPR30 in BV2 cells. B: bar graphs showing the effects of G1 (10 and 100 nmol/l) and 2-ME (10 and 100 nmol/l) on 2.5% FBS-induced DNA synthesis {[3H]thymidine incorporation in microglia in the presence and absence of G15 (100 nmol/l), a GPR30 antagonist}. Results are presented as means ± SE (n = 3). *P < 0.05 vs. 2.5% FBS alone; §P < 0.05 vs. Con (starved cells). C: effects of 2-ME (100 nmol/l) and G1 (100 nmol/l) on LPS-induced (1 μg/ml for 90 min) phagocytosing activity in microglia in the presence and absence of G15 (GPR30 antagonist, 100 nmol/l). In contrast to 2-ME, G1 was unable to inhibit phagocytosis by activated microglia under identical conditions. The inhibitory effects of 2-ME were not abrogated by GPR30 antagonist G15. Results are presented as means ± SE (n = 3). *P < 0.05 vs. LPS-treated cells.
DISCUSSION
Although microglia function is necessary for brain health, irreversible hyperactivation of microglia is associated with neurodegenerative disorders (4). Therefore, a therapeutic challenge is to limit the damaging inflammatory responses of microglia by modulating their properties. In the present study, we provide evidence that compared with estradiol, 2-ME, an endogenous metabolite of estradiol with little affinity for ERs, is a potent inhibitor of BV2 microglia cell proliferation. 2-ME also inhibits phagocytosis by BV2 cells and release of cytotoxic molecules from activated BV2 cells. Importantly, we demonstrate that sequential conversion of estradiol to 2-ME by CYP450 and COMT, respectively, plays a role in mediating ER-independent antiproliferative actions of estradiol.
The present study also shows that 2-OE and 2-ME are more potent than estradiol in inhibiting BV2 growth, suggesting that endogenous metabolism of estradiol to 2-ME may contribute to the effects of estradiol. Indeed, the inhibitory effect of estradiol on BV2 proliferation is enhanced by phenobarbital and 3-MC (CYP450 inducers) (14) and reduced by ABT (broad-spectrum CYP450 inhibitor) (14) and pyrene (selective inhibitor of CYP1B1) (13, 14). Moreover, the COMT inhibitor OR486 significantly abrogates the inhibitory effect of estradiol and completely blocks the effects of 2-OE. Importantly, the inhibitory effects of 2-OE and 2-ME on BV2 proliferation are not blocked by the ER antagonist ICI-182,780. This observation suggests that the inhibitory effects of 2-OE and 2-ME are ER independent. This notion is also supported by the fact that 2-ME has no binding affinity for ERs (13). The fact that the effects of 2-OE are completely blocked by the COMT inhibitor OR486 suggests that 2-ME mediates the inhibitory effect of 2-OE on BV2 growth. This hypothesis is further supported by our observation that BV2 cells metabolize 2-OE to 2-ME and that this metabolic step is blocked by OR486. Additionally, we find that 2-ME inhibits the upregulation of proinflammatory enzymes (iNOS and COX-2) and molecules (nitric oxide) in LPS-activated BV2 cells and abrogates the neuronal cytotoxicity of BV2-conditioned medium.
Our finding that 2-ME is more potent than estradiol in inhibiting glial cell proliferation and that low nanomolar concentrations of 2-ME inhibit LPS-induced generation of proinflammatory enzymes iNOS and COX-2 suggests that 2-ME (formed from the intermediate 2-OE) may play an important pathophysiological role in mediating the protective effects of estradiol via an ER-independent mechanism. This notion is further supported by the fact that the antiproliferative effects of estradiol are enhanced in BV2 cells stimulated by the CYP450 inducers phenobarbital and 3-MC, yet they are inhibited by the CYP450 inhibitor ABT and by the CYP1B1 inhibitor pyrene. Because neither ellipticine nor ketoconazole blocks the inhibitory effects of estradiol, CYP1A1/1A2 and CYP3A4 are likely not involved in metabolizing estradiol to 2-OE. This contention is futher supported by our observation that BV2 cells expressed CYP1B1 and pretreatment with 3-MC induced the expression of CYP1B1. Moreover, 3-MC stimulated 2-hydroxylation of radiolabeled estradiol to 2-OE and [3H]H2O in BV2 cells, and this effect was blocked by ABT and pyrene. The effects of estradiol are mimicked by 2-OE, the precursor of 2-ME, and these effects are abrogated when 2-OE's conversion to 2-ME is blocked by the COMT inhibitor OR486. Together, these findings suggest that 2-ME, rather than 2-OE, is the final effector molecule in mediating the ER-independent actions of estradiol on glial cells. This conclusion is supported by the rapid metabolism of 2-OE to 2-ME in the BV2 glial cells. In women, the circulating levels of 2-ME can be as high as 90 nmol/l during last months of pregnancy (3). Interestingly, the increase in 2-ME to 90 nmol/l is associated with remission of clinical symptoms of multiple sclerosis (27) and rheumatoid arthritis in these patients (28).
The current study also shows that the inhibitory effects of estradiol can be attenuated by a nonselective ER antagonist. This suggests that the antiproliferative actions of estradiol are mediated via both ER-dependent and ER-independent mechanisms. Indeed, many of the anti-inflammatory effects of estradiol on microglia have been described to be mediated via both ERα and ERβ (2, 5, 36). Our investigations on the roles of ERα and ERβ indicate that in BV2 cells ERβ is involved in the growth inhibitory effects of estradiol. In this regard, the antimitogenic effects of estradiol are mimicked by DPN (ERβ agonist) but not PPT (ERα agonist) and blocked by ICI-182,780 (ERα/β antagonist) but not by MPP (ERα antagonist). Under the culture conditions used, BV2 cells expressed ERβ but not ERα, and estradiol induced ERβ expression, indicating that ERβ is functionally active in our cellular system. Our finding that ERβ is also involved in estradiol action indicates that next to a metabolite-dependent mechanism, an ER-dependent mechanism may still play a prominent role in mediating specific protective actions of estradiol.
Our finding that both ER-independent and ERβ-dependent mechanisms mediate antiproliferative actions of estradiol in glial cells may be of clinical relevance. Until now, it was unclear why some studies suggested a negative effect of hormone replacement therapy or estradiol treatment on stroke (23), whereas other studies supported the concept that estradiol is neuroprotective (25, 31). The causes of the divergent results are a matter of debate. A recent study shows that the actual estradiol concentrations achieved by different administration methods might be responsible (31, 37). It is suggested that the specific hormone release profiles dictate the hormone's effectiveness on ischemic damage, since negative estradiol effects are seen mostly when a delivery method that results in early, very high estradiol concentrations is used (31, 37). Because estradiol concentrations achieved by different administration methods might also influence estradiol's metabolite profile (e.g., due to feedback inhibition of metabolizing enzymes) (10), it is possible that metabolite effects can only manifest when the appropriate estradiol serum concentration is achieved.
COMT plays a critical role in catecholamine catabolism. Since high levels of catecholamines induce cell toxicity, COMT-mediated catabolism of catecholamines is thought to protect the brain cells following injury. Indeed, catecholamine levels increase dramatically following stroke and traumatic brain injury and are implicated in the subsequent cell damage (34). Interestingly, COMT activity in microglial cells increases following traumatic brain injury (30), suggesting that microglia may protect the cells at the site of injury by catabolizing catecholamines. Our finding that COMT is involved in metabolizing 2-OE to 2-ME suggests that local formation of 2-ME may play a key role in mediating the protective effects of estradiol via an ER-independent mechanism. The fact that COMT serves as a common enzyme in the catabolism of catecholamines and formation of 2-ME may be of potential clinical relevance in the brain. We speculate that the equilibrium between catecholamine catabolism and 2-ME formation may define the protective or deleterious actions of estradiol following stroke. Our previous studies show that catecholamines inhibit the conversion of 2-OE to 2-ME and abrogate 2-OE's antimitogenic actions in human vascular smooth muscle cells (15). It is feasible that increases in catecholamines following stroke would result in decreased 2-ME formation and increased accumulation of catecholestrogens, which are genotoxic (18). This may explain the lack of protective actions of estradiol observed in many studies. The shift in equilibrium between catecholamine catabolism and 2-ME formation by COMT may play a critical role in defining the protective vs. deleterious actions of estradiol in stroke, and this hypothesis needs to be tested further. Increases in catecholestrogen levels and decreases in 2-ME levels are observed in patients with Parkinson's disease (18). The fact that COMT is a ubiquitous enzyme and 2-ME is protective and can be readily formed in the brain suggests that 2-ME may play an important role in the pathophysiology of stroke. Importantly, 2-ME administration may be more protective against stroke-induced brain damage, as it would eliminate the potential formation and accumulation of catecholestrogens and interference with catecholamine catabolism.
Phagocytosis and clearing of dead neurons and other debris by microglia within the brain is a critical process to maintain normal brain function (4). However, nonreversible activation of microglia following brain insult and cytokine release can result in exacerbated loss of neurons and lead to neurodegeneration (4). Inhibition of microglial phagocytosis prevents inflammatory neuronal death (26). Our finding that 2-ME inhibits LPS-induced phagocytosis by microglia suggests that 2-ME may protect against neuronal damage in part by suppressing microglial phagocytosis. In contrast to 2-ME, treatment of BV2 cells with estradiol under identical conditions does not inhibit phygocytosis, suggesting that 2-ME may be superior in preventing microglia-mediated neuronal damage. Consistent with our observations, studies by Bruce-Keller et al. (5) demonstrated that simultaneous treatment with estradiol is unable to prevent LPS-induced phagocytosis. We find that when cells are preincubated with 2-ME for 24 h, LPS-induced phagocytosis is significantly inhibited. This observation, together with the fact that BV2 cells express ERβ, estradiol upregulates ERβ expression, and DPN fails to inhibit phagocytosis, suggests that inhibition of phagocytosis is likely mediated via an ER-independent mechanism.
Compared to cell growth, the effects of 2-ME on phagocytosis seem to be direct and not due to the conversion of estradiol to 2-ME. This notion is supported by our observation that LPS-induced phagocytosis is not inhibited by the ERβ agonist DPN, which is not metabolized to 2-ME. Moreover, the role of ERβ and GPR30 can be ruled out, since the effects of 2-ME were not blocked by their respective antagonists. It is feasible that the formation of 2-ME by cultured cells is not sufficient to reach concentrations that inhibit phagocytosis and that circulating levels of 2-ME in vivo (∼30–90 nmol/l) may be more effective in inhibiting phagocytosis and are of pathophysiological relevance. Since unlike estradiol, 2-ME is known to inhibit tubulin polymerization (13), it is feasible that the inhibitory effects of 2-ME are mediated via this mechanism. Indeed, recent studies provide evidence that phagocytosis by BV2 cells is inhibited by inhibitors of microtubule polymerization (21).
Apart from phagocytosis, 2-ME also inhibits the generation NO, a precursor for the neurotoxic molecule peroxynitrite, and inhibits expression of COX-2, a proinflammatory enzyme, implying that 2-ME may prevent neuronal damage caused by these known proinflammatory molecules. Indeed, treatment of neuroblastoma cells with conditioned supernatant from LPS-treated microglia induces cell toxicity, and this effect is significantly abrogated in neuroblastoma cells treated with supernatants from microglia pretreated with LPS and 2-ME.
2-ME is protective against experimental autoimmune encephalomyelitis (16), neurodamage following traumatic brain injury (33), and brain infarction following middle cerebral artery occlusion-induced focal ischemia (8). These findings, along with our results, suggest that endogenous 2-ME may play an important role in mediating the protective effects of estradiol. Our finding that 2-ME dampens the proliferative response of BV2 glial cells, inhibits LPS-induced proinflammatory mechanisms, inhibits phagocytosis, and prevents toxicity to neuronal cells suggests that local metabolism of estradiol to 2-ME may play an important role in defining the neuroprotective effects of estradiol.
Although circulating estradiol derived from ovaries plays an important role, recent studies suggest that estradiol produced locally in the brain may also induce protective actions. Indeed, brain cells produce estradiol and express key enzymes (aromatase) and precursors (cholesterol) for estradiol formation (1). Based on the fact that estradiol is synthesized in the brain, we hypothesize that locally produced 2-ME may actively participate in mediating the protective actions of estradiol in the brain.
In conclusion, our findings support the hypothesis that microglia convert estradiol to 2-ME by the sequential actions of CYP1B1 and COMT and that 2-ME mediates in part the inhibitory effect of estradiol on microglia. Thus, metabolic disorders and interindividual differences in estradiol metabolism may affect the ability to recover from stroke and other diseases in which overactivation of microglia plays an important role. Additionally, estradiol metabolism may constitute an important natural defense mechanism against microglial overactivation.
GRANTS
This work was supported by Swiss National Science Foundation Grants IZERO-142213/1 and 31003A-138067 (to R. K. Dubey) and by National Institutes of Health Grants NS-087978, HL-109002, DK-091190, HL-069846, DK-068575, and DK-079307 (to E. K. Jackson).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.A.S., E.K.J., and R.K.D. conception and design of research; S.A.S., F.B., and D.G.G. performed experiments; S.A.S., M.R., F.B., and R.K.D. analyzed data; S.A.S., M.R., F.B., D.G.G., E.K.J., and R.K.D. interpreted results of experiments; S.A.S., E.K.J., and R.K.D. prepared figures; S.A.S., M.R., F.B., E.K.J., and R.K.D. drafted manuscript; S.A.S., M.R., E.K.J., and R.K.D. edited and revised manuscript; S.A.S., M.R., F.B., D.G.G., E.K.J., and R.K.D. approved final version of manuscript.
ACKNOWLEDGMENTS
This work was part of the Ph.D thesis of Dr. Sara Schaufelberger submitted to ETH Zurich (dissertation no. 18836), Switzerland.
REFERENCES
- 1.Azcoitia I, Yague JG, Garcia-Segura LM. Estradiol synthesis within the human brain. Neuroscience 191: 139–147, 2011. [DOI] [PubMed] [Google Scholar]
- 2.Baker AE, Brautigam VM, Watters JJ. Estrogen modulates microglial inflammatory mediator production via interactions with estrogen receptor β. Endocrinology 145: 5021–5032, 2004. [DOI] [PubMed] [Google Scholar]
- 3.Berg D, Sonsalla R, Kuss E. Concentrations of 2-methoxyoestrogens in human serum measured by a heterologous immunoassay with an 125I-labelled ligand. Acta Endocrinol 103: 282–288, 1983. [DOI] [PubMed] [Google Scholar]
- 4.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8: 57–69, 2007. [DOI] [PubMed] [Google Scholar]
- 5.Bruce-Keller AJ, Keeling JL, Keller JN, Huang FF, Camondola S, Mattson MP. Antiinflammatory effects of estrogen on microglial activation. Endocrinology 141: 3646–3656, 2000. [DOI] [PubMed] [Google Scholar]
- 6.Brueggemeier RW. Kinetics of rat liver microsomal estrogen 2-hydroxylase. Evidence for sex difference at initial velocity conditions. J Biol Chem 256: 10239–10242, 1981. [PubMed] [Google Scholar]
- 7.Carswell HV, Macrae IM, Gallagher L, Harrop E, Horsburgh KJ. Neuroprotection by a selective estrogen receptor β agonist in a mouse model of global ischemia. Am J Physiol Heart Circ Physiol 287: H1501–H1504, 2004. [DOI] [PubMed] [Google Scholar]
- 8.Chen C, Hu Q, Yan J, Lei J, Qin L, Shi X, Luan L, Yang L, Wang K, Han J, Nanda A, Zhou C. Multiple effects of 2ME2 and D609 on the cortical expression of HIF-1α and apoptotic genes in a middle cerebral artery occlusion-induced focal ischemia rat model. J Neurochem 102: 1831–1841, 2007. [DOI] [PubMed] [Google Scholar]
- 9.Chen Y, Won SJ, Xu Y, Swanson RA. Targeting microglial activation in stroke therapy: pharmacological tools and gender effects. Curr Med Chem 21: 2146–2155, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dawling S, Roodi N, Parl FF. Methoxyestrogens exert feedback inhibition on cytochrome P450 1A1 and 1B1. Cancer Res 63: 3127–3132, 2003. [PubMed] [Google Scholar]
- 11.Dubal DB, Zhu H, Yu J, Rau SW, Shughrue PJ, Merchenthaler I, Kindy MS, Wise PM. Estrogen receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury. Proc Natl Acad Sci USA 98: 1952–1957, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dubey RK, Imthurn B, Jackson EK. 2-Methoxyestradiol: a potential treatment for multiple proliferative disorders. Endocrinology 148: 4125–4127, 2007. [DOI] [PubMed] [Google Scholar]
- 13.Dubey RK, Jackson EK. Cardiovascular protective effects of 17β-estradiol metabolites. J Appl Physiol 91: 1868–1883, 2001. [DOI] [PubMed] [Google Scholar]
- 14.Dubey RK, Jackson EK, Gillespie DG, Rosselli M, Barchiesi F, Krust A, Keller H, Zacharia LC, Imthurn B. Cytochromes 1A1/1B1- and catechol-O-methyltransferase-derived metabolites mediate estradiol-induced antimitogenesis in human cardiac fibroblast. J Clin Endocrinol Metab 90: 247–255, 2005. [DOI] [PubMed] [Google Scholar]
- 15.Dubey RK, Jackson EK, Gillespie DG, Zacharia LC, Imthurn B. Catecholamines block the antimitogenic effect of estradiol on human coronary artery smooth muscle cells. J Clin Endocrinol Metab 89: 3922–3931, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Duncan GS, Brenner D, Tusche MW, Brustle A, Knobbe CB, Elia AJ, Mock T, Bray MR, Krammer PH, Mak TW. 2-Methoxyestradiol inhibits experimental autoimmune encephalomyelitis through suppression of immune cell activation. Proc Natl Acad Sci USA 109: 21034–21039, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dutheil F, Dauchy S, Diry M, Sazdovitch V, Cloarec O, Mellottee L, Bieche I, Ingelman-Sundberg M, Flinois JP, de Waziers I, Beaune P, Decleves X, Duyckaerts C, Loriot MA. Xenobiotic-metabolizing enzymes and transporters in the normal human brain: regional and cellular mapping as a basis for putative roles in cerebral function. Drug Metab Dispos 37: 1528–1538, 2009. [DOI] [PubMed] [Google Scholar]
- 18.Gaikwad NW, Murman D, Beseler CL, Zahid M, Rogan EG, Cavalieri EL. Imbalanced estrogen metabolism in the brain: possible relevance to the etiology of Parkinson's disease. Biomarkers 16: 434–444, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henn A, Lund S, Hedtjarn M, Schrattenholz A, Porzgen P, Leist M. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX 26: 83–94, 2009. [DOI] [PubMed] [Google Scholar]
- 20.Koellhoffer EC, McCullough LD. The effects of estrogen in ischemic stroke. Transl Stroke Res 4: 390–401, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koenigsknecht J, Landerth G. Microglial phagocytosis of fibrillar beta-amyloid through a beta1 integrin-dependent mechanism. J Neurosci 24: 9838–9846, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kognati S, Snyder R, Gumaste U, Karamyan VT, Thekkumkara. 2-methoxyestradiol binding of GPR30 down-regulates angiotensin AT(1) receptor. Eur J Pharmacol 723: 131–140, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lemaitre RN, Heckbert SR, Psaty BM, Smith NL, Kaplan RC, Longstreth WT Jr. Hormone replacement therapy and associated risk of stroke in postmenopausal women. Arch Intern Med 162: 1954–1960, 2002. [DOI] [PubMed] [Google Scholar]
- 24.Li R, Shen Y. Estrogen and brain: synthesis, function and diseases. Front Biosci 10: 257–267, 2005. [DOI] [PubMed] [Google Scholar]
- 25.Mikkola TS, Tuomikoski P, Lyytinen H, Korhonen P, Hoti F, Vattulainen P, Gissler M, Ylikorkala O. Estradiol-based postmenopausal hormone therapy and risk of cardiovascular and all-cause mortality. Menopause 22: 976–983, 2015. [DOI] [PubMed] [Google Scholar]
- 26.Neher JJ, Neniskyte U, Zhao JW, Bal-Price A, Tolkovsky AM, Brown GC. Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J Immunol 186: 4973–4983, 2011. [DOI] [PubMed] [Google Scholar]
- 27.Offner H, Polanczyk M. A potential role for estrogen in experimental autoimmune encephalomyelitis and multiple sclerosis. Ann NY Acad Sci 343–372, 2006. [DOI] [PubMed] [Google Scholar]
- 28.Ostensen M, Villiger PM. The remission of rheumatoid arthritis during pregnancy. Semin Immunopathol 29: 185–191, 2007. [DOI] [PubMed] [Google Scholar]
- 29.Patel AR, Ritzel R, McCullough LD, Liu F. Microglia and ischemic stroke: a double-edged sword. Int J Physiol Pathophysiol Pharmacol 5: 73–90, 2013. [PMC free article] [PubMed] [Google Scholar]
- 30.Redell JB, Dash PK. Traumatic brain injury stimulates hippocampal catechol-O-methyl transferase expression in microglia. Neurosci Lett 413: 36–41, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rocca WA, Grossardt BR, Miller VM, Shuster LT, Brown RD Jr. Premature menopause or early menopause and risk of ischemic stroke. Menopause 19: 272–277, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sampei K, Goto S, Alkayed NJ, Crain BJ, Korach KS, Traystman RJ, Demas GE, Nelson RJ, Hurn PD. Stroke in estrogen receptor-alpha-deficient mice. Stroke 31: 738–743, 2000. [DOI] [PubMed] [Google Scholar]
- 33.Schaible EV, Windschugl J, Bobkiewicz W, Kaburov Y, Dangel L, Kramer T, Huang C, Sebastiani A, Luh C, Werner C, Engelhard K, Thal SC, Schafer MK. 2-Methoxyestradiol confers neuroprotection and inhibits a maladaptive HIF-1α response after traumatic brain injury in mice. J Neurochem 129: 940–954, 2014. [DOI] [PubMed] [Google Scholar]
- 34.Schulze J, Vogelgesang A, Dressel A. Catecholamines, steroids and immune alterations in ischemic stroke and other acute diseases. Aging Dis 5: 327–339, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thiel A, Heiss WD. Imaging of microglia activation in stroke. Stroke 42: 507–512, 2011. [DOI] [PubMed] [Google Scholar]
- 36.Vegeto E, Belcredito S, Etteri S, Ghisletti S, Brusadelli A, Meda C, Krust A, Dupont S, Ciana P, Chambon P, Maggi A. Estrogen receptor-α mediates the brain antiinflammatory activity of estradiol. Proc Natl Acad Sci USA 100: 9614–9619, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wise PM, Dubal DB, Rau SW, Brown CM, Suzuki S. Are estrogens protective or risk factors in brain injury and neurodegeneration? Reevaluation after the Women's health initiative. Endocr Rev 26: 308–312, 2005. [DOI] [PubMed] [Google Scholar]
- 38.Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci USA 95: 15769–15774, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zacharia LC, Jackson EK, Gillespie DG, Dubey RK. Catecholamines block 2-hydroxyestradiol-induced antimitogenesis in mesangial cells. Hypertension 39: 854–859, 2002. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Z, Chopp M, Powers C. Temporal profile of microglial response following transient (2 h) middle cerebral artery occlusion. Brain Res 744: 189–198, 1997. [DOI] [PubMed] [Google Scholar]