

Abstract
Cystic fibrosis (CF) is the most common lethal genetic disease among Caucasians. It is caused by mutations in the CF Transmembrane Conductance Regulator (CFTR) gene, which encodes an apical membrane anion channel that is required for regulating the volume and composition of epithelial secretions. The most common CFTR mutation, present on at least one allele in >90% of CF patients, deletes phenylalanine at position 508 (F508del), which causes the protein to misfold. Endoplasmic reticulum (ER) quality control elicits the degradation of mutant CFTR, compromising its trafficking to the epithelial cell apical membrane. The absence of functional CFTR leads to depletion of airway surface liquid, impaired clearance of mucus and bacteria from the lung, and predisposes to recurrent infections. Ultimately, respiratory failure results from inflammation and bronchiectasis.
Although high throughput screening has identified small molecules that can restore the anion transport function of F508del CFTR, they correct less than 15% of WT CFTR activity, yielding insufficient clinical benefit. To date, most primary CF drug discovery assays have employed measurements of CFTR’s anion transport function, a method that depends on the recruitment of a functional CFTR to the cell surface, involves multiple wash steps, and relies on a signal that saturates rapidly. Screening efforts have also included assays for detection of extracellularly HA-tagged or HRP-tagged CFTR, which require multiple washing steps. We have recently developed tools and cell lines that report the correction of mutant CFTR trafficking by currently available small molecules, and have extended this assay to the 96-well format. This new and simple no-wash assay of F508del CFTR at the cell surface may permit the discovery of more efficacious drugs, and hopefully thereby prevent the catastrophic effects of this disease. In addition, the modular design of this platform should make it useful for other diseases where loss-of-function results from folding and/or trafficking defects in membrane proteins.
Keywords: F508del CFTR, Surface expression, Fluorogen activating protein tag, Corrector, High throughput screening
1. Introduction
Numerous human diseases arise from defects in the trafficking of proteins to the cell surface. Among them is cystic fibrosis (CF), which is caused by mutations in the gene encoding CFTR, an anion selective channel that normally resides at the apical membranes of epithelial cells. The most common mutation in CF, F508del, disrupts CFTR folding, elicits its premature degradation, and thereby blocks its trafficking to the apical membrane. Recent therapeutic discovery efforts for correctors of mutant CFTR trafficking have relied primarily on measurements that detect agonist stimulated CFTR channel function rather than the physical presence of the corrected protein at the cell surface. On this basis, prior high throughput screening (HTS) efforts have generated a number of small molecules, called correctors, which have demonstrated limited ability to improve F508del CFTR trafficking thus far. The most active compounds available at present correct the activity of F508del CFTR to between 10% and 15% of the wild type (WT) level [1], and initial clinical studies of one compound, while reducing sweat chloride by 8 mEq/L, failed to show improvements in the lung function of F508del CF patients [2]. In pre-clinical studies, corrector efficacy has varied, depending on the cell type examined [3], highlighting the importance of cell specific environment. As primary and secondary screens are generally performed using non-epithelial cells, this suggests that there may be a significant number of false negatives, which could represent missed opportunities. The use of correctors in combination can enhance rescue to levels greater than individual compound actions, and these may require more than one mechanism of action at the molecular level. Recently, it has been demonstrated in two phase 3 clinical trials that the combination of lumacaftor (VX-809), a CFTR corrector, and ivacaftor (VX-770), a CFTR potentiator, results in both improved FEV1 and a reduction of the rate of pulmonary exacerbations in CF patients homozygous for the F508del CFTR mutation when compared to placebo [4]. Even so, studies in cultured cells have demonstrated that VX-770 diminishes the corrective effect of VX-809, thus indicating the need for further optimization of corrector and potentiator compounds [5,6].
To date, most F508del CFTR corrector HTS have relied on measurements of restored CFTR function following small molecule treatment [7]. Halide sensitive yellow fluorescent proteins (YFPs) based assays where CFTR activity is assessed by measuring the rate of YFP signal decrease caused by iodide influx or assays utilizing FRET based voltage sensitive membrane dyes have been used to detect plasma membrane F508del CFTR function. These methods require multiple wash steps and rely on the recruitment of a functional CFTR to the surface membrane, which would eliminate from consideration compounds that directly interact with CFTR and interfere with its activity [7,8]. The defective trafficking of a CFTR-related, misfolded ABC transporter, P-glycoprotein, can be corrected by agents that act as inhibitors [9,10], and blockers of hERG potassium channel mutants also correct their trafficking [11,12]. In the latter case, it was possible to use medicinal chemistry to separate the channel blocking property from that for correction [11]. Thus, direct detection of F508del CFTR that has been rescued to the cell surface using a single labeling step would improve throughput and increase the dynamic range of corrector detection assays.
Here we report the development of a HTS using a fluorogen activating protein (FAP) fusion of F508del CFTR. The FAP platform has a modular design that allows for rapid construction of FAP fusions with other membrane proteins that are compromised by folding and/or trafficking defects. This growing list includes diseases such as type II diabetes (GLUT4 & insulin receptor), long QT syndrome (hERG), familial hypercholesterolemia (LDL receptor), diabetes insipidus (aquaporin-2), diseases of retinal degeneration (BEST1) and others [13].
2. Fluorogen activating protein tag
The FAP system consists of a genetically evolved engineered antibody fragment (scFv) dimer, constructed by screening a complex human scFv library for those that specifically elicited intense fluorescence enhancement from a small organic dye, Malachite Green (MG). Other than MG and its chemical derivatives, at this time we do not know of other compounds that bind the FAP domain and result in such increase in fluorescence [14].
The FAP technology is ‘all or nothing’; in solution, MG is completely dark, or non-fluorescent, but upon binding to the FAP, its fluorescence yield is increased 15,000–20,000-fold with a Cy5-like excitation–emission spectrum [14]. The bipartite FAP and cognate fluorogen system enables the sensitive and selective visualization of tagged proteins at the cell surface through the use of a cell impermeant sulfonated analog of MG, MG-B-TAU, without the need for any washing steps to remove the dye before imaging (Fig. 1). Alternatively, the total cellular distribution of a FAP tagged protein can be visualized using MG-ester, a fluorogen that passively diffuses across the cell membrane and is then trapped within the cell.
Fig. 1.
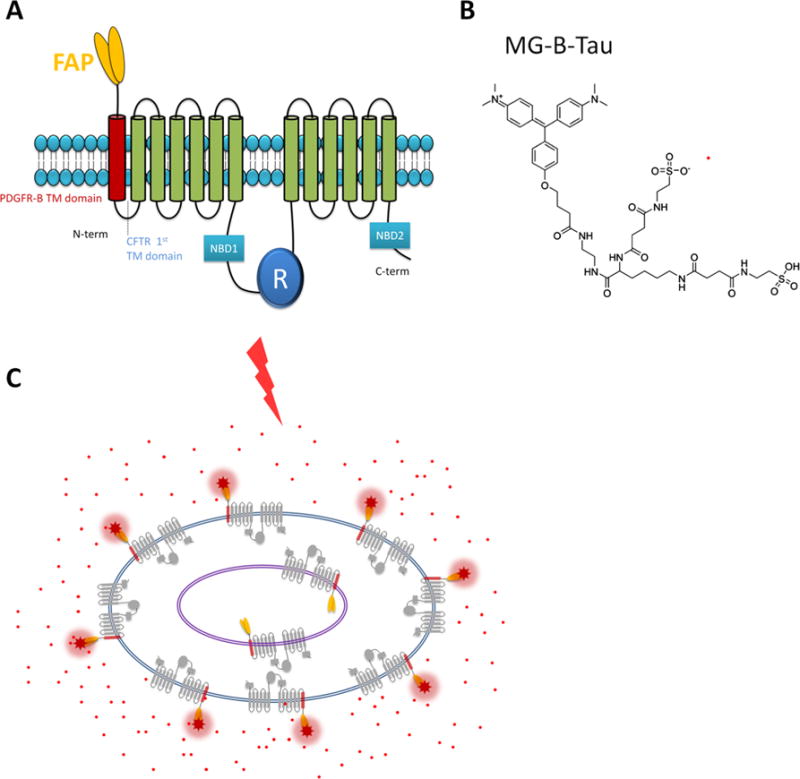
Fluorogen activating protein (FAP) technology. (A) Cartoon of the FAP-CFTR construct. In order to express the fluorogen activating protein (FAP) extracellularly on CFTR, a FAP/PDGFR-B transmembrane domain was fused to the N-terminus of CFTR. (B) The structure of MG-B-TAU dye. (C) Membrane-impermeant MG-B-TAU (red dots) binds to surface localized FAP and fluoresces when excited with 640 nM laser light. Internalized FAP is not labeled and unbound MG-B-TAU does not fluoresce.
As described previously [15], we have made a CFTR construct with a FAP tag inserted extracellularly via an extra transmembrane domain added to the N-terminus of CFTR (Fig. 1). This construct demonstrates characteristic CFTR functional activity and corrector trafficking responses that correlate closely with functional data from polarized human bronchial epithelia from CF patients. Specifically, it has been demonstrated that: (1) cAMP-stimulated anion transport activity of CFTR is maintained with FAP reporter modifications; (2) functional rescue of CFTR activity by corrector treatment occurs with FAP-tagged F508del-CFTR constructs; and (3) F508del-CFTR FAP demonstrates biochemical rescue in response to correctors [13,14].
3. Materials and reagents
MG-B-TAU was kindly provided by Dr. Marcel P. Bruchez, Carnegie Mellon University. MG-B-TAU is commercially available as se-Red-xc from Sharp Edge Labs (Pittsburgh, PA). Hoechst 33342 nuclear stain was from Life Technologies. Cell culture reagents were from Life Technologies. 96-well ViewPlates were from Perkin Elmer. HEK 293A cells were maintained in DMEM supplemented with 10% FBS (Life Technologies) and penicillin–streptomycin (50 U/mL, 50 μg/mL, Life Technologies) at 37 °C in a humidified tissue culture incubator with an atmosphere consisting of 95% air and 5% CO2. To create cells stably expressing FAP-F508del CFTR, HEK 293A cells were transfected with a linearized pIRES plasmid (Clontech) harboring a blasticidin resistance gene downstream of the IRES element and a FAP-F508del-CFTR expression cassette upstream of the IRES element. 20 μg/mL blasticidin in the media was used to select for cells with the plasmid stably integrated and clonal lines were isolated. One of the resulting clonal lines, 293A F508del 4C9, was used for the FAP-F508del-CFTR surface labeling assay. The stable 293A F508del 4C9 cell line as well as the FAP-F508del-CFTR expression plasmid can be obtained upon request.
4. Adapting the quantification of FAP-F508del-CFTR surface expression to a HTS in 96 well plates
4.1. Labeling of FAP-tagged F508del CFTR expressed on the cell surface in 96-well plates
We devised the following protocol to test the assay in 96-well plates:
293A F508del 4C9 cells were plated at a sub-confluent density in black 96-well plates with optically clear plastic bottom (Packard ViewPlate). They were left at room temperature for 1 h to let the cells attach and then transferred to a 37 °C humidified tissue culture incubator.
The following day media was exchanged for media containing test compounds and plates transferred to a 27 °C humidified tissue culture incubator in order to increase CFTR rescue.
The cells were ready for imaging the next day. The cell nuclei were labeled by adding 2 μg/mL Hoechst 33342 to the growth media 1–4 h before the imaging, to identify cells. 30 min before image acquisition 50 nM of MG-B-TAU was added to the media.
Using our Nikon ECLIPSE Ti-E microscopy system with an A1 resonant scanner, there were two strategies to get the correct focal plane for imaging in every well. The first option was to use the nuclear Hoechst staining for automatic autofocus and define an offset for MG-B-TAU image collection. The other option was to use the Perfect Focus System (PFS) that automatically adjusts the focal plane relative to the culture media/culture vessel interface. We have found the PFS system to be very reliable, hence we used it exclusively when imaging 96-well plates. We used a high NA (0.75) 20× air objective and a large pinhole (100 μm) to give maximal signal strength and enable the capture of both nuclear Hoechst signal and FAP/MG-B-TAU signal using the same focal plane for high speed imaging. Images were collected by sequential excitation of Hoechst 33342 and FAP/MG-B-TAU using 405 nm and 640 nm laser light, respectively. Using these parameters, a 96-well plate can be read in 3 min with one image per well or in 6 min with 4 images per well.
4.2. Dose-response curve for CFTR corrector VX-809
As an initial experiment in 96-well plates, we performed a dose–response experiment for the CFTR corrector VX-809 (Lumacaftor, Vertex Pharmaceuticals). VX-809 partially corrects the folding defect in F508del-CFTR which allows some of the mutant CFTR to escape ER-associated degradation, and thus causes an increase in F508del-CFTR surface expression. In Fig. 2A is shown the raw data from this experiment, and as expected, the FAP/MG-B-TAU signal (green) increased with increasing VX-809 concentration. We used Nikon Elements software to quantify the increase in FAP/MG-B-TAU signal by first generating a binary mask from the nuclear Hoechst signal and thickening it to include the whole cell (Fig. 2B). The binary mask was used to define the region-of-interest in which the average FAP/MG-B-TAU signal was then calculated. This was performed as an automated process available in the HCA module of the Nikon Elements software package and occurred concurrent with image acquisition. Using this readout versus using the total FAP/MG-B-TAU signal from the entire image served to minimize variability stemming from alternating cell numbers in each field of view. A heat map of the calculated FAP/MG-B-TAU signal is shown in Fig. 2C. The data was fitted to a non-linear dose–response curve and plotted in Fig. 2D. We calculated the EC50 value to be 0.13 μM for F508del CFTR correction, which is in agreement with previously published values using other assays [1].
Fig. 2.
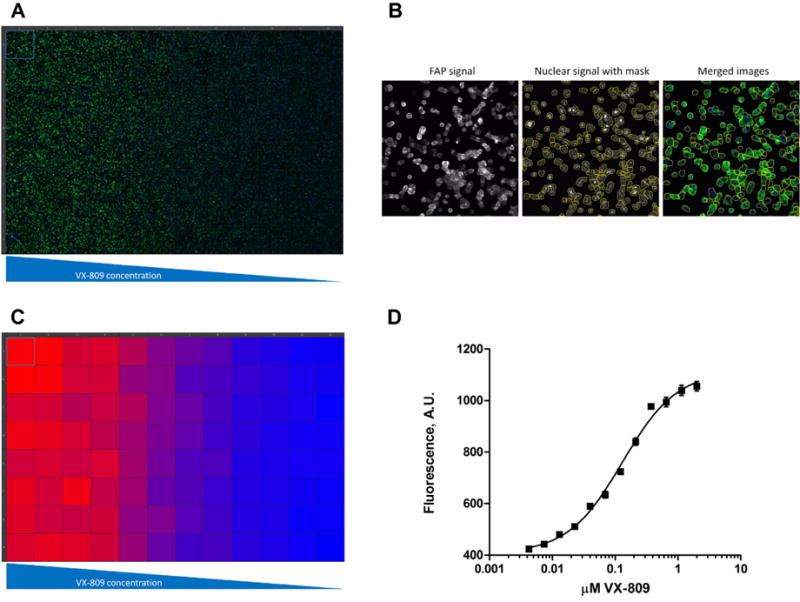
Dose–response curve for VX-809 correction of F508del CFTR surface expression. (A) Raw image data from a 96-well plate where increasing concentrations of VX-809 were added for 24 h to 293A cells stably transfected with FAP-F508del CFTR and imaged after Hoechst 33342 and MG-B-TAU addition. Hoechst 33342 signal is in blue and MG-B-TAU signal in green. (B) Here is shown an example of an image from one of the wells with the thickened nuclear binary mask that is used to define the region-of-interest in which MG-B-TAU signal is measured. (C) Heat map for MG-B-TAU signal of the raw data shown in (A). (D) Fitted dose–response curve for VX-809 activation of F508del CFTR surface expression. The VX-809 EC50 value was calculated to be 0.13 μM. Shown is average signal plus/minus SEM. The experiment was repeated 3 times with similar results.
4.3. Determining the Z′-factor for the assay
The Z′-factor [16] is a commonly accepted measure of the suitability and quality of an assay for HTS. It is defined by: 1 − ((3σc+ + 3σc−)/(|μc+ − μc−|)) where σc+ and σc− denote the standard deviation of the positive and negative controls, respectively, and μc+ and μc− denote the averages of the positive and negative controls, respectively. An assay with a Z′-factor between 0.5 and 1 is considered excellent for HTS [16].
To determine the Z′-factor for our assay, we used DMSO as a negative control and 2 μM VX-809 as the positive control for half a plate each in 96-well plates. The readout from this test is plotted in Fig. 3. From this, the Z′-factor was calculated to be 0.73 with 1 image taken per well – reading the plate with 4 images taken per well gave a calculated Z′-factor of 0.82. This indicates that the assay is of high quality and suitable for HTS even with only one image per well, allowing for higher throughput.
Fig. 3.
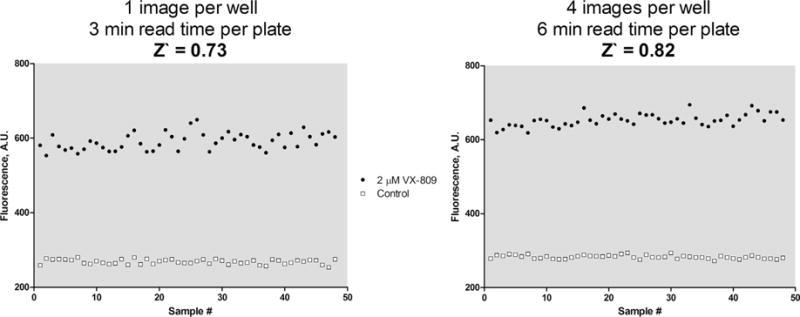
Determination of Z′-factor. The Z0-factor for the assay was determined by treating one half of a 96-well plate containing 293A cells stably transfected with FAP-F508del CFTR with 2 μM VX-809 and the other half with DMSO as a control for 24 h before adding Hoechst 33342 and MG-B-TAU followed by imaging. The resulting values are plotted and the Z′-factor calculated. The experiment was repeated twice with similar results.
4.4. The effect of DMSO and time dependency of the assay
We tested the effect of DMSO by incubating both control cells and cells with 2 μM VX-809 added with equal concentrations of up to 2% DMSO for 24 h prior to imaging the cells, and found that the assay was suitable for HTS with a Z′-factor above 0.7 under all DMSO concentrations tested (Fig. 4A). The signal of the assay increased during the first ~100 min after MG-B-TAU dye addition, presumably due to dye binding as well as CFTR trafficking to and from the surface, but was stable after that for several hours (Fig. 4B).
Fig. 4.
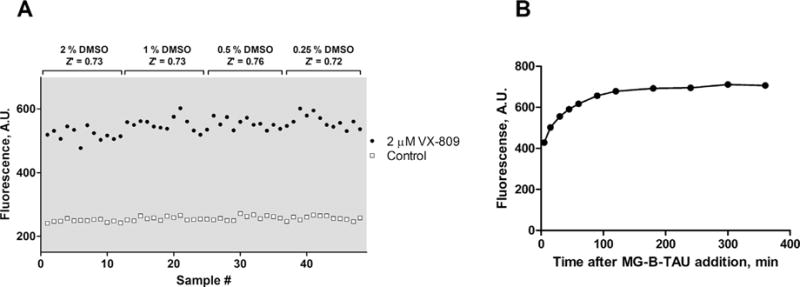
DMSO and time dependence of MG-B-TAU signal. (A) The DMSO dependence of the assay was determined by treating 293A cells stably transfected with FAP-F508del CFTR with the indicated concentration of DMSO plus/minus 2 μM VX-809 for 24 h before adding Hoechst 33342 and MG-B-TAU followed by imaging. The resulting values are plotted and the Z′-factors calculated. (B) The time dependence of fluorescence signal intensity after MG-B-TAU addition was determined by treating 293A cells stably transfected with FAP-F508del CFTR with 2 μM VX-809 for 24 h before adding Hoechst 33342 and MG-B-TAU followed by imaging at the indicated time points. The experiments were repeated with similar results.
5. Concluding remarks
Here we have described an image-based HTS assay for the surface expression of F508del-CFTR. The assay is very simple, requiring only the addition of a nuclear stain and the fluorogen MG-B-TAU to the cells without any washing steps and with Z′-factor values >0.7 making it ideal for HTS.
The assay should prove very useful in screening efforts to develop new small molecule F508del CFTR correctors. The assay could likewise be used in siRNA-based knockdown screening experiments to identify pathways and mechanisms regulating F508del CFTR degradation or biogenesis.
Another benefit to this assay is that it can be generalized to measure surface expression of any plasma membrane protein to which the FAP can be attached extracellularly. FAP can be added either to an extracellular N- or C-terminus (e.g. GPCRs) or added internally in an extracellular loop of the target protein. Alternatively, the FAP tag can be added by fusing an extra transmembrane domain containing FAP to the target protein, as used for CFTR in the present assay.
Acknowledgments
The work was supported by National Institutes of Health grants R03 MH09360601 (R.A. Frizzell and S.C. Watkins), 5R01EB017268-02 (S.C. Watkins) and P30 DK072506 (R.A. Frizzell and S.C. Watkins) and the Cystic Fibrosis Foundation grants R883-CR07 (R.A. Frizzell) and FRIZZE13XXO (R.A. Frizzell).
Contributor Information
Mads Breum Larsen, Email: mbl6@pitt.edu.
Jennifer Hu, Email: hu.jennifer@medstudent.pitt.edu.
Raymond A. Frizzell, Email: frizzell@pitt.edu.
References
- 1.Van Goor F, Hadida S, Grootenhuis PDJ, Burton B, Stack JH, Straley KS, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci USA. 2011;108:18843–18848. doi: 10.1073/pnas.1105787108. http://dx.doi.org/10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax. 2012;67:12–18. doi: 10.1136/thoraxjnl-2011-200393. http://dx.doi.org/10.1136/thoraxjnl-2011-200393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pedemonte N, Tomati V, Sondo E, Galietta LJV. Influence of cell background on pharmacological rescue of mutant CFTR. Am J Physiol Cell Physiol. 2010;298:C866–C874. doi: 10.1152/ajpcell.00404.2009. http://dx.doi.org/10.1152/ajpcell.00404.2009. [DOI] [PubMed] [Google Scholar]
- 4.Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, et al. Lumacaftor–ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373:220–231. doi: 10.1056/NEJMoa1409547. http://dx.doi.org/10.1056/NEJMoa1409547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Veit G, Avramescu RG, Perdomo D, Phuan PW, Bagdany M, Apaja PM, et al. Some gating potentiators, including VX-770, diminish ΔF508-CFTR functional expression. Sci Transl Med. 2014;6:246ra97. doi: 10.1126/scitranslmed.3008889. http://dx.doi.org/10.1126/scitranslmed.3008889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cholon DM, Quinney NL, Fulcher ML, Esther CR, Das J, Dokholyan NV, et al. Potentiator ivacaftor abrogates pharmacological correction of ΔF508 CFTR in cystic fibrosis. Sci Transl Med. 2014;6:246ra96. doi: 10.1126/scitranslmed.3008680. http://dx.doi.org/10.1126/scitranslmed.3008680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pedemonte N, Zegarra-Moran O, Galietta LJV. High-throughput screening of libraries of compounds to identify CFTR modulators. Methods Mol Biol (Clifton, NJ) 2011;741:13–21. doi: 10.1007/978-1-61779-117-8_2. http://dx.doi.org/10.1007/978-1-61779-117-8_2. [DOI] [PubMed] [Google Scholar]
- 8.Van Goor F, Straley KS, Cao D, González J, Hadida S, Hazlewood A, et al. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1117–L1130. doi: 10.1152/ajplung.00169.2005. http://dx.doi.org/10.1152/ajplung.00169.2005. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y, Loo TW, Bartlett MC, Clarke DM. Additive effect of multiple pharmacological chaperones on maturation of CFTR processing mutants. Biochem J. 2007;406:257–263. doi: 10.1042/BJ20070478. http://dx.doi.org/10.1042/BJ20070478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Loo TW, Bartlett MC, Clarke DM. Modulating the folding of P-glycoprotein and cystic fibrosis transmembrane conductance regulator truncation mutants with pharmacological chaperones. Mol Pharmacol. 2007;71:751–758. doi: 10.1124/mol.106.029926. http://dx.doi.org/10.1124/mol.106.029926. [DOI] [PubMed] [Google Scholar]
- 11.Zhou Z, Gong Q, January CT. Correction of defective protein trafficking of a mutant HERG potassium channel in human long QT syndrome. Pharmacological and temperature effects. J Biol Chem. 1999;274:31123–31126. doi: 10.1074/jbc.274.44.31123. [DOI] [PubMed] [Google Scholar]
- 12.Ficker E, Obejero-Paz CA, Zhao S, Brown AM. The binding site for channel blockers that rescue misprocessed human long QT syndrome type 2 ether-a-gogo-related gene (HERG) mutations. J Biol Chem. 2002;277:4989–4998. doi: 10.1074/jbc.M107345200. http://dx.doi.org/10.1074/jbc.M107345200. [DOI] [PubMed] [Google Scholar]
- 13.Aridor M, Hannan LA. Traffic jam: a compendium of human diseases that affect intracellular transport processes. Traffic. 2000;1:836–851. doi: 10.1034/j.1600-0854.2000.011104.x. [DOI] [PubMed] [Google Scholar]
- 14.Szent-Gyorgyi C, Schmidt BF, Schmidt BA, Creeger Y, Fisher GW, Zakel KL, et al. Fluorogen-activating single-chain antibodies for imaging cell surface proteins. Nat Biotechnol. 2008;26:235–240. doi: 10.1038/nbt1368. http://dx.doi.org/10.1038/nbt1368. [DOI] [PubMed] [Google Scholar]
- 15.Holleran JP, Glover ML, Peters KW, Bertrand CA, Watkins SC, Jarvik JW, et al. Pharmacological rescue of the mutant cystic fibrosis transmembrane conductance regulator (CFTR) detected by use of a novel fluorescence platform. Mol Med (Cambridge, MA) 2012;18:685–696. doi: 10.2119/molmed.2012.00001. http://dx.doi.org/10.2119/molmed.2012.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang JH, Chung TDY, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. http://dx.doi.org/10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]