

Abstract
Aims/Introduction
Src, a non‐receptor tyrosine kinase, regulates a wide range of cellular functions, and hyperactivity of Src is involved in impaired glucose metabolism in pancreatic β‐cells. However, the physiological role of Src in glucose metabolism in normal, unstressed β‐cells remains unclear. In the present study, we investigated the role of Src in insulin secretion and glucose metabolism.
Materials and Methods
Src was downregulated using small interfering ribonucleic acid in INS‐1 cells, and glucose‐induced insulin secretion, adenosine triphosphate content, intracellular calcium concentration, glucose utilization and glucokinase activity were measured. Expression levels of messenger ribonucleic acid and protein of glucokinase were examined by semiquantitative real‐time polymerase chain reaction and immunoblotting, respectively. Cells were fractionated by digitonin treatment, and subcellular localization of glucokinase was examined by immunoblotting. Interaction between glucokinase and neuronal nitric oxide synthase was estimated by immunoprecipitation.
Results
In Src downregulated INS‐1 cells, glucose‐induced insulin secretion was impaired, whereas insulin secretion induced by high K+ was not affected. Intracellular adenosine triphosphate content and elevation of intracellular calcium concentration by glucose stimulation were suppressed by Src downregulation. Src downregulation reduced glucose utilization in the presence of high glucose, which was accompanied by a reduction in glucokinase activity without affecting its expression. However, Src downregulation reduced glucokinase in soluble, cytoplasmic fraction, and increased it in pellet containing intaracellular organelles. In addition, interaction between glucokinase and neuronal nitric oxide synthase was facilitated by Src downregulation.
Conclusions
Src plays an important role in glucose‐induced insulin secretion in pancreatic β‐cells through maintaining subcellular localization and activity of glucokinase.
Keywords: Glucokinase, Insulin secretion, Src
Introduction
In pancreatic β‐cells, glucose metabolism regulates exocytosis of insulin granules through metabolism‐secretion coupling, in which glucose‐induced adenosine triphosphate (ATP) production in mitochondria plays an essential role1. Oxidative stress is one of the most significant factors that impairs glucose metabolism in β‐cells2. We previously reported that Src plays an important role in the production of reactive oxygen species (ROS)3. In particular, endogenous overproduction of ROS in pancreatic islets of diabetic Goto‐Kakizaki rats and oubain‐exposed islets of normoglycemic Wistar rats is reduced by treatment of 4‐amino‐5‐(4‐chlorophenyl)‐7‐(t‐butyl)pyrazolo[3, 4‐d]pyrimidine (PP2), a specific Src inhibitor4, while transient exendin‐4 treatment suppresses Src hyperactivity and reduces ROS overproduction in Goto‐Kakizaki rat islets5. In addition, exposure of palmitate increases ROS production with an increase in the activity of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, a pathological ROS source in plasma membrane, and reduces glucose‐induced insulin secretion (GIIS) in INS‐1 rat insulinoma cells, whereas Src inhibition reduces ROS production by decreasing its activity and restores insulin secretion6. Src is a non‐receptor tyrosine kinase and a member of Src family kinases (SFKs), which are expressed in various types of animal cells7. The activity of Src is regulated by tyrosine phosphorylation at two sites with opposite effect. Briefly, phosphorylation of Tyr527 at the COOH‐terminal regulatory domain results in the inactivation of Src, and phosphorylation of Tyr418 at the catalytic domain results in the activation of Src8. Src regulates a wide range of cellular events including cell growth, division, differentiation, survival and programed death, as well as specialized functions, such as immune responses, cell adhesion, migration and endocytosis, whereas dysregulation of Src has been implicated in the etiology of human diseases, especially cancers9. In addition, evidence that Src is related to pancreatic β‐cell function has also been reported10, 11, 12, 13. However, the physiological role of Src in glucose metabolism in normal, unstressed β‐cells remains unclear.
In the present study, we investigated the effects of Src downregulation in glucose metabolism in intact INS‐1 cells to characterize its role more precisely. We show here that Src regulates glucokinase activity to maintain glucose metabolism in pancreatic β‐cells.
Materials and Methods
Chemicals, cell culture and small interfering ribonucleic acid transfection
ATP, adenosine diphosphate, poly‐L‐ornithine and diadenosine pentaphosphate were purchased from Sigma‐Aldrich (St. Louis, MO, USA). PP2 was purchased from Tocris (Ellisville, MO, USA). All other reagents were purchased from Nacalai Tesque (Kyoto, Japan), unless otherwise noted. INS‐1 cells were cultured in RPMI 1640 medium as previously described14. Stealth™ small interfering ribonucleic acids (siRNAs) were synthesized by Life Technologies (Carlsbad, CA, USA). The sequences of siRNA specific for rat Src, called Src siRNA, and a control siRNA with the nonsense sequence are shown in Table S1. Transfection of siRNAs was carried out as previously reported14, except that the reacted cell number was 5 × 105 cells/mL in the present study. All experiments using siRNA‐transfected INS‐1 cells were carried out 48 h after transfection, unless otherwise noted.
Isolation of total RNA and semiquantitative real‐time polymerase chain reaction
Total RNA was isolated using RNeasy Mini Kit (Qiagen, Hilden, Germany), and complementary deoxyribonucleic acid was synthesized as previously described14. Semiquantitative real‐time polymerase chain reaction (PCR) was carried out using SYBR Green PCR Master Mix (Life Technologies) as previously reported14, and results were normalized by the expression of β‐actin. The rat sequences of forward and reverse primers to detect Src, glucokinase, glucose transporter 2 (Glut2) and β‐actin are shown in Table S2.
Immunoprecipitation and immunoblotting
Immunoprecipitation, sodium dodecyl sulfate polyacrylamide gel electrophoresis and immunoblotting was carried out as previously described5. Primary antibodies used were mouse monoclonal anti‐Src antibody from Merck KGaA (Darmstadt, Germany); mouse monoclonal anti‐complex I (39 kDa subunit), anti‐complex III (core II), anti‐complex IV (subunit I) and anti‐complex V (subunit α) of mitochondrial respiratory chain antibody from Invitrogen (Eugene, OR, USA); rabbit anti‐glucokinase antibody and anti‐GLUT2 antibody from Abcam (Cambridge, UK); rabbit anti‐Lyn, anti‐Lck, anti‐Fgr, anti‐Blk, anti‐C‐terminal Src kinase (Csk), anti‐calnexin and goat anti‐Hck antibody from Santa Cruz Biotechnology (Santa Cruz, CA, USA); rabbit anti‐neuronal nitric oxide synthase (nNOS) antibody from Cell Signaling Technology (Danvers, MA, USA); mouse monoclonal anti‐β‐actin antibody from Sigma‐Aldrich; and mouse monoclonal anti‐glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) antibody from Merck KGaA. Secondary antibodies used were horseradish peroxidase‐conjugated anti‐rabbit and mouse antibody from GE Healthcare (Buckinghamshire, UK), and anti‐goat antibody from Merck KGaA. Band intensities were quantified with Multi Gauge software (Fuji Film, Tokyo, Japan).
Insulin secretion, ATP content and intracellular calcium concentration
Insulin secretion, ATP content and intracellular calcium concentration ([Ca2+]i) were measured as previously described14. Krebs‐Ringer bicarbonate HEPES (KRBH) buffer was composed of 140 mmol/L NaCl, 3.6 mmol/L KCl, 0.5 mmol/L MgSO4, 0.5 mmol/L NaH2PO4, 1.5 mmol/L CaCl2, 2 mmol/L NaHCO3, 0.1% bovine serum albumin and 10 mmol/L HEPES (pH 7.4).
Mitochondrial ATP production
Measurement of ATP production in the mitochondrial fraction was carried out as previously described14, except 3 mmol/L KH2PO4 was included in the solution used for mitochondria suspension and reaction. ATP production was corrected by mitochondrial protein content.
Glucose utilization
Glucose utilization was measured using the previously described method with slight modifications15. Briefly, INS‐1 cells cultured on 24‐well plates were incubated in KRBH buffer with 2 mmol/L glucose at 37°C for 30 min, followed by incubation in 500 μL of KRBH buffer containing 2 or 10 mmol/L glucose and 1.5 mCi [5‐3H] glucose (GE Healthcare) for 60 min. Subsequently, 150 μL of supernatant of each well was transferred into a micro tube containing 50 μL of HCl to completely stop the reaction. The tube was then incubated in the capped vial containing 500 μL of distilled water overnight at 34°C to allow 3H2O (GE Healthcare) in the tube to equilibrate with the water in the vial. During the overnight incubation period, the micro tubes were kept standing with their caps opened. Each tube was then removed, and the disintegrations per minute of 3H2O in the water were counted.
Measurement of glucokinase activity
Glucokinase activity was measured as previously described with slight modifications16. Briefly, after preincubation in KRBH buffer with 2 mmol/L glucose for 60 min, INS‐1 cells were lysed and subjected to glucose‐6‐phosphate dehydrogenase‐coupled reaction at two (0.5 and 50 mmol/L) concentrations of glucose for 60 min at 37°C. The glucose phosphorylation rate was estimated as the increase of NADH concentration, which was measured by fluorometric plate reader with excitation 360 nm and emission 460 nm. Glucokinase activity was determined by subtracting hexokinase activity measured at 0.5 mmol/L glucose subtracted from the activity measured at 50 mmol/L glucose.
Subcellular localization of glucokinase
Fractionation of digitonin‐permiabilized INS‐1 cells was carried out as described previously17. Briefly, INS‐1 cells were collected and incubated in 50 mmol/L HEPES buffer (pH 7.2) containing 125 mmol/L KCl, 20 mmol/L NaCl, 0.5 mmol/L CaCl2, 0.5 mmol/L MgCl2, along with 20 μg/mL digitonin for 10 min at 4°C. After centrifugation in a microcentrifuge (20 min at 12,000 g), both supernatants, the soluble fraction containing cytoplasmic proteins, and the pellet, the insoluble fraction containing any organelles including membrane or nuclei, were collected and used for immunoblotting. The intensities of glucokinase were measured and normalized by those of GAPDH in soluble fraction, and by those of calnexin in pellet.
Statistical analysis
The data are expressed as means ± standard error of the mean. Statistical significance was determined by unpaired Student's t‐test. P < 0.05 was considered statistically significant.
Results
Silencing effects of Src in INS‐1 cells
Src is a member of SFKs, and several SFKs are expressed in rat pancreatic islets5. We first confirmed which SFKs are expressed in INS‐1 cells, and Src, Lyn, Blk, Hck, Lck and Fgr were detected, but Fyn and Yes were not detected by immunoblotting (data not shown). Src siRNA was used to achieve highly specific Src downregulation and avoid unexpected cross‐reactivity or chemical effects. A total of 48 h after transfection of siRNAs, semiquantitative real‐time PCR assays and immunoblotting analysis showed 67.0% reduction of Src messenger RNA (Figure 1a) and 55.7% reduction of Src protein by Src siRNA (Figure 1b) compared with negative control siRNA. Expression levels of other SFKs or Csk, a negative regulator of Src18, were not affected by Src downregulation (Figure 1c).
Figure 1.
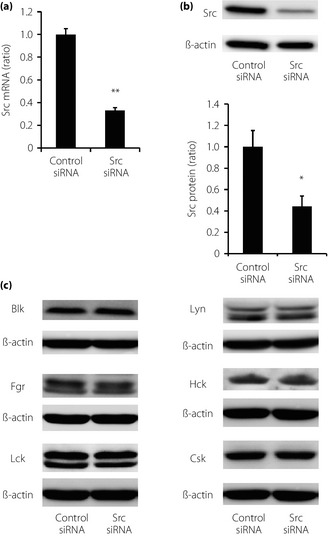
Silencing effects of Src in INS‐1 cells. (a) Effects of transfection of Src small interfering ribonucleic acid (siRNA) on the expression of Src messenger RNA. Data were normalized using β‐actin messenger RNA (n = 5 in each group). Values are expressed as mean ± standard error of the mean. **P < 0.01 compared with control siRNA. (b) Effects of transfection of Src siRNA on the expression of Src protein. Data were normalized by the expression of β‐actin (n = 6 in each group). Values are expressed as mean ± standard error of the mean. *P < 0.05 compared with control siRNA. (c) Effects of transfection of Src siRNA on the protein expressions of other Src family kinases and C‐terminal Src kinase. Representative blots were presented out of five independent examinations.
Insulin secretion, intracellular ATP content and [Ca2+]i
At the beginning of the analysis, we investigated the role of Src in insulin secretion. Downregulation of Src decreased GIIS, whereas insulin secretion induced by high K+ was not affected (Figure 2a). Insulin content was not affected by Src downregulation (Figure 2b). In addition, exposure of PP2 for 48 h reduced GIIS in INS‐1 cells (Figure 2c), whereas transient PP2 treatment for 30 min did not change GIIS in INS‐1 cells, as previously reported6.
Figure 2.
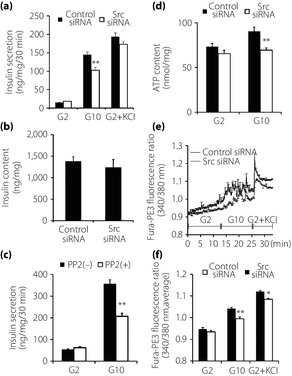
Effects of Src downregulation on insulin secretion, insulin content, adenosine triphosphate (ATP) content and intracellular calcium concentration. (a) Effects of Src downregulation on insulin secretion. Insulin secretion was measured after 30 min incubation in Krebs‐Ringer bicarbonate HEPES (KRBH) buffer with 2 mmol/L glucose (G2), 10 mmol/L glucose (G10) or 2 mmol/L glucose and 30 mmol/L KCl. Data were normalized by protein content (n = 4). Values are expressed as mean ± standard error of the mean. **P < 0.01 compared with control small interfering ribonucleic acid (siRNA) at the corresponding condition. (b) Effects of Src downregulation on insulin content. Insulin content in INS‐1 cells was measured 48 h after transfection. Data were normalized by protein content (n = 4). Values are expressed as mean ± standard error of the mean. (c) Effects of 4‐amino‐5‐(4‐chlorophenyl)‐7‐(t‐butyl)pyrazolo[3, 4‐d]pyrimidine (PP2) treatment on insulin secretion. INS‐1 cells were cultured with or without 10 μmol/L PP2 for 48 h, and insulin secretion was measured after 30 min incubation in KRBH buffer with G2 or G10. Data are expressed as ratio of the value of PP2(–) group at 2 mmol/L glucose (n = 4). Values are expressed as mean ± standard error of the mean. **P < 0.01 compared with the PP2(–) group at the corresponding condition. (d) Effects of Src downregulation on ATP content. ATP content was measured after 30‐min incubation in KRBH buffer with G2 or G10. Data were normalized by protein content (n = 4). Values are expressed as mean ± standard error of the mean. **P < 0.01 compared with control siRNA at the corresponding condition. (e) Effects of Src downregulation on intracellular calcium concentration. As an indicator of intracellular calcium concentration, fura‐PE3 fluorescence ratio (340:380 nm) in INS‐1 cells was monitored during incubation in KRBH buffer with G2, G10 or 2 mmol/L glucose and 30 mmol/L KCl (n = 10). Values are expressed as mean ± standard error of the mean. (f) Average values calculated from the data from (e). Values are expressed as mean ± standard error of the mean. *P < 0.05 and **P < 0.01 compared with control siRNA at the corresponding condition.
We then evaluated the glucose‐induced increase of intracellular ATP content and [Ca2+]i. Downregulation of Src reduced approximately 30% of intracellular ATP content at a stimulating level of 10 mmol/L glucose (Figure 2d). The elevation of [Ca2+]i in response to 10 mmol/L glucose was suppressed and delayed by downregulation of Src. In addition, the rate and amplitude of glucose‐induced [Ca2+]i oscillation were reduced. When the stimulus was changed from 10 mmol/L glucose to 30 mmol/L K+, [Ca2+]i in Src downregulated INS‐1 cells increased as quickly as that in control. [Ca2+]i evoked by high K+ was also reduced by Src downregulation, whereas the suppressive effect was milder than that evoked by high glucose (Figure 2e,f).
Glucose utilization, glucokinase activity, GLUT2 expression and mitochondrial ATP production
We then evaluated the functions proximal to membrane depolarization. Src downregulation reduced glucose utilization, which reflects the velocity of glucose metabolism in glycolysis, in the presence of 10 mmol/L glucose (Figure 3a), as well as the activity of glucokinase, a rate‐limiting enzyme in glycolysis in β‐cells (Figure 3b).
Figure 3.
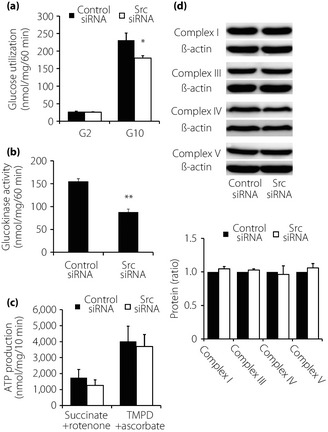
Effects of Src downregulation on glycolysis and mitochondrial metabolism. (a) Effects of Src downregulation on glycolysis. Glucose utilization was measured after 60‐min incubation in Krebs‐Ringer bicarbonate HEPES buffer with 2 mmol/L (G2) and 10 mmol/L (G10) glucose. Data were normalized by protein content (n = 4). Values are expressed as mean ± standard error of the mean. *P < 0.05 compared with control small interfering ribonucleic acid (siRNA) at the corresponding condition. (b) Effects of Src downregulation on glucokinase activity. Glucokinase activity of whole cell extracts of INS‐1 cells was measured. Data were normalized by protein content (n = 3). Values are expressed as mean ± standard error of the mean. **P < 0.01 compared with control siRNA at the corresponding condition. (c) Effects of Src downregulation on adenosine triphosphate (ATP) production. ATP production in mitochondria fraction from INS‐1 cells was measured in the presence of the combination of 1 mmol/L succinate and 1 μmol/L rotenone, and in the presence of 0.5 mmol/L N,N,N',N'‐tetramethyl‐p‐phenylenediamine (TMPD) and 2 mmol/L ascorbate (n = 4). Values are expressed as mean ± standard error of the mean. (d) Effects of Src downregulation on expression levels of mitochondrial proteins. Lysates of whole INS‐1 cells were used for immunoblotting analysis using antibodies against complex I, III, IV and V. Quantification data were obtained from four independent experiments and normalized with β‐actin level. Values are expressed as mean ± standard error of the mean.
Downregulation of Src did not significantly affect expression levels of Glut2 mRNA or GLUT2 protein (Figure S1). ATP production from mitochondria fraction in the presence of mitochondrial substrates (Figure 3c) or expression levels of mitochondrial respiratory chain complex proteins (Figure 3d), by which oxidative phosphorylation is carried out to produce ATPs, were not altered by Src downregulation.
Expression levels and subcellular localization of glucokinase
We then attempted to determine the mechanism of reduction of glucokinase activity by Src downregulation. The expression levels of glucokinase messenger RNA and protein were not significantly affected by Src downregulation (Figure 4a,b). Therefore, subcellular localization of glucokinase, which affects enzyme activity independent of expression level, was investigated. INS‐1 cells were separated to soluble fraction containing cytoplasmic proteins and pellet containing any organelles including membrane or nuclei, and immunoblotting analysis was carried out. Src downregulation was shown to reduce the expression level of glucokinase in soluble fraction and increase that in pellet (Figure 4c). This finding suggests that Src downregulation suppresses glucokinase activity by shifting its subcellular localization.
Figure 4.
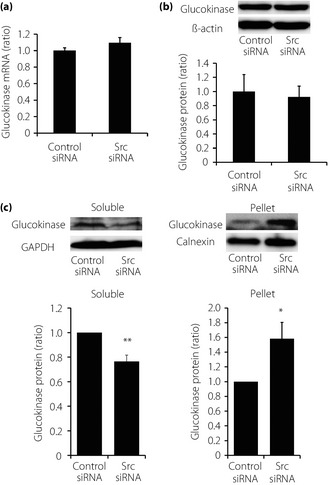
Expression levels and subcellular localization of glucokinase. (a) Effects of Src downregulation on expression level of glucokinase messenger ribonucleic acid (mRNA). Expression level of glucokinase was evaluated with semiquantitative real‐time polymerase chain reaction. Data were normalized using β‐actin mRNA (n = 5 in each group). Values are expressed as mean ± standard error of the mean. (b) Effects of Src downregulation on expression level of glucokinase protein. Lysates of whole INS‐1 cells were used for immunoblotting analysis with antibodies against glucokinase. Quantification data were obtained from four independent experiments and normalized with β‐actin level. Values are expressed as mean ± standard error of the mean. (c) Effects of Src downregulation on subcellular localization of glucokinase. Lysates of whole INS‐1 cells were separated to soluble fraction and pellet. Both fractions were used for immunoblotting analysis with antibodies against glucokinase. Quantification data for each fraction were obtained from four independent experiments and normalized with glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) levels for soluble fraction and calnexin levels for pellet. Values are expressed as mean ± standard error of the mean. *P < 0.05 and **P < 0.01 compared with control small interfering ribonucleic acid (siRNA).
Interaction of glucokinase with nNOS
It has been reported that glucokinase localization and activity is regulated by nNOS19. Therefore, we examined whether Src regulates the association of glucokinase with nNOS by immunoprecipitation using anti‐nNOS antibody, followed by immunoblotting using anti‐glucokinase and anti‐nNOS antibody. Interaction between glucokinase and nNOS was facilitated by Src downregulation without affecting the protein expression level of nNOS (Figure 5a,b).
Figure 5.
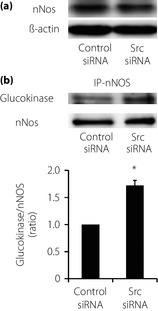
Effects of Src downregulation on interaction of glucokinase and neuronal nitric oxide synthase (nNOS). (a) Expression level of nNOS in INS‐1 cells. Lysates of whole INS‐1 cells were used for immunoblotting analysis with anti‐nNOS antibody. Representative blots were presented out of four independent examinations. (b) Interaction of glucokinase and nNOS was estimated by immunoprecipitation. Lysates of whole INS‐1 cells were used for immunoprecipitation by anti‐nNOS antibody, followed by immunoblotting using anti‐glucokinase antibody or anti‐nNOS antibody (n = 3). Values are expressed as mean ± standard error of the mean. *P < 0.05 compared with control small interfering ribonucleic acid (siRNA).
Discussion
We previously reported that Src inhibition by short exposure to PP2 for 30–60 min reduced overproduction of ROS, and restored impaired glucose metabolism and GIIS in pathophysiological conditions, such as that in diabetic Goto‐Kakizaki rat islets and INS‐1 cells exposed to palmitate, but showed little effect on these parameters in physiological conditions3, 4, 5, 6. However, the more prolonged effects of Src inhibition on pancreatic β‐cells in physiological conditions are unknown. The aim of the present study was to clarify the physiological role of Src in pancreatic β‐cells in an unstressed condition.
To estimate the role of Src in β‐cell function, we first examined insulin secretion. Src downregulation reduced GIIS, but had no effect on high K+‐induced insulin secretion (Figure 2a,c), and the suppressive effect of Src downregulation on [Ca2+]i elevation during high K+ exposure was milder than that during glucose exposure (Figure 2e,f). Glucose‐induced increase in intracellular ATP content was also decreased by Src downregulation (Figure 2d). These results show that Src has some effects on glucose metabolism.
To narrow down the candidate site for Src in β‐cells, we examined the activity of glycolysis and mitochondrial function. In other cells, Src is reported to be involved in the glycolysis pathway20, 21 and in the expression of glucose transporters22, 23. Src downregulation suppressed glucose utilization in the medium containing 10 mmol/L glucose significantly, and reduced the activity of glucokinase, a rate‐limiting enzyme in glycolysis, which determines glucose metabolism in β‐cells (Figure 3a,b), but Src downregulation did not affect GLUT2 expression (Figure S1). Src also plays an important role in mitochondrial function in other cells24, 25, 26, and mitochondrial oxidative phosphorylation is essential in ATP production, resulting in inactivation of ATP sensitive potassium channels and membrane depolarization. Mitochondrial ATP production and protein expression levels of mitochondrial respiratory chain complex were therefore examined. However, Src downregulation did not affect ATP production significantly when mitochondrial fraction was incubated in the presence of succinate plus rotenone or N,N,N',N'‐tetramethyl‐p‐phenylenediamine plus ascorbate, which renders electrons at complex II or complex IV, respectively (Figure 3c). Protein expression levels of several mitochondrial respiratory chain complex proteins were not altered prominently by Src downregulation in the present study, which implies that mitochondrial mass was not affected (Figure 3d). Apparently, the suppressive effects of Src downregulation on glucose metabolism are derived from decrease in glycolysis as a result of reduced glucokinase activity and not from a decrease in capacity to produce ATP in mitochondria.
In β‐cells, glucose metabolism is regulated mainly by reaction at glucokinase in glycolysis, which has far less velocity than those of reactions at other enzymes in glycolysis other than glucokinase and glucose transport27. Glucokinase activity is often regulated by its expression level28, 29, 30, but it was not affected by Src downregulation in the present study. Glucokinase activity is modified by various post‐translational mechanisms including translocation17, 31, 32, S‐nitrosylation19, 33, 34 and interaction with other molecules35, 36. In β‐cells, glucokinase is localized to insulin granules17, 37; it has been reported that glucokinase translocates from the membrane of insulin granules to the cytosol in response to glucose or insulin stimuli, and that glucokinase activity is enhanced by translocation. A fractionation study was therefore carried out, and it was found that Src downregulation reduced and increased protein levels of glucokinase in soluble, cytoplasmic fraction and pellet containing intracellular organelles, respectively (Figure 4c), which is consistent with the results that suppression of glucokinase activity in whole cell extracts is accompanied with a decrease in cytosolic glucokinase17, 32. It is possible that Src downregulation shifted glucokinase from cytosol to membrane of insulin granules, which resulted in suppression of its activity.
As mentioned, Src is a non‐receptor tyrosine kinase. Glucokinase is reported to contain serine phosphorylation sites, but not tyrosine38, 39, and glucokinase has not been considered as a substrate of Src7, 40, 41, 42. Because interaction of Src with glucokinase was not observed by immunoprecipitation (data not shown), Src is not likely to interact with glucokinase directly. Therefore, glucokinase would be regulated by Src through an indirect mechanism.
Regarding the mechanism of glucokinase translocation and activation, Rizzo et al.19 elucidated detail. Subcellular localization of glucokinase is determined by nNOS localization, and insulin treatment increases glucokinase S‐nitorosylation nNOS dependently, resulting in dissociation from insulin granules19. Naturally occurring mutations of glucokinase cause defects in S‐nitrosylation and block glucokinase activation in β‐cells33. Glucagon‐like peptide‐1, an incretin, nitrosylates and activates glucokinase34. Hao et al.32 also showed that cholesterol exposure increases glucokinase–nNOS binding accompanied by reduced glucokinase translocation from membrane‐bound fraction to cytoplasmic fraction and inactivation of the enzyme. Src regulates nNOS activity and NO production in neurons43. Facilitation of glucokinase‐nNOS binding by Src downregulation (Figure 5) implies Src regulation of glucokinase localization and activation in a nNOS dependent manner, and that S‐nitrosylation of glucokinase might be maintained at a low level while involvement of glucokinase S‐nitrosylation is unclear. Whether Src regulates nNOS directly or indirectly is also still unclear.
In the present study, the suppressive effects of Src downregulation on glucose metabolism and insulin secretion were as a result of reduction of glucokinase activity, an underlying mechanism that differs from our previous reports that Src hyperactivation‐related oxidative stress decreased glucose metabolism3, 4, 5, 6. These differences illustrate the diversity of Src function, which is especially dependent on cellular environments or conditions. How Src‐dependent glucokinase activation is affected in the diabetic condition will be investigated in future study.
In conclusion, Src‐involved regulation of glucokinase activity by shifting its subcellular localization to cytosol plays an important role in GIIS in pancreatic β‐cells. Further examination of Src signaling could provide further insight into the mechanisms of glycolysis and glucose metabolism.
Disclosure
The authors declare no conflict of interest.
Supporting information
Figure S1| Expression levels of glucose transporter 2.
Table S1| Small interfering ribonucleic acid (siRNA) sequences.
Table S2| Primer sequences used in semiquantitative real‐time polymerase chain reaction (PCR).
Acknowledgments
We thank M Okada and C Oneyama (Department of Oncogene Research, Research Institute for Microbial Diseases, Osaka University, Osaka, Japan) for useful discussions, and C Kotake, S Yasui and M Akazawa for technical assistance. This work was supported by JSPS KAKENHI Grant Numbers 25461346; a Grant‐in‐Aid for Scientific Research on Innovative Areas “Oxygen Biology: a new criterion for integrated understanding of life” (No. 26111004) of The Ministry of Education, Culture, Sports, Science and Technology, Japan; grants for project research (Development of Fundamental Technology for Analysis and Evaluation of Functional Agricultural Products and Functional Foods) from the Ministry of Agriculture, Forestry and Fisheries of Japan; grants from the Ministry of Health, Labor, and Welfare of Japan; a grant from the Advanced Research for Medical Products Mining Program of the National Institute of Biomedical Innovation (NIBIO); a grant from Core Research for Evolutional Science and Technology (CREST) of Japan Science and Technology Cooperation; and grants from Kyoto University Global COE Program “Center for Frontier Medicine”.
J Diabetes Investig 2016; 7: 171–178
References
- 1. Maechler P, Wollheim CB. Mitochondrial function in normal and diabetic β‐cells. Nature 2001; 414: 807–812. [DOI] [PubMed] [Google Scholar]
- 2. Montane J, Cadavez L, Novials A. Stress and the inflammatory process: a major cause of pancreatic cell death in type 2 diabetes. Diabetes Metab Syndr Obes 2014; 7: 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fujimoto S, Mukai E, Inagaki N. Role of endogenous ROS production in impaired metabolism‐secretion coupling of diabetic pancreatic β cells. Prog Biophys Mol Biol 2011; 107: 304–310. [DOI] [PubMed] [Google Scholar]
- 4. Kominato R, Fujimoto S, Mukai E, et al Src activation generates reactive oxygen species and impairs metabolism‐secretion coupling in diabetic Goto‐Kakizaki and ouabain treated rat pancreatic islets. Diabetologia 2008; 51: 1226–1235. [DOI] [PubMed] [Google Scholar]
- 5. Mukai E, Fujimoto S, Sato H, et al Exendin‐4 suppresses SRC activation and reactive oxygen species production in diabetic Goto‐Kakizaki rat islets in an Epac‐dependent manner. Diabetes 2011; 60: 218–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sato Y, Fujimoto S, Mukai E, et al Palmitate induces reactive oxygen species production and β‐cell dysfunction by activating nicotinamide adenine dinucleotide phosphate oxidase through Src signaling. J Diabetes Investig 2014; 5: 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brown MT, Cooper JA. Regulation, substrates and functions of src. Biochim Biophys Acta 1996; 1287: 121–149. [DOI] [PubMed] [Google Scholar]
- 8. Yeatman TJ. A renaissance for SRC. Nat Rev Cancer 2004; 4: 470–480. [DOI] [PubMed] [Google Scholar]
- 9. Hunter T. Tyrosine phosphorylation: thirty years and counting. Curr Opin Cell Biol 2009; 21: 140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Broca C, Quoyer J, Costes S, et al β‐Arrestin 1 is required for PAC1 receptor‐mediated potentiation of long‐lasting ERK1/2 activation by glucose in pancreatic β‐cells. J Biol Chem 2008; 284: 4332–4342. [DOI] [PubMed] [Google Scholar]
- 11. Zhang F, Zhang Q, Tengholm A, et al Involvement of JAK2 and Src kinase tyrosine phosphorylation in human growth hormone‐stimulated increases in cytosolic free Ca2+ and insulin secretion. Am J Physiol Cell Physiol 2006; 291: C466–C475. [DOI] [PubMed] [Google Scholar]
- 12. Weaver JR, Taylor‐Fishwick DA. Regulation of NOX‐1 expression in beta cells: a positive feedback loop involving the Src‐kinase signaling pathway. Mol Cell Endocrinol 2013; 369: 35–41. [DOI] [PubMed] [Google Scholar]
- 13. Buteau J, Foisy S, Joly E, et al Glucagon‐like peptide 1 induces pancreatic β‐cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes 2003; 52: 124–132. [DOI] [PubMed] [Google Scholar]
- 14. Nishi Y, Fujimoto S, Sasaki M, et al Role of mitochondrial phosphate carrier in metabolism‐secretion coupling in rat insulinoma cell line INS‐1. Biochem J 2011; 435: 421–430. [DOI] [PubMed] [Google Scholar]
- 15. Sasaki M, Fujimoto S, Sato Y, et al Reduction of reactive oxygen species ameliorates metabolism‐secretion coupling in islets of diabetic GK rats by suppressing lactate overproduction. Diabetes 2013; 62: 1996–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shimodahira M, Fujimoto S, Mukai E, et al Rapamycin impairs metabolism‐secretion coupling in rat pancreatic islets by suppressing carbohydrate metabolism. J Endocrinol 2010; 204: 37–46. [DOI] [PubMed] [Google Scholar]
- 17. Rizzo MA, Magnuson MA, Drain PF, et al A functional link between glucokinase binding to insulin granules and conformational alterations in response to glucose and insulin. J Biol Chem 2002; 277: 34168–34175. [DOI] [PubMed] [Google Scholar]
- 18. Okada M. Regulation of the SRC family kinases by Csk. Int J Biol Sci 2012; 8: 1385–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rizzo MA, Piston DW. Regulation of β cell glucokinase by S‐nitrosylation and association with nitric oxide synthase. J Cell Biol 2003; 161: 243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gupte RS, Floyd BC, Kozicky M, et al Synergistic activation of glucose‐6‐phosphate dehydrogenase and NAD(P)H oxidase by Src kinase elevates superoxide in type 2 diabetic, Zucker fa/fa, rat liver. Free Radic Biol Med 2009; 47: 219–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang W, Xia Y, Ji H, et al Nuclear PKM2 regulates β‐catenin transactivation upon EGFR activation. Nature 2011; 480: 118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee JO, Lee SK, Kim JH, et al Metformin regulates glucose transporter 4 (GLUT4) translocation through AMP‐activated protein kinase (AMPK)‐mediated Cbl/CAP signaling in 3T3‐L1 preadipocyte cells. J Biol Chem 2012; 287: 44121–44129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Matsouka PT, Flier JS. Relationship between c‐src tyrosine kinase activity and the control of glucose transporter gene expression. Mol Endocrinol 1989; 3: 1845–1851. [DOI] [PubMed] [Google Scholar]
- 24. Livigni A, Scorziello A, Agnese S, et al Mitochondrial AKAP121 links cAMP and src signaling to oxidative metabolism. Mol Biol Cell 2006; 17: 263–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hebert‐Chatelain E. Src kinases are important regulators of mitochondrial functions. Int J Biochem Cell Biol 2013; 45: 90–98. [DOI] [PubMed] [Google Scholar]
- 26. Tibaldi E, Brunati AM, Massimino ML, et al Src‐Tyrosine kinases are major agents in mitochondrial tyrosine phosphorylation. J Cell Biochem 2008; 104: 840–849. [DOI] [PubMed] [Google Scholar]
- 27. Meglasson MD, Matschinsky FM. Pancreatic islet glucose metabolism and regulation of insulin secretion. Diabetes Metab Rev 1986; 2: 163–214. [DOI] [PubMed] [Google Scholar]
- 28. Liang Y, Najafi H, Smith RM, et al Concordant glucose induction of glucokinase, glucose usage, and glucose‐stimulated insulin release in pancreatic islets maintained in organ culture. Diabetes 1992; 41: 792–806. [DOI] [PubMed] [Google Scholar]
- 29. Jung H, Joo J, Jeon Y, et al Advanced glycation end products downregulate glucokinase in mice. Diabetes Metab Res Rev 2011; 27: 557–563. [DOI] [PubMed] [Google Scholar]
- 30. Weinhaus AJ, Stout LE, Bhagroo NV, et al Regulation of glucokinase in pancreatic islets by prolactin: a mechanism for increasing glucose‐stimulated insulin secretion during pregnancy. J Endocrinol 2007; 193: 367–381. [DOI] [PubMed] [Google Scholar]
- 31. Noma Y, Bonner‐Weir S, Latimer JB, et al Translocation of glucokinase in pancreatic β‐cells during acute and chronic hyperglycemia. Endocrinology 1996; 137: 1485–1491. [DOI] [PubMed] [Google Scholar]
- 32. Hao M, Head WS, Gunawardana SC, et al Direct effect of cholesterol on insulin secretion: a novel mechanism for pancreatic β‐cell dysfunction. Diabetes 2007; 56: 2328–2338. [DOI] [PubMed] [Google Scholar]
- 33. Ding SY, Tribble ND, Kraft CA, et al Naturally occurring glucokinase mutations are associated with defects in posttranslational S‐nitrosylation. Mol Endocrinol 2010; 24: 171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ding SY, Nkobena A, Kraft CA, et al Glucagon‐like peptide 1 stimulates post‐translational activation of glucokinase in pancreatic β‐Cells. J Biol Chem 2011; 286: 16768–16774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shiraishi A, Yamada Y, Tsuura Y, et al A novel glucokinase regulator in pancreatic β cells: precursor of propionyl‐CoA carboxylase β subunit interacts with glucokinase and augments its activity. J Biol Chem 2001; 276: 2325–2328. [DOI] [PubMed] [Google Scholar]
- 36. Massa L, Baltrusch S, Okar DA, et al Interaction of 6‐phosphofructo‐2‐kinase/fructose‐2, 6‐bisphosphatase (PFK‐2/FBPase‐2) with glucokinase activates glucose phosphorylation and glucose metabolism in insulin‐producing cells. Diabetes 2004; 53: 1020–1029. [DOI] [PubMed] [Google Scholar]
- 37. Toyoda Y, Yoshie S, Shironoguchi H, et al Glucokinase is concentrated in insulin‐secretory granules of pancreatic β‐cells. Histochem Cell Biol 1999; 112: 35–40. [DOI] [PubMed] [Google Scholar]
- 38. Ekman P, Nilsson E. Phosphorylation of glucokinase from rat liver in vitro by protein kinase A with a concomitant decrease of its activity. Arch Biochem Biophys 1988; 261: 275–282. [DOI] [PubMed] [Google Scholar]
- 39. Liu Q, Shen Y, Liu S, et al Crystal structure of E339K mutated human glucokinase reveals changes in the ATP binding site. FEBS Lett 2011; 585: 1175–1179. [DOI] [PubMed] [Google Scholar]
- 40. Courtneidge SA. Isolation of novel Src substrates. Biochem Soc Trans 2003; 31: 25–28. [DOI] [PubMed] [Google Scholar]
- 41. Amanchy R, Zhong J, Molina H, et al Identification of c‐Src tyrosine kinase substrates using mass spectrometry and peptide microarrays. J Proteome Res 2008; 7: 3900–3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ferrando IM, Chaerkady R, Zhong J, et al Identification of targets of c‐Src tyrosine kinase by chemical complementation and phosphoproteomics. Mol Cell Proteomics 2012; 11: 355–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gingerich S, Krukoff TL. Activation of ERβ increases levels of phosphorylated nNOS and NO production through a Src/PI3K/Akt‐dependent pathway in hypothalamic neurons. Neuropharmacology 2008; 55: 878–885. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1| Expression levels of glucose transporter 2.
Table S1| Small interfering ribonucleic acid (siRNA) sequences.
Table S2| Primer sequences used in semiquantitative real‐time polymerase chain reaction (PCR).