

Abstract
Aims/Introduction
According to some authors, in type 2 diabetes there is a reduced postprandial action of glucagon‐like peptide‐1 (GLP‐1) and glucose‐dependent insulinotropic polypeptide (GIP). However, little is known about the role of fasting incretins in glucose homeostasis. Our aim was to evaluate, through a two‐step cluster analysis, the possibility of phenotyping patients with type 2 diabetes at onset on the basis of fasting GLP‐1, GIP and ghrelin.
Materials and Methods
A total of 96 patients with type 2 diabetes within 6 months of onset (mean age 62.40 ± 6.36 years) were cross‐sectionally studied. Clinical, anthropometric and metabolic parameters were evaluated. At fasting the following were carried out: assay of GLP‐1, GIP, ghrelin, insulin, C‐peptide, glucagon and a panel of adipocytokines (visfatin, resistin, leptin, soluble leptin receptor and adiponectin).
Results
The analysis resulted in two clusters: cluster 1 (63 patients) had significantly lower levels of GLP‐1 (4.93 ± 0.98 vs 7.81 ± 1.98 pmol/L; P < 0.001), GIP (12.73 ± 9.44 vs 23.88 ± 28.56 pmol/L; P < 0.001) and ghrelin (26.54 ± 2.94 vs 39.47 ± 9.84 pmol/L; P < 0.001) compared with cluster 2 (33 patients). Between the two clusters, no differences in age, duration of disease, sex, clinical‐anthropometric parameters, insulin sensitivity and adipocytokines were highlighted. However, cluster 1 was associated with significantly higher levels of glycated hemoglobin (7.4 ± 0.61 vs 6.68 ± 0.57%, P = 0.007), glucagon (232.02 ± 37.27 vs 183.33 ± 97.29 ng/L; P = 0.001), fasting glucose (7.85 ± 1.60 vs 6.93 ± 1.01 mmol/L; P = 0.003) and significantly lower levels of C‐peptide (0.12 ± 0.11 vs 0.20 ± 0.20 nmol/L; P = 0.017).
Conclusions
The present study suggests that fasting incretins play an important role in the pathophysiology of type 2 diabetes, which requires to further investigation.
Keywords: Ghrelin, Glucagon‐like peptide‐1, Glucose‐dependent insulinotropic polypeptide
Introduction
It is known that in normal subjects, oral glucose administration elicits a higher insulin response than intravenous glucose load at identical plasma glucose concentrations; this phenomenon, the incretin effect, is mainly attributed to the postprandial action of glucagon‐like peptide‐1 (GLP‐1) and glucose‐dependent insulinotropic polypeptide (GIP)1, 2, 3, 4. However, in recent years, it has been debated whether post‐load GLP‐1 and GIP secretion really are lower in patients with type 2 diabetes mellitus compared with matched healthy controls5, 6. A recent meta‐analysis suggested that patients with type 2 diabetes, in general, do not show reduced GLP‐1 secretion in response to an oral glucose tolerance test or meal test, and that it is the deterioration of glycemic control that is associated with reduced GLP‐1 secretion7.
Furthermore, in addition to GLP‐1 and GIP, another incretin, ghrelin, produced in the gastrointestinal tract, though not involved in the postprandial incretin effect described, plays an important role in glucose homeostasis8. The ghrelin effect on plasma insulin and glucose levels in humans, which is produced mainly in the fasting state, is still a matter of debate. Some groups have found no changes9, 10, 11, whereas others have found increased fasting blood glucose and decreased plasma insulin after administration of ghrelin12.
To date, there is a great deal of literature, although controversial, on the postprandial incretin effect and its role in the pathogenesis of type 2 diabetes; conversely, little is known about the role of fasting incretin tone, and especially about the impact of this on the pathophysiology of type 2 diabetes.
In the present study, through a two‐step cluster analysis we evaluated the possibility of phenotyping patients with type 2 diabetes at onset on the basis of fasting serum levels of GLP‐1, GIP and ghrelin.
Materials and Methods
A total of 96 consecutive Caucasian patients with type 2 diabetes within 6 months of onset (mean age 62.40 ± 6.36 years, range 51–75 years), followed up in our dedicated outpatients clinic at Unit of Endocrinology, Diabetology and Metabolism, University of Palermo, Palermo, Italy (from 1 March 2012 to 31 December 2013), were cross‐sectionally studied. Inclusion criteria were type 2 diabetes known for <6 months and in stable treatment for the last 3 months with metformin (1.5–2 g/day). Patients with glycated hemoglobin (HbA1c) ≥8 (64 mmol/mol) were excluded, in order to avoid the effects of severe glucotoxicity on incretins and adipocytokines. The following were excluded: patients with type 1 diabetes and patients with previous treatment with other antidiabetic drugs taken in the past 6 months. To avoid interfering with the Visceral Adiposity Index, patients with morbid obesity, pendulous abdomen, severe hypertriglyceridemia and/or or use of fibrates were excluded13, 14. Patients with macro‐ or microvascular complications were excluded (except 8 patients with simple preproliferative retinopathy). Patients were not excluded if they had essential hypertension (62 patients). A complete medical history, a complete physical examination, bodyweight, height, and waist and hip circumference were recorded for the patients included. Waist circumference (WC) was measured midway between the inferior margin of the last rib and the crest of the ilium in a horizontal plane by the measurer sitting by the subject and fitting the tape snugly, but not compressing soft tissues. Hip circumference (HP) was measured around the pelvis at the point of maximal protrusion of the buttocks. Fasting blood samples and a morning spot urine sample were collected for biochemical analyses. All participants were instructed to carry out home glucose monitoring (one full‐day glucose profile per week: fasting, 2 h after breakfast, 2 h after lunch and 2 h after dinner).
The study was approved by the institutional review board at the Faculty of Medicine of the University of Palermo. At the time of observation, all patients regularly signed an informed consent for scientific use of their data.
Anthropometric Indices
Body mass index (BMI) was calculated as bodyweight (in kilograms) divided by the square of height (in meters). Waist‐to‐hip ratio was calculated by dividing the waist circumference by the hip circumference. The Body Adiposity Index (BAI) was calculated using the following formula15:
Hip circumference (HC) is expressed in centimeters and height in meters.
The Visceral Adiposity Index (VAI) was calculated as described13, 14 using the following sex‐specific equation, where triglyceride (TG) levels are expressed in mmol/L and high‐density lipoprotein (HDL) cholesterol levels expressed are mmol/L: Females:
Males:
Secretory Units of Islets in Transplantation
As a measure of β‐cell function, the secretory units of islets in transplantation (SUIT)16 were calculated from the fasting plasma glucose (mg/dL) and fasting C‐peptide (ng/mL) levels using the formula:
Assay
The BioPlex Pro Human Diabetes 10‐plex assay (BioRad, Milan, Italy) was used to quantitate the following diabetes‐related analytes: C‐peptide (nmol/L), ghrelin (pg/mL), total GLP‐1 (pmol/L), total GIP (pmol/L), leptin (ng/mL), resistin (ng/mL) and visfatin (pg/mL). The BioPlex Pro Human Diabetes Adipsin and Adiponectin duplex assay (BioRad) was used for adipsin (ng/mL) and adiponectin (μg/mL) assays. The intra‐ and interassay coefficients of variability were 4% and 2% for ghrelin, 3% and 4% for GIP, and 6% and 3% for GLP‐1, respectively.
All these assays were run on a BioPlex 200 workstation (BioRad). Human soluble leptin receptor (sOb‐R; ng/mL) was assayed using an enzyme‐linked immunosorbent assay sandwich enzyme immunoassay (Human leptin receptor ELISA; BioVendor, Heidelberg, Germany). Serum non‐esterified fatty acids (mmol/L) were assayed after overnight fasting using an enzymatic colorimetric method (Randox NEFA assay FA115; Randox Laboratories, County Antrim, UK). Blood glucose levels (mg/dL) were measured using an electrochemical system (Glucocard; Menarini Diagnostics, Florence, Italy). Total cholesterol, HDL cholesterol, triglycerides, and urinary albumin and creatinine were measured in our laboratory using standard assays. HbA1c was determined by high‐pressure liquid chromatography with ion‐exchange resin (Bio‐Rad D‐10 HPLC analyzer). Low‐density lipoprotein cholesterol levels were calculated with Friedewald's formula. The conversion factors for the International System were the following: glucose (mg/dL vs mmol/L; ×0.0555), insulin (mUI/L vs pmol/L; ×6.945), total cholesterol (mg/dL vs mmol/L; ×0.0259), visfatin (pg/mL vs ng/mL; ×1000), Ghrelin (pg/mL vs pmol/L; ×0.296).
Statistical Analysis
The Statistical Packages for Social Sciences, spss version 17 (SPSS, Chicago, IL, USA), was used for data analysis. Baseline characteristics were presented as mean ± standard deviation for continuous variables; rates and proportions were calculated for categorical data. Normality of distribution for quantitative variables was assessed by the Shapiro–Wilk test. Some variables (leptin, leptin/sOb‐R ratio, adiponectin and resistin) did not show normal distribution, and the natural logarithmic transformed values of each adipocytokine for statistical analysis were used. Differences between the two groups were detected by the unpaired Student's t‐test for continuous variables (after testing for equality of variance: Levene test) and by the chi square‐test and Fisher's exact test (when appropriate) for categorical variables.
In order to identify the phenotypes of type 2 diabetes based on fasting incretin tone, we carried out a two‐step cluster analysis using log‐likelihood distance measures. Cluster analysis, also called segmentation analysis, is an explorative analysis that tries to identify structures within the data. More specifically, it tries to identify homogenous groups of cases based on the distribution of some variables (input variables). Cluster analysis is used to identify groups of cases if the grouping is not previously known. In particular, a two‐step cluster analysis is more of a tool than a single analysis. It identifies the groupings by running pre‐clustering first and then by hierarchical methods. In this respect, it combines the best of both approaches. This technique can detect latent relationships within a complex dataset between patients with multiple distinct characteristics. Cluster analysis was applied using an a priori number of fixed clusters (cluster 1 and cluster 2), and the three following continuous variables were included in the analysis (input variables): fasting GLP‐1, GIP and ghrelin. The three variables included produced a silhouette coefficient >0.5, indicative of good data partitioning (Figure 1a). In this model, the most important variables in the creation of the two clusters were GLP‐1 and GIP (Figure 1a).
Figure 1.
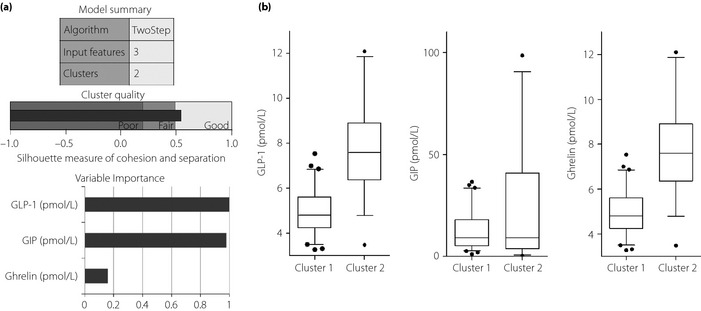
(a) Model summary of the two‐step cluster analysis with a graph of the quality of clusters and importance of the three input variables. (b) Differences in serum levels of glucagon‐like peptide‐1 (GLP‐1), glucose‐dependent insulinotropic polypeptide (GIP) and ghrelin between cluster 1 and cluster 2; the ends of the whiskers represent the 5th and 95th percentiles.
For comparison between cluster 1 and cluster 2, the group sizes gave 74.7% power to detect a moderate effect size (Cohen's d = 0.5) using unpaired t‐test, with alpha at 0.05. Post‐hoc power analysis was carried out using g*power Version 3.1.6 software (Heinrich‐Heine‐Universität Düsseldorf, Düsseldorf, Germany). A P‐value of <0.05 was considered statistically significant.
Results
The 96 patients studied were 51 women and 45 men; the clinical characteristics, and the incretin and adipocytokine patterns of the patients are summarized in Table 1.
Table 1.
Clinical‐anthropometric characteristics, incretin and adipocytokine patterns in 96 patients with type 2 diabetes
| Clinical‐anthropometric characteristics | n (%) |
|---|---|
| Women | 51 (53.1) |
| Men | 45 (46.9) |
| Mean ± SD | |
|---|---|
| Age (years) | 62.40 ± 6.36 |
| Duration of disease (months) | 4.41 ± 1.66 |
| BMI (kg/m2) | 29.29 ± 4.65 |
| Waist circumference (cm) | 102.08 ± 11.31 |
| Hip circumference (cm) | 107.23 ± 10.28 |
| Waist‐to‐hip ratio | 0.95 ± 0.08 |
| Body Adiposity Index | 33.94 ± 6.47 |
| Visceral Adiposity Index | 2.28 ± 1.33 |
| HbA1c | |
| % | 6.92 ± 0.62 |
| mmol/mol | 52 ± 6.80 |
| HOMA‐IR | 3.08 ± 3.22 |
| Fasting insulin (pmol/L) | 64.32 ± 67.26 |
| Fasting C‐peptide (nmol/L) | 0.15 ± 0.15 |
| Fasting glucagon (ng/L) | 215.28 ± 68.08 |
| Urinary albumin‐to‐creatinine ratio (mg/g) | 16.69 ± 38.10 |
| Fasting lipids | |
| Total cholesterol (mmol/L) | 4.58 ± 0.85 |
| HDL cholesterol (mmol/L) | 1.25 ± 0.32 |
| LDL cholesterol (mmol/L) | 2.76 ± 0.84 |
| Triglycerides (mmol/L) | 1.41 ± 0.56 |
| NEFA (mmol/L) | 0.46 ± 0.27 |
| Fasting incretins | |
| GLP‐1 (pmol/L) | 5.92 ± 1.96 |
| GIP (pmol/L) | 16.56 ± 19.01 |
| Ghrelin (pmol/L) | 30.99 ± 8.74 |
| Fasting adipocytokines | |
| Leptin (ng/dL) | 8.83 ± 10.45 |
| sOb‐R (ng/mL) | 21.70 ± 6.56 |
| Leptin/sOb‐R ratio | 0.55 ± 0.88 |
| Adiponectin (μg/mL) | 11.75 ± 12.94 |
| Visfatin (ng/mL) | 0.62 ± 0.52 |
| Resistin (ng/mL) | 4.01 ± 4.68 |
BMI, body mass index; GIP, glucose‐dependent insulinotropic polypeptide; GLP‐1, glucagon‐like peptide‐1; HbA1c, glycated hemoglobin; HDL, high‐density lipoprotein; HOMA‐IR, homeostatic model addessment of insulin resistance; LDL, low‐density lipoprotein; NEFA, non‐esterified fatty acids; SD, standard deviation; sOB‐R, soluble leptin receptor.
The two clusters derived from the cluster analysis showed the following fasting incretin patterns: compared with cluster 2 (33 patients), cluster 1 (63 patients) showed significantly lower levels of GLP‐1 (4.93 ± 0.98 vs 7.81 ± 1.98 pmol/L; P < 0.001), GIP (12.73 ± 9.44 vs 23.88 ± 28.56 pmol/L; P < 0.001) and ghrelin (26.54 ± 2.94 vs 39.47 ± 9.84 pmol/L; P < 0.001; Figure 1b).
Regarding the clinical and anamnestic characteristics of the patients, we did not find any significant differences between the two clusters, except for a greater prevalence of patients practicing physical activity in cluster 2 (27.3 vs 11.1%; P = 0.044; Table 2). Similarly, no significant differences in anthropometric parameters were found (Table 2).
Table 2.
Differences in clinical and anthropometric characteristics between cluster 1 and cluster 2
| Cluster 1 n (%) | Cluster 2 n (%) | P | |
|---|---|---|---|
| n = 63 | n = 33 | ||
| Clinical characteristics | |||
| Sex | |||
| Male | 30 (47.6) | 15 (45.5) | 0.840 |
| Female | 33 (52.4) | 18 (54.5) | |
| Family history of diabetes | 51 (81) | 24 (72.7) | 0.437 |
| Dietary compliance | 43 (68.3) | 21 (63.6) | 0.819 |
| Physical activity | 7 (11.1) | 9 (27.3) | 0.044 |
| Smoking | |||
| Current | 10 (15.9) | 4 (12.1) | 0.845 |
| Former | 13 (20.6) | 8 (24.2) | |
| Metabolic syndromea | 46 (73) | 22 (66.7) | 0.516 |
| High blood pressure | 42 (66.7) | 20 (60.6) | 0.555 |
| High triglycerides | 20 (31.7) | 6 (18.2) | 0.155 |
| Low HDL cholesterol | 40 (63.5) | 21 (63.6) | 0.989 |
| Increased WC | 41 (65.1) | 19 (57.6) | 0.471 |
| Mean ± SD | Mean ± SD | ||
|---|---|---|---|
| Age (years) | 61.94 ± 6.14 | 63.27 ± 6.78 | 0.348 |
| Duration of disease (months) | 4.51 ± 1.66 | 4.21 ± 1.67 | 0.413 |
| Anthropometric parameters | |||
| BMI (kg/m2) | 29.56 ± 4.49 | 28.79 ± 4.98 | 0.461 |
| WC (cm) | 103.63 ± 10.62 | 99.12 ± 12.13 | 0.076 |
| Hip circumference (cm) | 107.60 ± 9.92 | 106.54 ± 11.05 | 0.647 |
| Waist‐to‐hip ratio | 0.96 ± 0.09 | 0.93 ± 0.08 | 0.067 |
| Body Adiposity Index | 33.90 ± 6.20 | 34.02 ± 7.04 | 0.931 |
| Visceral Adiposity Index | 2.36 ± 1.43 | 2.12 ± 1.12 | 0.364 |
Student's t‐test after testing for equality of variance (Levene test).
According to Adult Treatment Panel (ATP) III criteria. BMI, body mass index; HDL, high‐density lipoprotein; SD, standard deviation; WC, waist circumference.
Cluster 1 showed a worse glyco‐metabolic profile compared with cluster 2: higher levels of HbA1c (7.41 ± 0.61 vs 6.68 ± 0.57%; P = 0.007) and fasting plasma glucose (7.85 ± 1.60 vs 6.93 ± 1.01 mmol/L; P = 0.003) were found; no difference was found between the two clusters in regard to postprandial glucose levels. Regarding pancreatic function, cluster 1 showed significantly higher levels of fasting flucagon (232.02 ± 37.27 vs 183.33 ± 97.29 ng/L; P = 0.001), lower levels of fasting C‐peptide (0.12 ± 0.11 vs 0.20 ± 0.20 nmol/L; P = 0.017) and secretory units of islets in transplantation (7.66 ± 6.35 vs 16.00 ± 16.78; P = 0.001; Table 3).
Table 3.
Differences in glycometabolic profiles and adipocytokine pattern between cluster 1 and cluster 2
| Cluster 1 | Cluster 2 | P | |
|---|---|---|---|
| n = 63 | n = 33 | ||
| Mean ± SD | Mean ± SD | ||
| Glycometabolic profile | |||
| HbA1c (%) | 7.41 ± 0.61 | 6.68 ± 0.57 | 0.007 |
| HbA1c (mmol/mol) | 57 ± 6.7 | 49 ± 6.2 | 0.007 |
| HOMA‐IR | 2.94 ± 3.08 | 3.35 ± 3.50 | 0.566 |
| Fasting insulin (pmol/L) | 59.07 ± 60.32 | 74.34 ± 78.88 | 0.293 |
| Fasting glucagon (ng/L) | 232.02 ± 37.27 | 183.33 ± 97.29 | 0.001 |
| Fasting C‐peptide (nmol/L) | 0.12 ± 0.11 | 0.20 ± 0.20 | 0.017 |
| Fasting glucose (mmol/L) | 7.85 ± 1.60 | 6.93 ± 1.01 | 0.003 |
| SUIT | 7.66 ± 6.35 | 16.00 ± 16.78 | 0.001 |
| 2 h after breakfast glucose (mmol/L) | 6.96 ± 1.69 | 6.47 ± 0.93 | 0.124 |
| 2 h after lunch glucose (mmol/L) | 7.74 ± 1.88 | 7.23 ± 1.11 | 0.133 |
| 2 h after dinner glucose (mmol/L) | 7.98 ± 2.06 | 7.33 ± 1.48 | 0.082 |
| Urinary albumin‐to‐creatinine ratio (mg/g) | 19.18 ± 45.61 | 11.93 ± 15.62 | 0.257 |
| Fasting lipids | |||
| Total cholesterol (mmol/L) | 4.55 ± 0.86 | 4.65 ± 0.84 | 0.603 |
| HDL cholesterol (mmol/L) | 1.24 ± 0.30 | 1.26 ± 0.36 | 0.753 |
| LDL cholesterol (mmol/L) | 2.71 ± 0.84 | 2.85 ± 0.83 | 0.445 |
| Triglycerides (mmol/L) | 1.45 ± 0.58 | 1.33 ± 0.50 | 0.324 |
| NEFA (mmol/L) | 0.50 ± 0.29 | 0.39 ± 0.21 | 0.051 |
| Fasting adipocytokine profile | |||
| Leptin (ng/dL)a | 9.77 ± 9.61 | 7.03 ± 11.84 | 0.257 |
| sOb‐R (ng/mL)a | 22.20 ± 6.71 | 20.76 ± 6.28 | 0.077 |
| Leptin/sOb‐R ratioa | 0.56 ± 0.69 | 0.54 ± 1.17 | 0.928 |
| Adiponectin (μg/mL)a | 10.46 ± 11.14 | 14.22 ± 15.70 | 0.953 |
| Visfatin (ng/mL) | 0.60 ± 0.51 | 0.64 ± 0.55 | 0.788 |
| Resistin (ng/mL)a | 4.28 ± 5.05 | 3.49 ± 3.90 | 0.089 |
Student's t‐test after testing for equality of variance (Levene test).
The natural logarithmic transformed (Ln) values were used for the Student's t‐test. BMI, body mass index; HbA1c, glycated hemoglobin; HDL, high‐density lipoprotein; HOMA‐IR, homeostatic model addessment of insulin resistance; LDL, low‐density lipoprotein; NEFA, non‐esterified fatty acids; SD, standard deviation; sOB‐R, soluble leptin receptor; SUIT, secretory units of islets in transplantation.
The lipid pattern appeared comparable between the two clusters. No difference was found between the two clusters in regard to adipocytokine profile (Table 3).
Discussion
In the present study we showed that it is possible to phenotype patients with type 2 diabetes at onset based on the values of fasting GLP‐1, GIP and ghrelin; patients with reduced fasting incretin tone are characterized by worse HbA1c and fasting glucose, probably secondary to reduced fasting β‐cell activity associated with increased α‐cell activity.
Regarding the importance of the three variables in the process of clusterization, the results of the cluster analysis show that we should give priority to GLP‐1 and GIP. In this connection, fasting ghrelin, although it proved to be significantly lower in the group with reduced fasting incretin tone, was not decisive in the formation of the two clusters. At present, the effects of fasting plasma ghrelin on fasting plasma insulin and glucose levels in humans are still a matter of debate. Some authors have found no changes9, 10, 11, whereas others have found that acyl ghrelin increased fasting blood glucose and decreased plasma insulin after its administration12, 17, 18. Unfortunately, the present study is not able to contribute to the clarification of these aspects, both because of the slight weight of ghrelin in the formation of the clusters, and because of the limit of having assayed total ghrelin and not acyl ghrelin.
Instead, a more important role in glucose homeostasis in patients with type 2 diabetes seems to be played by fasting levels of GLP‐1 and GIP. It is known that the secretion of GLP‐1 (mainly synthesized by L‐cells in the duodenum, and small and large intestine) in the gastrointestinal tract is influenced by glucose and fatty acids after food ingestion or as a result of vagus nerve stimulation19. The main GLP‐1 action mechanisms involve stimulating insulin secretion by β‐cells in the islets of Langerhans and inhibiting glucagon secretion by α‐cells; increased insulin secretion is the result of its increased synthesis20, 21, 22. The present study has shown that the basal secretion of GLP‐1 also plays an important role in glucose homeostasis, although with our data we were not able to show whether this is the result of secretion by L‐cells or of degradation by dipeptidyl peptidase‐4 (DPP‐4)23, 24. Future studies evaluating the concentration or enzyme activity of DPP‐4 in patients with type 2 diabetes might be clarifiers. Analogous considerations must be made for GIP (a peptide secreted by K‐cells in the mucosa of the duodenum, jejunum and the proximal portion of the ileum) about its incretin activity stimulating food intake‐mediated insulin secretion by β‐cells in the islets of Langerhans24, 25. In this case too, although the postprandial levels of GIP depend on the basic nutrient content of a meal (higher values are observed after the ingestion of carbohydrates, compared with proteins)26, regarding the fasting serum levels we do not know if they are the result of secretion by K‐cells or of degradation by DPP‐423.
In the present study, what we believe to be of particular interest is the similarity between the two groups in regard to the clinical‐anthropometric characteristics and the insulin‐sensitivity.
Even adipose tissue function was similar, as shown by the lack of significant differences between the two clusters in relation to the VAI values, leptin, sOb‐R, leptin/sOb‐R ratio, adiponectin, visfatin and resistin. Although today it is known that adipocytokines form an important part of an ‘adipoinsular axis,’ dysregulation of which could contribute to β‐cell failure and hence to type 2 diabetes27, the present data confirm that the fasting level of incretin hormones do not influence this axis; we still cannot exclude a possible role in postprandial phases. Indeed, it is known that postprandial GLP‐1 amplifies insulin‐mediated glucose uptake in adipocytes27, and this certainly contributes to normal adipocyte function. The same goes for GIP, which appears to provide a benefit by not only stimulating postprandial intestinal glucose transport and releasing insulin to facilitate nutrient storage, but also through its insulin‐mimetic properties, including enhanced uptake of glucose by adipocytes28.
In conclusion, although the physiological importance of the decreased fasting level of incretins in patients with type 2 diabetes requires further clarification, the present study suggests that a simple phenotyping of patients at onset is possible based on these parameters. In the future, similar studies of larger size could also suggest the appropriate cut‐off of GLP‐1 and GIP pointing to reduced incretin tone, in order to better select patients who require treatment with GLP‐1 analogs or DPP4‐inhibitors.
Disclosure
The authors declare no conflict of interest.
Acknowledgments
In memory of our great teacher Professor Aldo Galluzzo. We thank Dr Gabriella Misiano and Dr Salvatore Milano for past collaboration. This study was partially supported by the University of Palermo ‘Fondo Ricerca di Ateneo 2012’ (ID: 2012‐ATE‐0302).
J Diabetes Investig 2016; 7: 219–225
References
- 1. Vilsbøll T, Holst JJ. Incretins, insulin secretion and type 2 diabetes mellitus. Diabetologia 2004; 47: 357–366. [DOI] [PubMed] [Google Scholar]
- 2. Nauck MA, Homberger E, Siegel EG, et al Incretin effects of increasing glucose loads in man calculated from venous insulin and C‐peptide responses. J Clin Endocrinol Metab 1986; 63: 492–498. [DOI] [PubMed] [Google Scholar]
- 3. Muscelli E, Mari A, Casolaro A, et al Separate impact of obesity and glucose tolerance on the incretin effect in normal subjects and type 2 diabetic patients. Diabetes 2008; 57: 1340–1348. [DOI] [PubMed] [Google Scholar]
- 4. Drucker DJ, Nauck MA. The incretin system: glucagon‐like peptide‐1 receptor agonists and dipeptidyl peptidase‐4 inhibitors in type 2 diabetes. Lancet 2006; 368: 1696–1705. [DOI] [PubMed] [Google Scholar]
- 5. Meier JJ, Nauck MA. Is secretion of glucagon‐like peptide‐1 reduced in type 2 diabetes mellitus? Nat Clin Pract Endocrinol Metabol 2008; 4: 606–607. [DOI] [PubMed] [Google Scholar]
- 6. Nauck MA, Vardarli I, Deacon CF, et al Secretion of glucagon‐like peptide‐1 (GLP‐1) in type 2 diabetes: what is up, what is down? Diabetologia 2011; 54: 10–18. [DOI] [PubMed] [Google Scholar]
- 7. Calanna S, Christensen M, Holst JJ, et al Secretion of glucagon‐like peptide‐1 in patients with type 2 diabetes mellitus: systematic review and meta‐analyses of clinical studies. Diabetologia 2013; 56: 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Heppner KM, Tong J. Mechanisms in endocrinology: regulation of glucose metabolism by the ghrelin system: multiple players and multiple actions. Eur J Endocrinol 2014; 171: R21–R32. [DOI] [PubMed] [Google Scholar]
- 9. Tong J, Prigeon RL, Davis HW, et al Ghrelin suppresses glucose‐stimulated insulin secretion and deteriorates glucose tolerance in healthy humans. Diabetes 2010; 59: 2145–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tong J, Prigeon RL, Davis HW, et al Physiologic concentrations of exogenously infused ghrelin reduces insulin secretion without affecting insulin sensitivity in healthy humans. J Clin Endocrinol Metab 2013; 98: 2536–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lucidi P, Murdolo G, Di Loreto C, et al Metabolic and endocrine effects of physiological increments in plasma ghrelin concentrations. Nutr Metab Cardiovasc Dis 2005; 15: 410–417. [DOI] [PubMed] [Google Scholar]
- 12. Broglio F, Arvat E, Benso A, et al Ghrelin, a natural GH secretagogue produced by the stomach, induces hyperglycemia and reduces insulin secretion in humans. J Clin Endocrinol Metab 2001; 86: 5083–5086. [DOI] [PubMed] [Google Scholar]
- 13. Amato MC, Giordano C, Galia M, et al Visceral Adiposity Index: a reliable indicator of visceral fat function associated with cardiometabolic risk. Diabetes Care 2010; 33: 920–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Amato MC, Giordano C. Clinical indications and proper use of Visceral Adiposity Index. Nutr Metab Cardiovasc Dis 2013; 23: e31–e32. [DOI] [PubMed] [Google Scholar]
- 15. Bergman RN, Stefanovski D, Buchanan TA, et al A better index of body adiposity. Obesity 2011; 19: 1083–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yamada Y, Fukuda K, Fujimoto S, et al SUIT, secretory units of islets in transplantation: an index for therapeutic management of islet transplanted patients and its application to type 2 diabetes. Diabetes Res Clin Pract 2006; 74: 222–226. [DOI] [PubMed] [Google Scholar]
- 17. Broglio F, Benso A, Gottero C, et al Non‐acylated ghrelin does not possess the pituitaric and pancreatic endocrine activity of acylated ghrelin in humans. J Endocrinol Invest 2003; 26: 192–196. [DOI] [PubMed] [Google Scholar]
- 18. Broglio F, Gottero C, Prodam F, et al Non‐acylated ghrelin counteracts the metabolic but not the neuroendocrine response to acylated ghrelin in humans. J Clin Endocrinol Metab 2004; 89: 3062–3065. [DOI] [PubMed] [Google Scholar]
- 19. Maffeis C, Surano MG, Cordioli S, et al High‐fat vs. a moderate‐fat meal in obese boys: nutrient balance, appetite, and gastrointestinal hormone changes. Obesity 2010; 18: 449–455. [DOI] [PubMed] [Google Scholar]
- 20. Kreymann B, Williams G, Ghatei MA, et al Glucagon‐like peptide‐1 7‐36: a physiological incretin in man. Lancet 1987; 5: 1300–1304. [DOI] [PubMed] [Google Scholar]
- 21. Gautier JF, Choukem SP, Gerard J. Physiology of incretins (GIP and GLP‐1) and abnormalities in type 2 diabetes. Diabetes Metab 2008; 34: 65–72. [DOI] [PubMed] [Google Scholar]
- 22. D'Alessio DA. Sandoval Da, Salley RJ. New ways in which GLP‐1 can regulate glucosae homeostasis. J Clin Invest 2000; 115: 3406–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kieffer TJ, McIntosh CH, Pederson RA. Degradation of glucose‐dependent insulinotropic polypeptide and truncated glucagon‐like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology 1995; 136: 3585–3596. [DOI] [PubMed] [Google Scholar]
- 24. Baggio LL, Drucker DJ. Biology of incretins: GLP‐1 and GIP. Gastroenterology 2007; 132: 2131–2157. [DOI] [PubMed] [Google Scholar]
- 25. Choukem SP, Gerard J. Physiology of incretins (GIP and GLP‐1) and abnormalities in type 2 diabetes. Diabetes Metab 2008; 34: 65–72. [DOI] [PubMed] [Google Scholar]
- 26. Karamanlis A, Chaikomin R, Doran S, et al Effects of protein on glycemic and incretin responses and gastric emptying after oral glucose in healthy subjects. Am J Clin Nutr 2007; 86: 1364–1368. [DOI] [PubMed] [Google Scholar]
- 27. Dunmore SJ, Brown JE. The role of adipokines in betacell failure of type 2 diabetes. J Endocrinol 2013; 216: T37–T45. [DOI] [PubMed] [Google Scholar]
- 28. Wolfe MM, Boylan MO. Obesity and the gastrointestinal tract: you are what you eat. J Clin Gastroenterol 2014; 48: 817–822. [DOI] [PubMed] [Google Scholar]