

Abstract
Background/Aims:
There has been controversy about the role of Toll-like receptor 2 (TLR2) in renal injury following ureteric obstruction. Although inhibition of the renin angiotensin system (RAS) reduces TLR2 expression in mice, the exact relationship between TLR2 and RAS is not known. The aim of this study was to determine whether the RAS modulates TLR2.
Methods:
We used 8-week-old male wild type (WT) and TLR2-knockout (KO) mice on a C57Bl/6 background. Unilateral ureteral obstruction (UUO) was induced by complete ligation of the left ureter. Angiotensin (Ang) II (1,000 ng/kg/min) and the direct renin inhibitor aliskiren (25 mg/kg/day) were administrated to mice using an osmotic minipump. Molecular and histologic evaluations were performed.
Results:
Ang II infusion increased mRNA expression of TLR2 in WT mouse kidneys (p < 0.05). The expression of renin mRNA in TLR2-KO UUO kidneys was significantly higher than that in WT UUO kidneys (p < 0.05). There were no differences in tissue injury score or mRNA expression of monocyte chemotactic protein 1 (MCP-1), osteopontin (OPN), or transforming growth factor β (TGF-β) between TLR2-KO UUO and WT UUO kidneys. However, aliskiren decreased the tissue injury score and mRNA expression of TLR2, MCP-1, OPN, and TGF-β in WT UUO kidneys (p < 0.05). Aliskiren-treated TLR2-KO UUO kidneys showed less kidney injury than aliskiren-treated WT UUO kidneys.
Conclusions:
TLR2 deletion induced activation of the RAS in UUO kidneys. Moreover, inhibition of both RAS and TLR2 had an additive ameliorative effect on UUO injury of the kidney.
Keywords: Toll-like receptor 2, Renin-angiotensin system, Unilateral ureteral obstruction
INTRODUCTION
Toll-like receptors (TLRs) recognize endogenous danger molecules such as hyaluronan fragments that have been altered from their native state or accumulated in non-physiologic sites or amounts during cellular injury [1-5]. Min et al. [6] reported that TLR inhibition may have renoprotective effects on obesity-related kidney disease through its anti-inflammatory properties. TLR2 and TLR4 play crucial roles in the induction of acute inflammation and early tubular injury in the kidney in a reversible model of acute renal injury [7-9]. Leemans et al. [10] reported that unilateral ureteral obstruction (UUO) increases renal expression of TLR2. However, Chowdhury et al. [11] reported that TLR2 is not involved in renal injury following ureteric obstruction, and deletion of TLR2 does not have a beneficial effect on kidneys with UUO [10,11]. Although deletion of TLR2 reduces renal injury and infiltration of interstitial myofibroblasts, it does not affect the development of progressive renal fibrosis in UUO mice [10]. It is not clear why deletion of TLR2 does not reduce the renal injury induced by UUO, and the role of TLR2 in renal injury following ureteric obstruction remains controversial.
The renin angiotensin system (RAS) has been reported to play a crucial role in renal inflammation and fibrosis in UUO kidneys [12,13] and inhibition of RAS attenuates renal injury in UUO mice [14,15]. Olmesartan reduces TLR2 in the aortic roots of mice, resulting in a decrease in intimal neovascularization and plaque growth. Losartan decreases the expression of TLR2 mRNA and protein that is upregulated in cyclosporin-induced renal injury [16]. Although these reports suggest that inhibition of RAS reduces TLR2 signaling, it is not clear whether RAS modulates TLR signaling. This study was performed to investigate whether concomitant inhibition of RAS and TLR2 attenuates inflammation and fibrosis in UUO mice and to evaluate the relationship between RAS and TLR2.
METHODS
Animals and drug treatments
All experiments were performed on 8-week-old male C57BL/6 mice (weight 20 to 25 g, Samtako, Osan, Korea) and TLR2 knockout (KO) mice (weight 20 to 25 g, Korea Research Institute of Bioscience and Biotechnology, Daejeon, Korea). The mice were given a standard laboratory diet and water and were cared for under a protocol approved by the Institutional Animal Care and Use Committee of the Chungnam National University Medical School.
Two separate studies were performed. First, we evaluated the association between the RAS and TLR2. Wild type (WT) and TLR2-KO mice (n = 8 per group) were infused with angiotensin (Ang) II to activate RAS. Second, we divided the mice into eight groups: vehicle (Vh)-treated sham (n = 7), aliskiren-treated sham (n = 7), TLR2-KO (n = 8), aliskiren-treated TLR2-KO (n = 8), Vh-treated UUO (n = 8), aliskiren-treated WT UUO (n = 8), TLR2-KO UUO (n = 8), and aliskiren-treated TLR2-KO UUO (n = 8). To establish UUO, complete ligation of the left ureter at the ureteropelvic junction was performed using double silk sutures. Mice were anesthetized with intraperitoneal ketamine (2 mg/kg, Ketalar, Bayer, Leverkusen, Germany) and xylazine (200 µL/kg, Rompun, Bayer). Aliskiren powder was supplied by Novartis Co. (East Hanover, NJ, USA). To continuously deliver solutions over the course of 8 days, an Alzet osmotic minipump (model 2004, Durect Corp., Cupertino, CA, USA) was implanted subcutaneously in each mouse 1 day before UUO or sham operation. The pumps were filled with either normal saline or saline containing aliskiren. Both were administered at a dose of 25 mg/kg/day. Ang II was infused at a rate of 1,000 ng/kg/min via the subcutaneously implanted osmotic minipumps. Mice were sacrificed on day 7 after UUO or on day 12 after Ang II infusion.
Tissue preparation
Tissue preparation was performed as described previously [14]. Briefly, at the end of the study (7 days after UUO and 12 days after Ang II infusion), mice were sacrificed, and both kidneys were excised immediately and cut into three coronal sections. Two pieces were snap-frozen in liquid nitrogen and stored at –70°C for subsequent RNA extraction and protein analysis. The third piece (central section) was fixed in 10% buffered formaldehyde at room temperature and then embedded in Paraplast (Sherwood Medical, St. Louis, MO, USA) for light microscopy and immunohistochemical staining.
Light microscopy
Tissue staining was performed as described previously [14]. Briefly, paraffin-embedded kidney pieces were cut into sections 4-µm thick and mounted on glass slides. The sections were deparaffinized with xylene, stained with hematoxylin and eosin (H&E) and Masson’s trichrome (MT), and examined under an Olympus BX51 microscope (Olympus, Tokyo, Japan). The tubulointerstitial injury score was evaluated based on morphological changes in tubuels, such as dilatation, distortion of tubular basement membranes and atrophy: grade 0, no morphological deformities; grade 1, less than 10%; grade 2, less than 25%; grade 3, less than 50%; grade 4, less than 75%; and grade 5, 75% or greater involved. Ten fields of the outer medulla were evaluated. Renal fibrotic areas were quantified using morphometric analysis with blue stained area in MT staining. The size of the blue-stained (fibrotic) area in MT staining (as a percentage of the total area in 10 different fields of each section under ×200 magnification) was determined using a digital camera-based image analyzer (Metamorpho version 6.3, Olympus).
Immunohistochemistry
Immunohistochemistry was performed as described previously [14]. Paraffin-embedded tissues were cut into sections 4-µm thick, mounted on glass slides, and stained using indirect immunoperoxidase. The slides were processed for immunodetection of transforming growth factor β (TGF-β) and angiotensin II type 1 receptor (AT1R) with antibodies specific for TGF-β (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and anti-AT1R (Santa Cruz Biotechnology) respectively. Diaminobenzidine (Sigma Chemical Co., St. Louis, MO, USA) was used as a chromogen. All samples were evaluated under an Olympus BX51 microscope. The size of the area stained positively for TGF-β (as a percentage of the total area in 10 separate fields of each section under ×200 magnification) was determined using a digital camera-based image analyzer (Metamorpho version 6.3, Olympus).
Western blot
Western blot analysis was performed to measure TLR2, renin, and AT1R protein expression. Kidney sections were homogenized in PRO-PREP protein extraction solution (iNtRON, Seongnam, Korea). Total protein (40 µg) was loaded. Samples were wet transferred onto 0.2 µm nitrocellulose membranes (Amersham Pharmacia, Piscataway, NJ, USA). Blots were blocked for 1 hour with 5% nonfat dry milk in Tris-buffered saline-Tween buffer compose of 20 mM Tris-HCl (pH 7.6), 0.8% NaCl and 0.05% Tween 20, and incubated overnight at 4°C with anti-TLR2 antibody (Abcam, Cambridge, MA, USA), AT1R (Santa Cruz Biotechnology), anti-renin (Santa Cruz Biotechnology) and anti-β-actin antibody (Santa Cruz Biotechnology). Blots were incubated with horseradish peroxidase-conjugated secondary anti-rabbit immunoglobulin G antibody (Cell Signaling Technology) for 1 hour. Bands were detected by enhanced chemiluminescence (Millipore, Billerica, MA, USA) and exposed to film. The optical density for quantification was determined using Gel-Pro Analyzer version 3.1 (Media Cybernetics, Bethesda, MD, USA).
RNA extraction, and quantitative real-time polymerase chain reaction
Total RNA was extracted from whole kidney using an RNeasy Mini Kit (Qiagen, Hilden, Germany). cDNA was synthesized from 2 µg total RNA using an oligo dT primer (Amersham Pharmacia), deoxynucleotide tirphosphates (Amersham Pharmacia), moloney murine leukemia virus reverse transcriptase (Gibco-BRL, Grand Island, NY, USA), 0.1 M dithiothreitol, and buffers in a volume of 20 µL. The cDNA reaction mix was diluted to a total volume of 40 µL and polymerase chain reaction (PCR) was performed to amplify the following specific cDNAs: glyceraldehyde 3-phosphate dehydrogenase (GAPDH; primers: sense 5′-CC CAG ACC CCA TAA CAA CAG-3′; antisense 5′-TGA GGG TGC AGC GAA CTT TA-3′); monocyte chemotactic protein 1 (MCP-1; primers: sense 5′-ACT GCA TCT GCC CTA AGG TCT TCA-3′; antisense 5′-AGA AGT GCT TGA GGT GGT TGT GGA-3′); osteopontin (OPN; primers: sense 5′-GGC ATT GCC TCC TCC CTC-3′; antisense 5′-CGA GGC TGT AAA GCT TCT CC-3′); TGF-β (primers: sense 5′-AAC TAT TGC TTC AGC TCC AGA GAG A-3′; antisense 5′-AGT TGG ATG GTA GCC CTT G-3′); TLR2 (primers: sense 5′-GCC ACC ATT TCC ACG GAC T-3′; antisense 5′-GGC TTC CTC TTG GCC TGG-3′); AT1 receptor (primers: sense 5′-AGA ACA CCA ATA TCA CTG TTT G-3′; antisense 5′-TAG CTG GTA AGA ATG ATT AGG A-3′); and renin (primers: sense 5′-ATG AAG GGG GTG TCT GTG GGG TC-3′; antisense 5′-ATG TCG GGG AGG GTG GGC ACC TG-3′). PCR was performed using SYBR Green PCR mastermix (Qiagen). The amplification reaction volume was 20 µL, consisting of 10 µL iQ SYBR Green PCR mastermix, 2 µL primers, 2 µL cDNA, and 6 µL water. Amplification and detection were performed using a thermal cycler (Rotor-Gene 6000, Corbett Research, Mortlake, Australia). PCR conditions were as follows: denaturation at 95°C for 10 minutes followed by 40 cycles of 10 seconds at 95°C, 15 seconds at the annealing temperature (55°C for GAPDH, TGF-β, MCP-1), and 20 seconds at 72°C. SYBR green fluorescence was measured at the end of each cycle using the comparative threshold cycle (Ct) method: 2–ΔΔCt = 2 – [(Ct of target gene – Ct of GAPDH in treated mice) – (Ct of target gene – Ct of GAPDH in sham mouse)].
Statistical analysis
Data are reported as the mean ± SD. Multiple comparisons among groups were performed by one-way analysis of variance with the post hoc Bonferroni test correction (SPSS version 11.0, SPSS Inc., Chicago, IL, USA). The difference between groups was considered statistically significant at p < 0.05.
RESULTS
RAS and TLR2 expression in the kidneys of sham-operated and UUO mice
The kidneys of Ang II-infused WT mice showed increased expression of TLR2 mRNA compared with those of saline-infused WT controls (p < 0.05) (Fig. 1A). To evaluate the association between the RAS and TLR2, we evaluated renin and AT1R expression in TLR2-KO mouse kidneys. TLR2-KO mouse kidneys showed little TLR2 expression (Fig. 1B). Renal mRNA expression of renin and AT1R tended to be higher in TLR2-KO mouse kidneys than in WT mice (Fig. 1C and 1D). However there were no significant differences.
Figure 1.
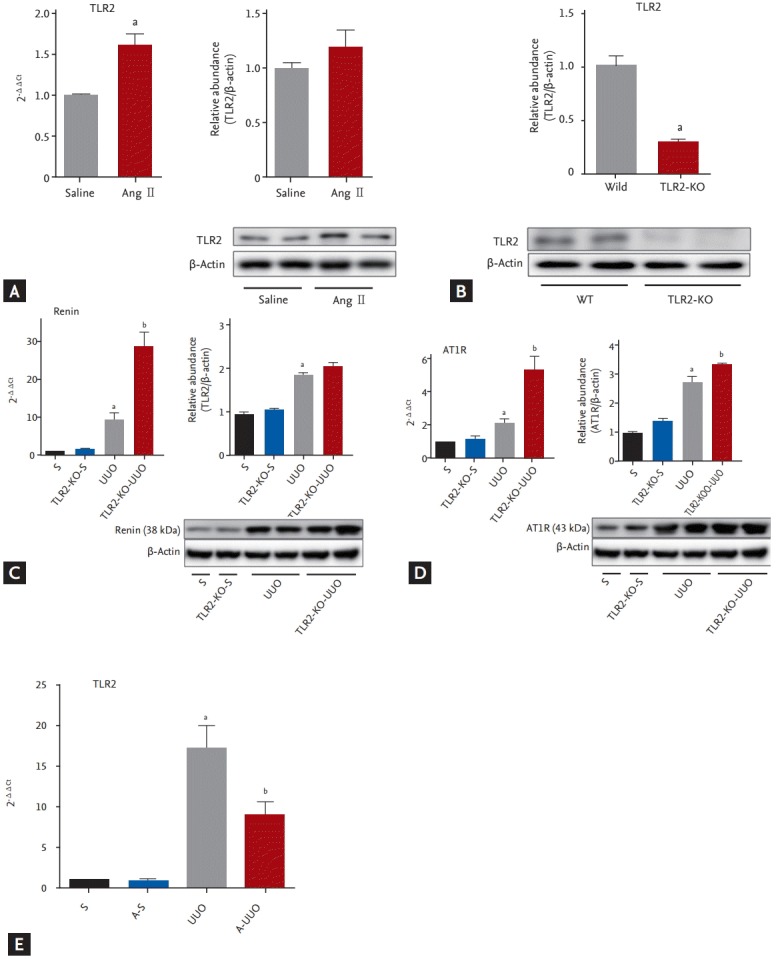
(A) Representative effects of Ang II infusion on TLR2 mRNA expression. Representative kidneys from Ang II-infused WT sham-operated mice (n = 8) showed higher mRNA expression of TLR2 than saline-infused WT sham-operated mice (n = 7). (B) Representative immunoblots from Ang II-infused WT sham-operated mice (n = 4) and saline-infused WT sham-operated mice (n = 4) TLR2 expression in WT and TLR2-KO mouse kidneys. (C, D) Representative immunoblots from WT mice (n = 4) and TLR2-KO mice (n = 4) renin and AT1R expression. Renin and AT1R mRNA expression levels were significantly higher in UUO kidneys than in sham kidneys (C, D), and in TLR2-KO UUO kidneys compared with WT UUO kidneys (C, D). There was no significant difference in the basal mRNA expression of renin and AT1R between sham and TLR2-KO kidneys. Representative immunoblotting results indicating significantly higher renin and AT1R expression in UUO kidneys (n = 4) compared to sham-operated kidneys (n = 3) (C, D). The protein level of AT1R was significantly increased in TLR2-KO UUO kidneys compared with WT UUO kidneys (D). TLR2 mRNA expression in UUO kidneys was significantly higher than that in sham-operated kidneys (D). Aliskiren treatment reduced the mRNA level of TLR2 in UUO kidneys (E). Ang, angiotensin; TLR2, Toll-like receptor 2; WT, wild type; KO, knockout; AT1R, angiotensin II type 1 receptor; UUO, unilateral ureteral obstruction; S, sham-operated mice (n = 7); A-S, aliskiren treated sham-operated mice (n = 7); TLR2-KO, TLR2-KO mice (n = 8); TLR2-KO UUO, TLR2-KO UUO mice (n = 8); A UUO, aliskren treated WT UUO mice (n = 8). a,bp < 0.05.
To evaluate RAS activation, we measured renin and AT1R mRNA expression in UUO kidneys. In the TLR2-KO UUO kidney, mRNA expression of renin and AT1 receptor was significantly increased compared with that in WT UUO kidneys (p < 0.05) (Fig. 1C and 1D). Protein level of AT1R in TLR2-KO UUO kidneys was significantly increased compared with that in the WT UUO kidneys (p < 0.05) (Fig. 1D). Protein level of renin showed increased tendency in TRL2-KO UUO kidneys, but there was no statistical significance (Fig. 1C). TLR2 mRNA expression was significantly higher in UUO kidneys than in sham-operated kidneys (p < 0.05) (Fig. 1E). Aliskiren treatment reduced renal mRNA expression of TLR2 in UUO kidneys (p < 0.05) (Fig. 1E). There was no significant difference in renal mRNA expression of TLR2 between Vhand aliskiren-treated sham mice.
Inflammation and fibrosis of kidneys in TLR2-KO UUO mice
The expression of OPN, MCP-1, and TGF-β mRNA were increased in UUO kidneys compared with sham-operated kidneys. However, there were no significant differences in renal mRNA expression of OPN, MCP-1, and TGF-β between TLR2 KO and WT UUO kidneys (Fig. 2A-2C). Light microscopic examination of WT UUO kidneys showed tissue injury, including infiltration of mononuclear cells into the interstitium and tubules, and desquamation of tubular epithelial cells. There was no significant difference in tissue injury score between WT and TLR2-KO UUO kidneys. TLR2-KO UUO kidneys showed no significant difference in the area positive for MT or TGF-β compared with WT controls (Fig. 2D).
Figure 2.
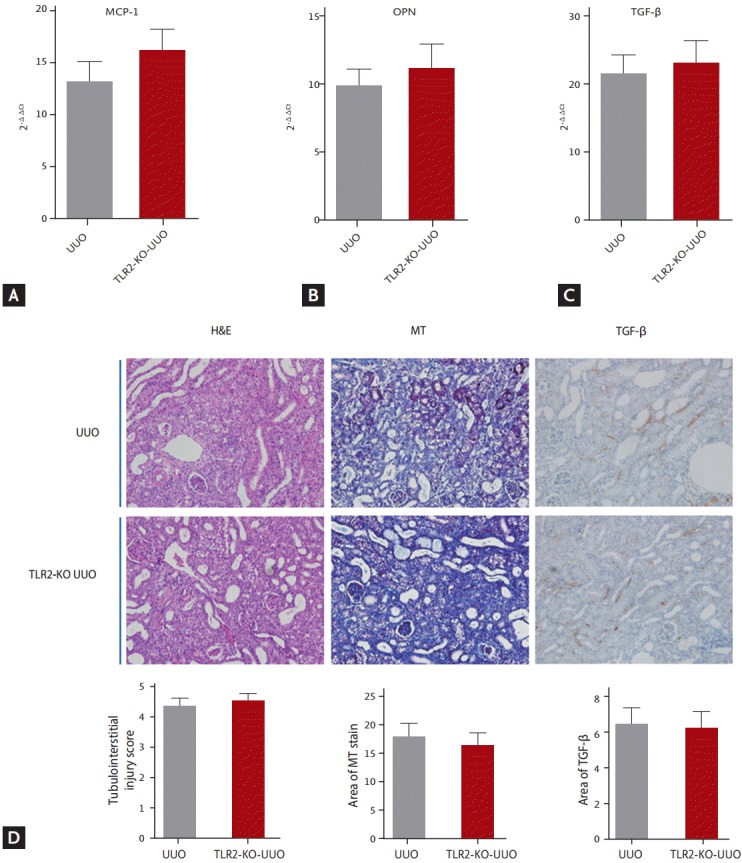
The effects of TLR2-KO on UUO-induced injury. Renal mRNA expression of (A) MCP-1, (B) OPN, or (C) TGF-β were similar in TLR2-KO and WT UUO mice. There were no significant differences in renal expression of MCP-1 (A, p = 0.299), OPN (B, p = 0.549), and TGF-β (C, p = 0.654) between WT and TLR2-KO UUO kidneys. (D) There was no significant difference in tubulointerstitial injury sore, MT stained area, and TGF-β positive area between WT and TLR2-KO UUO kidneys (×200). UUO: UUO WT mice (n = 8), TLR2-KO UUO mice (n = 8). TLR2, Toll-like receptor 2; KO, knockout; UUO, unilateral ureteral obstruction; MCP-1, monocyte chemotactic protein 1; OPN, osteopontin; TGF-β, transforming growth factor β; WT, wild type; MT, Masson’s trichrome.
Inhibition of RAS in TLR2 KO UUO mice
Aliskiren treatment improved the renal tubulointerstitial injury score in UUO kidneys. Renal tubulointerstitial injury was significantly decreased in aliskiren-treated TLR2-KO UUO kidneys compared to WT UUO kidneys treated with this agent (p < 0.05) (Fig. 3). Aliskiren treatment significantly reduced renal mRNA expression of OPN and MCP-1 in UUO kidneys (p < 0.05) (Fig. 4). Moreover, renal mRNA expression of OPN and MCP-1 was significantly decreased in aliskiren-treated TLR2-KO UUO kidneys compared to WT UUO kidneys treated with this agent (p < 0.05) (Fig. 4).
Figure 3.
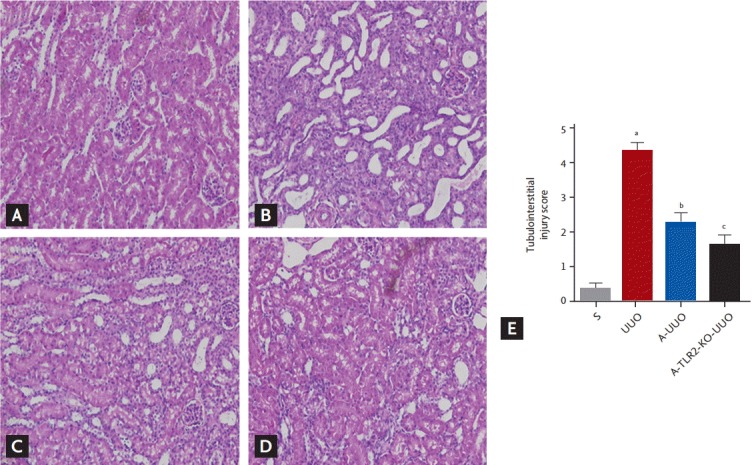
The effects of inhibition of renin angiotensin system and TLR on renal histology 7 days after UUO in (A) S, (B) vehicle-treated WT UUO, (C) aliskiren-treated WT UUO, and (D) aliskiren-treated TLR2-KO UUO mice (×200). Vehicle-treated obstructed kidneys showed marked injury, with mononuclear cells in the interstitium and tubules, and dilatation and desquamation of tubular epithelial cells. In contrast, aliskiren-treated WT UUO mice showed significantly less tubulointerstitial damage. (E) Aliskiren-treated TLR2-KO UUO kidneys showed a significant decrease in tissue injury score compared with aliskiren-treated WT UUO kidneys. TLR, Toll-like receptor; UUO, unilateral ureteral obstruction; WT, wild type; KO, knockout; S, sham; A, aliskiren; S, sham-operated mice (n = 7); UUO, UUO mice (n = 8); A-UUO, aliskiren-treated WT UUO mice (n = 8); A-TLR2-KO UUO, aliskiren-treated TLR2-KO UUO mice (n = 8). ap < 0.05 vs. sham operation, bp < 0.05 vs. UUO, cp < 0.05 vs. A-UUO.
Figure 4.
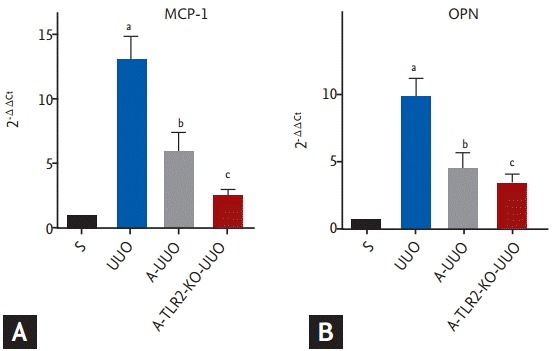
The effects of inhibition of renin angiotensin system and TLR on MCP-1 and OPN expression 7 days after UUO. (A) MCP-1 and (B) OPN mRNA expression were increased in UUO kidneys compared with sham-operated kidneys. Aliskiren treatment reduced renal mRNA expression of MCP-1 and OPN in UUO kidneys. Aliskiren-treated TLR2-KO UUO kidneys showed a significant decrease in mRNA expression of MCP-1 and OPN compared with aliskiren-treated WT UUO kidneys. TLR, Toll-like receptor; MCP-1, monocyte chemotactic protein 1; OPN, osteopontin; UUO, unilateral ureteral obstruction; KO, knockout; WT, wild type; S, sham-operated mice (n = 7); UUO, UUO operated mice (n = 8); A-UUO, aliskiren-treated UUO WT mice (n = 8); A-TLR2-KO-UUO, aliskiren-treated TLR2-KO UUO mice (n = 8). a,b,cp < 0.05.
Aliskiren improved renal interstitial fibrosis in UUO kidneys, as indicated by decreased Masson’s trichome staining and TFG-β expression (Fig. 5). Aliskiren-treated TLR2-KO UUO kidneys showed significant decreases in areas positive for MT and TGF-β compared with aliskiren-treated WT UUO kidneys (p < 0.05) (Fig. 5 Ad and Bd). Also, renal mRNA expression of TGF-β was significantly decreased in aliskiren-treated TLR2-KO UUO kidneys compared to with WT UUO kidneys treated with this agent (p < 0.05) (Fig. 5Bf). The expression of renin was increased in UUO kidneys (p < 0.05) (Fig. 5C). Although not statistically significant, aliskiren treatment showed increase tendency in the renin expression of both WT UUO and TLR2-KO UUO kidneys compared with untreated UUO kidneys (Fig. 5C). The expression of AT1R was increased in UUO kidneys (p < 0.05) (Fig. 5D). Aliskiren treatment significantly decreased the AT1R expression in WT UUO kidneys compared with untreated UUO kidneys (p < 0.05) (Fig. 5D). Although there was no statistical significance, AT1R expression showed the increase tendency in aliskiren-treated TLR2-KO kidneys compared to WT UUO kidneys treated with this agent (Fig. 5D).
Figure 5.
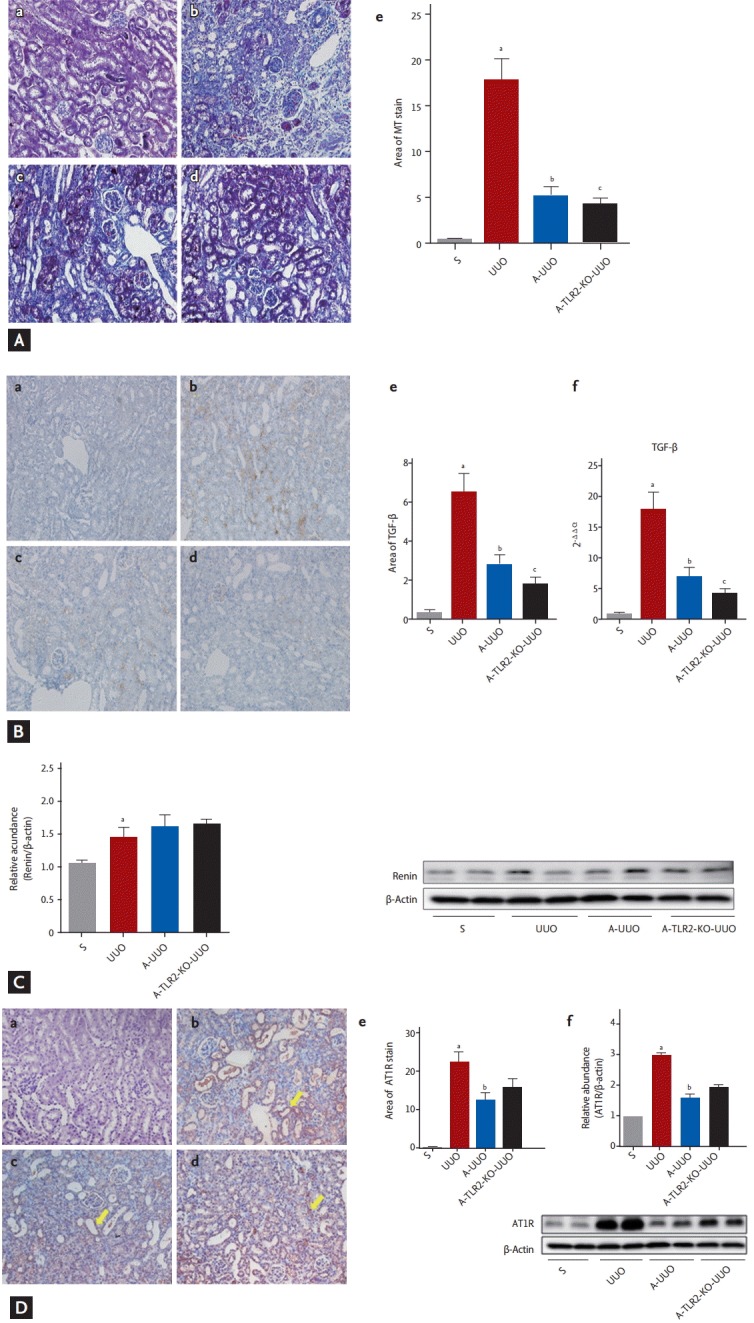
The effects of inhibition of renin angiotensin system and TLR on renal fibrosis at 7 days after UUO. (A) Masson’s trichrome (MT) staining and (B) TGF-β staining of (a) sham-operated, (b) UUO, (c) aliskiren-treated WT UUO, and (d) aliskiren-treated TLR2-KO UUO kidneys (×200). Ae and Be show quantification of the data in each panel. (Ab) MT-stained and (Bb) TGF-β stained areas were larger in UUO kidneys compared with sham-operated kidneys. Aliskiren treatment reduced the MT-stained area and TGF-β stained area in UUO kidneys (Ac, Bc). Aliskiren-treated TLR2-KO UUO kidneys showed a significant decrease in MT-stained and TGF-β stained areas compared with aliskiren-treated WT UUO kidneys (Ad, Bd). Aliskiren treatment reduced the renal mRNA expressions of TGF-β in UUO kidneys. Aliskiren-treated TLR2-KO UUO kidneys showed significant decreases in renal mRNA expression of TGF-β compared with aliskiren-treated WT UUO kidneys by RT-PCR (Bf). (C) Renin expression. The expression of renin was increased in UUO, A-UUO, and A-TRL2-KO UUO kidney (p < 0.05). (D) AT1R expression. (a) Sham-operated, (b) WT UUO, (c) aliskiren-treated WT UUO, (d) aliskiren-treated TLR2-KO UUO kidneys, (e) area of AT1R stain, (f) representative AT1R immunoblots: sham-operated (n = 3), WT-UUO (n = 3), A-UUO (n = 4), A-TLR2-KO-UUO (n = 4) (×200). The expression of AT1R was increased in UUO kidney. The area of AT1R stain in tubulointerstitium (yellow arrows). However, aliskiren treatment significantly decreased the AT1R expression in both WT UUO and TLR2-KO UUO kidneys compared with aliskiren-untreated WT UUO kidneys (Da-Dd and Df, p < 0.05). Although AT1R expression showed the increase tendency in aliskiren-treated TLR2-KO kidney compared with aliskiren-treated WT UUO kidney, there were no significant difference between two groups (p = 0.075). TLR, Toll-like receptor; UUO, unilateral ureteral obstruction; MT, Masson’s trichrome; TGF-β, transforming growth factor β; WT, wild type; KO, knockout; RT-PCR, real-time polymerase chain reaction; A-UUO, aliskiren-treated UUO WT mice (n = 8); A-TLR2-KO UUO, aliskiren-treated TLR2-KO UUO mice (n = 8); AT1R, angiotensin II type 1 receptor; S, sham-operated mice (n = 7); UUO, UUO operated mice (n = 8). a,b,cp < 0.05.
DISCUSSION
The results of the present study indicated that deletion of TLR2 induces RAS activation in the UUO kidney, and that inhibition of the RAS reduces renal injury in the TLR2-KO UUO kidney.
TLRs are thought to be crucial cellular sentinels that detect “danger” signals released by tissue damage [17], and they may also be important initiators of inflammation and fibrosis. TLR2 is involved in the activation of TGF-β and migration of macrophages and myofibroblasts [10,11]. Deletion of TLR2 reduces apoptosis of tubular cells in renal ischemia reperfusion injury. However, the role of TLR2 in renal injury by UUO is not clear [10,11]. Although TLR2 recruits inflammatory cytokines, macrophages, and fibroblasts in the kidney, most studies have reported that deletion of TLR2 does not reduce the induction of inflammatory cytokines or progression of renal fibrosis [10,11]. Similar to these findings, our study showed no significant difference in renal inflammation or fibrosis induced by UUO between WT and TLR2-KO mice.
The renin and AT1R mRNA expression levels are increased in UUO kidneys [14,18,19]. In general, renin is expressed in the macular densa and AT1R is expressed in tubules and interstitial area. We did not separate the tubular area, but used the whole tissue for immunoblotting. Therefore, we could not determine the precise locations and changes in expression of AT1R. Chronic renal injury results in elevated renin and AT1R expression [20]. In addition, it is well known that activation of RAS also plays an important role in kidney injury induced by UUO. Direct renin inhibition and AT1R KO showed improvement of renal injury in UUO kidneys [12,14].
We showed that deletion of TLR2 tends to increase the basal renal renin and AT1R mRNA expression compared to those in WT mice. Moreover, TLR2-KO UUO mice showed significant increases in renal renin and AT1R mRNA expression compared with WT UUO mice. The enhanced activation of RAS accompanied by inhibition of TLR2 may offset the anti-inflammatory effects of the TLR2 KO and may explain why blocking of TLR2 did not reduce the renal injury induced by UUO. Various kidney injury models have shown that RAS activation is important in the inflammatory process, including increased levels of OPN and MCP-1 expression [21-23]. TGF-β plays a major role in renal fibrosis induced by UUO [24], and RAS activation is involved in TGF-β expression in the obstructed kidney [25].
Although there have been no reports that TLR2 modulates the RAS, there is evidence that the RAS may increase activation of TLRs. It has been reported that Ang II induces activation of TLR4 signaling in cultured fibroblasts and mesangial cells [26,27]. Lim et al. [28] reported that activation of RAS increased renal TLR2 expression whereas inhibition of RAS by an Ang receptor blocker decreased renal TLR2 expression in cyclosporine A-induced renal injury. Our study showed that RAS activation induced by Ang treatment increased TLR2 mRNA expression. Moreover, the induction of renal TLR2 by UUO was reduced by aliskiren treatment. Taken together, these findings indicate that the RAS modulates TLR2 activation. Although the mechanism by which deletion of TLR2 activates RAS is not known, there are a number of possibilities. Deletion of TLR2 may activate a different inflammatory pathway [29]. It was recently revealed that TLR2 plays a role in innate immunity and angiogenesis by sensing the oxidation status and that deletion of TLR2 reduces wound healing in the dermis of mice [30]. TLR2 is expressed at high levels in renal tubules and the vessel endothelium of normal kidneys [31]. It is possible that deletion of TLR2 reduces the healing process of the kidney, thus potentiating other inflammatory processes including activation of RAS.
In summary, the present study indicated that TLR2 deletion results in RAS activation in UUO kidneys. Moreover, concomitant inhibition of RAS and TLR2 has an additive benefit on the amelioration of UUO-induced kidney injury.
KEY MESSAGE
1. Toll-like receptor (TLR) 2 deletion results in activation of renin angiotensin system (RAS) in unilateral ureteral obstruction (UUO) kidneys.
2. Concomitant inhibition of RAS and TLR2 has an additive benefit on the amelioration of UUO-induced kidney injury.
Footnotes
No potential conflict of interest relevant to this article was reported.
REFERENCES
- 1.Taylor KR, Trowbridge JM, Rudisill JA, Termeer CC, Simon JC, Gallo RL. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem. 2004;279:17079–17084. doi: 10.1074/jbc.M310859200. [DOI] [PubMed] [Google Scholar]
- 2.Saemann MD, Weichhart T, Zeyda M, et al. Tamm-Horsfall glycoprotein links innate immune cell activation with adaptive immunity via a Toll-like receptor-4-dependent mechanism. J Clin Invest. 2005;115:468–475. doi: 10.1172/JCI22720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaefer L, Babelova A, Kiss E, et al. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J Clin Invest. 2005;115:2223–2233. doi: 10.1172/JCI23755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scheibner KA, Lutz MA, Boodoo S, Fenton MJ, Powell JD, Horton MR. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J Immunol. 2006;177:1272–1281. doi: 10.4049/jimmunol.177.2.1272. [DOI] [PubMed] [Google Scholar]
- 5.Miyake K. Innate immune sensing of pathogens and danger signals by cell surface Toll-like receptors. Semin Immunol. 2007;19:3–10. doi: 10.1016/j.smim.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 6.Min HS, Kim JE, Lee MH, et al. Effects of Toll-like receptor antagonist 4,5-dihydro-3-phenyl-5-isoxasole acetic acid on the progression of kidney disease in mice on a high-fat diet. Kidney Res Clin Pract. 2014;33:33–44. doi: 10.1016/j.krcp.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leemans JC, Stokman G, Claessen N, et al. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J Clin Invest. 2005;115:2894–2903. doi: 10.1172/JCI22832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu H, Chen G, Wyburn KR, et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117:2847–2859. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pulskens WP, Teske GJ, Butter LM, et al. Toll-like receptor-4 coordinates the innate immune response of the kidney to renal ischemia/reperfusion injury. PLoS One. 2008;3: doi: 10.1371/journal.pone.0003596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leemans JC, Butter LM, Pulskens WP, et al. The role of Toll-like receptor 2 in inflammation and fibrosis during progressive renal injury. PLoS One. 2009;4: doi: 10.1371/journal.pone.0005704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chowdhury P, Sacks SH, Sheerin NS. Endogenous ligands for TLR2 and TLR4 are not involved in renal injury following ureteric obstruction. Nephron Exp Nephrol. 2010;115:e122–e130. doi: 10.1159/000313493. [DOI] [PubMed] [Google Scholar]
- 12.Esteban V, Lorenzo O, Ruperez M, et al. Angiotensin II, via AT1 and AT2 receptors and NF-kappaB pathway, regulates the inflammatory response in unilateral ureteral obstruction. J Am Soc Nephrol. 2004;15:1514–1529. doi: 10.1097/01.asn.0000130564.75008.f5. [DOI] [PubMed] [Google Scholar]
- 13.Chevalier RL, Forbes MS, Thornhill BA. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 2009;75:1145–1152. doi: 10.1038/ki.2009.86. [DOI] [PubMed] [Google Scholar]
- 14.Choi DE, Jeong JY, Lim BJ, et al. Aliskiren ameliorates renal inflammation and fibrosis induced by unilateral ureteral obstruction in mice. J Urol. 2011;186:694–701. doi: 10.1016/j.juro.2011.03.122. [DOI] [PubMed] [Google Scholar]
- 15.Kaneto H, Morrissey J, McCracken R, Reyes A, Klahr S. Enalapril reduces collagen type IV synthesis and expansion of the interstitium in the obstructed rat kidney. Kidney Int. 1994;45:1637–1647. doi: 10.1038/ki.1994.215. [DOI] [PubMed] [Google Scholar]
- 16.Ahn KO, Lim SW, Li C, et al. Influence of angiotensin II on expression of Toll-like receptor 2 and maturation of dendritic cells in chronic cyclosporine nephropathy. Transplantation. 2007;83:938–947. doi: 10.1097/01.tp.0000258589.39006.94. [DOI] [PubMed] [Google Scholar]
- 17.Johnson GB, Brunn GJ, Tang AH, Platt JL. Evolutionary clues to the functions of the Toll-like family as surveillance receptors. Trends Immunol. 2003;24:19–24. doi: 10.1016/s1471-4906(02)00014-5. [DOI] [PubMed] [Google Scholar]
- 18.Wu WP, Chang CH, Chiu YT, et al. A reduction of unilateral ureteral obstruction-induced renal fibrosis by a therapy combining valsartan with aliskiren. Am J Physiol Renal Physiol. 2010;299:F929–F941. doi: 10.1152/ajprenal.00192.2010. [DOI] [PubMed] [Google Scholar]
- 19.Yoo KH, Norwood VF, el-Dahr SS, Yosipiv I, Chevalier RL. Regulation of angiotensin II AT1 and AT2 receptors in neonatal ureteral obstruction. Am J Physiol. 1997;273(2 Pt 2):R503–R509. doi: 10.1152/ajpregu.1997.273.2.R503. [DOI] [PubMed] [Google Scholar]
- 20.Yoon HE, Ghee JY, Piao S, et al. Angiotensin II blockade upregulates the expression of Klotho, the anti-ageing gene, in an experimental model of chronic cyclosporine nephropathy. Nephrol Dial Transplant. 2011;26:800–813. doi: 10.1093/ndt/gfq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amann B, Tinzmann R, Angelkort B. ACE inhibitors improve diabetic nephropathy through suppression of renal MCP-1. Diabetes Care. 2003;26:2421–2425. doi: 10.2337/diacare.26.8.2421. [DOI] [PubMed] [Google Scholar]
- 22.Wolak T, Kim H, Ren Y, Kim J, Vaziri ND, Nicholas SB. Osteopontin modulates angiotensin II-induced inflammation, oxidative stress, and fibrosis of the kidney. Kidney Int. 2009;76:32–43. doi: 10.1038/ki.2009.90. [DOI] [PubMed] [Google Scholar]
- 23.Persy VP, Verhulst A, Ysebaert DK, De Greef KE, De Broe ME. Reduced postischemic macrophage infiltration and interstitial fibrosis in osteopontin knockout mice. Kidney Int. 2003;63:543–553. doi: 10.1046/j.1523-1755.2003.00767.x. [DOI] [PubMed] [Google Scholar]
- 24.Huang Y, Wongamorntham S, Kasting J, et al. Renin increases mesangial cell transforming growth factorbeta1 and matrix proteins through receptor-mediated, angiotensin II-independent mechanisms. Kidney Int. 2006;69:105–113. doi: 10.1038/sj.ki.5000011. [DOI] [PubMed] [Google Scholar]
- 25.Burns WC, Velkoska E, Dean R, Burrell LM, Thomas MC. Angiotensin II mediates epithelial-to-mesenchymal transformation in tubular cells by ANG 1-7/MAS-1-dependent pathways. Am J Physiol Renal Physiol. 2010;299:F585–F593. doi: 10.1152/ajprenal.00538.2009. [DOI] [PubMed] [Google Scholar]
- 26.Wolf G, Bohlender J, Bondeva T, Roger T, Thaiss F, Wenzel UO. Angiotensin II upregulates Toll-like receptor 4 on mesangial cells. J Am Soc Nephrol. 2006;17:1585–1593. doi: 10.1681/ASN.2005070699. [DOI] [PubMed] [Google Scholar]
- 27.Ji Y, Liu J, Wang Z, Liu N. Angiotensin II induces inflammatory response partly via Toll-like receptor 4-dependent signaling pathway in vascular smooth muscle cells. Cell Physiol Biochem. 2009;23:265–276. doi: 10.1159/000218173. [DOI] [PubMed] [Google Scholar]
- 28.Lim SW, Li C, Ahn KO, et al. Cyclosporine-induced renal injury induces Toll-like receptor and maturation of dendritic cells. Transplantation. 2005;80:691–699. doi: 10.1097/01.tp.0000173594.69089.a0. [DOI] [PubMed] [Google Scholar]
- 29.Rivera CA, Gaskin L, Allman M, et al. Toll-like receptor-2 deficiency enhances non-alcoholic steatohepatitis. BMC Gastroenterol. 2010;10:52. doi: 10.1186/1471-230X-10-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.West XZ, Malinin NL, Merkulova AA, et al. Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature. 2010;467:972–976. doi: 10.1038/nature09421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shigeoka AA, Holscher TD, King AJ, et al. TLR2 is constitutively expressed within the kidney and participates in ischemic renal injury through both MyD88-dependent and -independent pathways. J Immunol. 2007;178:6252–6258. doi: 10.4049/jimmunol.178.10.6252. [DOI] [PubMed] [Google Scholar]