

Abstract
Nonalcoholic fatty liver disease (NAFLD) and obesity are characterized by altered gut microbiota, inflammation, and gut barrier dysfunction. Here, we investigated the role of mucin-2 (Muc2) as the major component of the intestinal mucus layer in the development of fatty liver disease and obesity. We studied experimental fatty liver disease and obesity induced by feeding wild-type and Muc2-knockout mice a high-fat diet (HFD) for 16 wk. Muc2 deficiency protected mice from HFD-induced fatty liver disease and obesity. Compared with wild-type mice, after a 16-wk HFD, Muc2-knockout mice exhibited better glucose homeostasis, reduced inflammation, and upregulated expression of genes involved in lipolysis and fatty acid β-oxidation in white adipose tissue. Compared with wild-type mice that were fed the HFD as well, Muc2-knockout mice also displayed higher intestinal and plasma levels of IL-22 and higher intestinal levels of the IL-22 target genes Reg3b and Reg3g. Our findings indicate that absence of the intestinal mucus layer activates the mucosal immune system. Higher IL-22 levels protect mice from diet-induced features of the metabolic syndrome.
Keywords: microbiome, metabolic syndrome, endotoxin, Reg3
obesity has become a major health issue in recent decades, and estimates indicate that more than two-thirds of adults in the United States are either overweight or obese (27). Obesity leads to a substantially higher risk of developing features of the metabolic syndrome such as hypertension; dyslipidemia; type 2 diabetes; coronary heart disease; nonalcoholic fatty liver disease (NAFLD); and endometrial, breast, prostate, and colon cancer (25, 31).
Obesity is associated with chronic, low-grade white adipose tissue inflammation in response to metabolic stimuli (24). Adipose tissue macrophages secrete proinflammatory cytokines and chemokines such as tumor necrosis factor-α (TNF-α), interleukin (IL)-6, IL-1β, and monocyte chemotactic protein-1 (MCP-1 or Ccl2) (12, 28). Elevated levels of TNF-α, IL-1β, and IL-6 are associated with insulin resistance and diabetes, and neutralization of TNF-α improves insulin sensitivity in obese rodents (18, 19, 37).
It is known that the gut microbiota also contributes to metabolic disease. Toll-like receptor-5 (TLR5)-deficient mice develop hallmark features of the metabolic syndrome such as hyperlipidemia, hypertension, insulin resistance, and weight gain (12, 40). In the gastrointestinal tract, TLR5 is activated by bacterial products such as flagellin, which induces the production of IL-1β, IL-6, IL-12, IL-18, and IL-23 in dendritic cells (30). These cytokines lead to the production of IL-22 from Th17, Th1, innate lymphoid, natural killer cells, and γδ T cells (30). IL-22 is involved in the production of the antimicrobial peptides known as regenerating islet-derived (Reg)-3 beta and gamma (Reg3b and Reg3g, respectively) in Paneth cells; furthermore, IL-22 induces goblet cells to produce mucins (30). It was recently discovered that the metabolically beneficial cytokine IL-22 is impaired in obese mice (42). Administration of IL-22 to obese mice improves insulin sensitivity, decreases chronic inflammation, and modulates lipid metabolism in liver and adipose tissue (42). Therefore, IL-22 is not only crucial for maintaining the mucosal barrier function and mediating innate immunity, but it also has important metabolic functions (30). Fatty liver disease depends on intestinal microbiota as well (35). Administration of IL-22 protects from various liver diseases (20, 32, 46).
The intestinal mucus layer as a physical barrier protects the epithelium against noxious agents, viruses, and pathogenic bacteria (15). Goblet cells secrete the gel-forming mucins 2, 5AC, and 6 (Muc2, Muc5AC, and Muc6, respectively) in the gastrointestinal tract. These mucins contribute to the viscous properties of the intestinal mucus layer (23). Muc2 is the predominant secreted mucin in the small and large intestine (39). Muc2 deficiency in mice is associated with disruption of epithelial homeostasis and the development of colon cancer (39). On the other hand, Muc2-knockout mice fed alcohol are protected from bacterial overgrowth due to increased killing of commensal bacteria, and they exhibit reduced endotoxemia and liver injury (15).
So far it has been reported that obesity and NAFLD are characterized by altered gut microbiota (1, 16, 35, 38), inflammation (29, 35), and gut barrier disruption (3, 4, 9, 35). However, to our knowledge, no patient data or experimental studies exist that have assessed the function of the intestinal mucus layer in obesity and NAFLD. We studied the role of Muc2 as the major component of the intestinal mucus layer using a high-fat diet (HFD) feeding model to induce obesity and obesity-related liver disease in Muc2-knockout and wild-type (WT) mice.
MATERIALS AND METHODS
Mice.
Muc2-deficient mice (back-crossed to C57BL/6J mice for more than 10 generations) were kindly provided by Dr. Anna Velcich (Albert Einstein College of Medicine, Yeshiva University, New York, NY) (15, 39). Heterozygous mice were bred to obtain WT and knockout littermate mice. Three nonlittermate knockout mice that ate regular chow were added to the control group for hepatic triglyceride measurement, real-time quantitative polymerase chain reactions (qPCRs) procedures, and IL-22 ELISA. Animals were maintained in a temperature-controlled room (22°C) on a 12:12-h light-dark cycle. Age-matched male and female mice were used for this study (as indicated in the figure legends) and were fed HFD (59% energy by fat; Bio-Serv, Frenchtown, NJ) or regular chow diet (13% energy by fat; 1816971-206; LabDiet, Brentwood, MO) for 16 wk. The caloric profile of the HFD consisted of 3.24 kcal/g from fat (lard), 1.43 kcal/g from carbohydrates (maltodextrin, sucrose), and 0.82 kcal/g from proteins (casein) (total 5.49 kcal/g), whereas the control diet contained 0.45 kcal/g from fat, 2.12 kcal/g from carbohydrates, and 0.84 kcal/g from proteins (total 3.41 kcal/g). It is this regular lard-derived HFD that is referred to throughout this manuscript unless HFD with another qualifier is specifically mentioned. For the plant-derived HFD experiments, age-matched male mice were used and fed HFD (41.5% energy by fat; S3282; TestDiet, St. Louis, MO) for 14 wk. The caloric profile of the plant-derived HFD consisted of 1.80 kcal/g from fat (corn oil, hydrogenated coconut oil, cholesterol), 1.837 kcal/g from carbohydrates (sucrose, maltodextrin, corn starch, powdered cellulose), and 0.695 kcal/g from proteins (soy protein concentrate) (total 4.33 kcal/g). For intestinal decontamination, age-matched male mice were fed the lard-derived HFD for 21 wk. During the last 5 wk they were gavaged with 4 mg/kg vancomycin, 8 mg/kg ampicillin, 8 mg/kg neomycin, 8 mg/kg metronidazole, and 8 mg/kg gentamicin. The gavage was carried out twice daily 5 days/wk and once daily 2 days/wk. Mice had ad libitum access to water. Food intake was quantified in week 3 and week 12. This was carried out by forming a solid round lump of HFD and weighing it on the first day of the week and then 7 days later; the difference was the absolute food intake. Similarly, the mouse body weight was determined on the first day of the week and then 7 days later. The daily food intake in gram per gram of body weight was then calculated by dividing the absolute food intake per day by the averaged mouse weight. Body weight was measured once a week for the duration of the respective experiment. All animals studies were reviewed and approved by the Institutional Animal Care and Use Committee of the University of California, San Diego.
Administration of flagellin.
Blood was taken from male HFD-fed WT and Muc2−/− mice to determine IL-22 plasma levels at baseline. After 7 days, 200 μl of vehicle (sterile H2O) or flagellin (10 μg; InvivoGen, San Diego, CA) was injected intraperitoneally and blood was taken 3 h after the injection.
Metabolic studies.
For the glucose tolerance test (GTT), following 16–17 h of fasting an intraperitoneal (ip) dose of 1 g/kg glucose was administered to mice after they had consumed HFD for 13 wk. Blood glucose concentration was measured before glucose injection and 15, 30, 60, 90, and 120 min after injection. For the insulin tolerance test (ITT), following 6–7 h of fasting an ip dose of 0.35 U/kg insulin was administered after 12 wk of HFD, and blood glucose was measured at the indicated time points. In the antibiotic intervention experiment, GTT and ITT were repeated in the third and fourth weeks of the antibiotic intervention.
Measurement of activity level.
Mice were observed over a period of 1 h in their respective week 15 of HFD feeding. A movement equivalent to the length of the shorter side of the cage (∼18 cm) was counted as one horizontal movement. A movement of both front paws above the shoulders was counted as one vertical movement. To account for external factors affecting the activity level (such as noise sources and slight time differences during the day of the activity level measurement), the sum of the horizontal and vertical movements of each knockout mouse was standardized to the averaged sum of the WT mice for each activity measurement experiment, respectively. The final graph hence shows the activity ratio.
Hepatic and fecal triglyceride measurement.
Hepatic triglyceride levels were measured using the Triglyceride Liquid Reagents Kit (Pointe Scientific, Canton, MI) as previously described (15). For fecal triglyceride measurement, stool samples were mixed with PBS in a 1:10 dilution and centrifuged at 12,000 g for 5 min to separate solid particles. Triglyceride concentrations in supernatants were determined as previously described (6).
Fecal protein and carbohydrate measurement.
For fecal protein and carbohydrate measurement, stool samples were mixed with PBS in a 1:10 dilution, and centrifuged to separate solid particles (12,000 g for 5 min). For protein quantitation, the supernatant of these samples was measured with the Pierce BCA Protein Assay (Thermo Scientific, Waltham, MA) according to the manufacturer's instructions. For carbohydrate quantitation, the phenol-sulfuric method was employed: 100 μl of the supernatant of the prepared fecal samples was mixed with 100 μl of 7% phenol solution (diluted in water) and 1.2 ml of concentrated sulfuric acid (36 N). To measure the monosaccharide content, the absorbance of 200 μl of this mix was determined at 490 nm wavelength. To measure the total carbohydrate content, the whole mix was further digested at 100°C for 10 min; then the absorbance of 200 μl of this mix was determined at 490 nm wavelength.
Staining procedures.
Formalin-fixed, paraffin-embedded liver, small intestinal, and colonic samples were stained with hematoxylin and eosin (Surgipath, Buffalo Grove, IL) using standard staining protocols. For hepatic lipid accumulation analysis, frozen sections were cut and stained with oil red O.
Real-time quantitative polymerase chain reaction.
RNA was extracted from mouse tissue using TRIzol (Invitrogen, Carlsbad, CA). RNA was digested with DNase using the DNA-free DNA removal kit (Ambion, Carlsbad, CA) and reverse transcribed using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA). Gene expression was detected with Universal SYBR Green Supermix (Bio-Rad, Hercules, CA) as described (36, 41) using primer sets specific for IL-1β, TNF-α, Ccl2, F4/80, ACOX1, HSL, ATGL, IL-22, Reg3b, Reg3g, HMGCL, HMGCS2, TBP, and 18S genes (all from the National Institutes of Health qPrimerDepot). The relative expression of the target genes was obtained by using a standard curve and normalized to TBP as an internal control in adipose tissue, and 18S as an internal control in all nonadipose tissues. To quantify intestinal bacteria, DNA was extracted from adherent and luminal intestinal contents as described (15). DNA was amplified using published 16S rRNA primer sets for universal bacteria (17) and SYBR Green. To determine the total bacterial load present in the cecum, the qPCR value for each sample was multiplied by the total amount of DNA per gram of cecal content. Firmicutes, Bacteroidetes, Lactobacillus, and Akkermansia muciniphila were amplified using published primer sets (7, 13, 26, 33) and SYBR Green.
Protein expression analysis.
Immunoblot analysis was performed as described (15) using anti-UCP1 (Abcam) and anti-β-actin (Sigma-Aldrich) or anti-HSP90 (Cell Signaling) antibodies as loading controls. Densitometry of immunoblot analysis was performed using ImageJ software (National Institutes of Health, Bethesda, MD).
Plasma assays.
IL-22 in mouse plasma was measured using an ELISA Development Kit (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions. All material used for harvesting blood and measuring endotoxin was pyrogen free. The LPS level in the plasma was determined by employing an ELISA Kit (Cloud-Clone, Houston, TX) according to the manufacturer's protocol.
Statistical analysis.
The unpaired Student's t-test was used for statistical analysis (Prism, GraphPad Software, La Jolla, CA). Results are presented as means ± SE. A value of P < 0.05 was selected as the level of significance.
RESULTS
Muc2-deficient mice fed HFD showed significantly less body weight gain than their WT counterparts.
Feeding mice HFD for 16 wk is an established model for diet-induced obesity, which is accompanied by hepatic steatosis and insulin resistance (10). After 16 wk eating HFD, WT mice exhibited a significantly higher body weight compared with Muc2-deficient mice (Fig. 1, A and B). Similarly, the latter displayed significantly decreased weights of subcutaneous and epididymal white adipose tissue and brown adipose tissue relative to WT mice also given HFD (Fig. 1C); we also noticed a whitening of the brown adipose tissue in WT mice fed the HFD (Fig. 1C). These differences cannot be explained by a different amount of HFD ingested by these mouse strains, because food intake was comparable between the groups as exemplified by the quantity of HFD consumed in week 3 and week 12 of the experiment (Fig. 1D). We next examined whether the absence of a mucus layer would affect intestinal absorption of fat, proteins, or carbohydrates. WT and Muc2-deficient mice showed similar concentrations of triglycerides, proteins, and simple and total carbohydrates in the feces (Fig. 1E). Thus Muc2-deficient mice are protected from diet-induced obesity, which could not be explained by differences in food intake or malabsorption of fat, proteins, or carbohydrates.
Fig. 1.

Mucin-2 (Muc2)-deficient mice gain less body weight than wild-type (WT) mice after 16 wk of being fed a high-fat diet (HFD). WT and Muc2−/− mice were fed regular chow (n = 2–4) or HFD (n = 6–8) for 16 wk. A: body weights in grams (top) and % relative to starting weight (bottom). B: representative mice after 16 wk of HFD. C: subcutaneous fat weight (left), epididymal fat weight (middle), brown adipose tissue weight (right) with representative photographs. D: daily food intake during week 3 and week 12 on HFD. E: fecal triglyceride, protein, monosaccharide, and total carbohydrate concentration in groups of mice fed HFD. *P < 0.05.
Muc2 deficiency results in improved glucose homeostasis following HFD.
The intraperitoneal GTT demonstrated that Muc2−/− mice exhibited significantly lower glucose blood levels and hence improved glucose tolerance after HFD relative to WT mice (Fig. 2A). Consistent with this, mice lacking Muc2 revealed a greater responsiveness to insulin than WT mice fed HFD, as shown by the intraperitoneal ITT (Fig. 2B). Hence the better glycemic control further demonstrates that Muc2 deficiency results in improved features of the metabolic syndrome. There is at least one published report of sex-specific differences in glucose tolerance and insulin responsiveness after HFD (11). Female Muc2-deficient mice fed a lard-derived HFD were not protected from obesity and they showed the same glucose tolerance and insulin sensitivity as female WT mice fed a lard-derived HFD (Fig. 3, A and B). However, loss of protection from features of the metabolic syndrome could be due to a significantly higher food intake in female Muc2−/− mice during the entire feeding period (Fig. 3C).
Fig. 2.
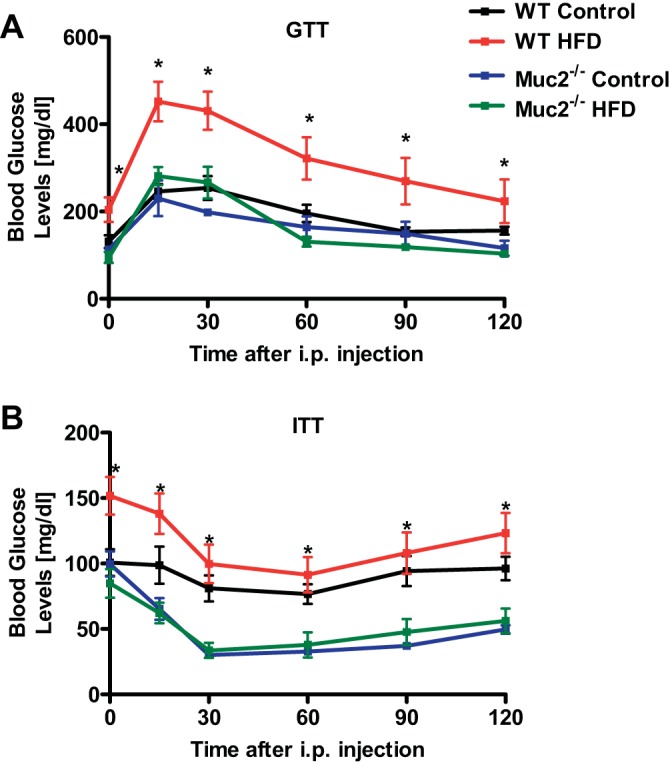
Muc2−/− mice fed HFD show a better glucose tolerance and insulin sensitivity than WT mice fed HFD. WT and Muc2−/− mice were fed regular chow (n = 3–4) or HFD (n = 8) for 16 wk. A: glucose tolerance test (GTT). B: insulin tolerance test (ITT). *P < 0.05 between WT mice and Muc2−/− mice both fed HFD.
Fig. 3.

Female Muc2−/− mice have higher HFD intake and are not protected from regular lard-derived HFD-induced obesity, glucose intolerance, and insulin insensitivity. A–C: female WT and Muc2−/− mice were fed a lard-derived HFD (n = 3–8) for 16 wk. A: body weights in grams (left) and % relative to starting weight (right). B: GTT (left) and ITT (right). C: daily food intake during week 3 and week 12 on a lard-derived HFD. *P < 0.05.
Hepatic steatosis is significantly ameliorated in Muc2−/− mice following HFD.
Because fatty liver disease is part of the metabolic syndrome and is linked to the onset of insulin resistance, we next investigated hepatic steatosis. Livers showed a trend toward lower weight in Muc2−/− mice following a 16-wk HFD, but this was not significantly different (Fig. 4A). Livers of WT mice appeared more steatotic than livers from Muc2−/− mice following HFD, as evidenced macroscopically by a different color and texture (Fig. 4A). This was confirmed on liver sections stained with hematoxylin and eosin (Fig. 4B) and oil red O (Fig. 4C), as WT mice showed impressively more and larger fat droplets than Muc2−/− mice following 16 wk of HFD. Finally, hepatic levels of triglycerides were significantly reduced in mice deficient in Muc2 and fed HFD (Fig. 4D). Thus Muc2 deficiency largely prevents the development of a fatty liver following HFD.
Fig. 4.

HFD results in a fatty liver in WT mice, but not in Muc2−/− mice. WT and Muc2−/− mice were fed regular chow (n = 2–5) or HFD (n = 8) for 16 wk. A: liver weights and representative photographs of fatty liver from mice in groups fed the HFD. B: representative liver sections after hematoxylin and eosin staining, ×40 magnification. C: photographs of representative liver sections after oil red O staining, ×100 magnification. D: hepatic triglyceride levels. *P < 0.05.
White adipose tissue showed less inflammation and an upregulated expression of genes involved in lipolysis and fatty acid β-oxidation in Muc2-deficient mice fed HFD.
Inflammation in adipose tissue has been described as a driver for the development of obesity-associated liver disease and insulin resistance in humans (2, 18) and animals (19, 45). As stated earlier, white adipose tissue was significantly more prominent in WT mice than in Muc2−/− mice (Fig. 1C). Markers of inflammation such as IL-1β, TNF-α, Ccl2 (MCP-1), and F4/80 were elevated in white adipose tissue in WT mice fed HFD relative to WT mice fed regular chow; but most importantly, they were significantly higher in WT mice that were fed HFD than in their Muc2-deficient littermates (Fig. 5A).
Fig. 5.

Gene expression in white and brown adipose tissue, and energy expenditure in WT and Muc2−/− mice after 16 wk of HFD. WT or Muc2−/− mice were fed regular chow (n = 3–5) or HFD (n = 7–8) for 16 wk. A–C: gene expression by quantitative polymerase chain reaction (qPCR) in epididymal fat. A: inflammatory markers IL-1β, TNF-α, monocyte chemotactic protein-1 (MCP-1 or Ccl2), and F4/80. B: lipolysis enzymes adipose triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL). C: fatty acid β-oxidation enzyme palmitoyl acyl-CoA oxidase 1 (ACOX1). D and E: WT and Muc2−/− mice were fed regular chow (n = 2–4) or HFD (n = 3–8) for 16 wk. D: level of physical activity. E: hepatic gene expression by qPCR, 3-hydroxy-3-methylglutaryl-CoA lyase (or HMG-CoA lyase, HMGCL) and the rate-limiting enzyme of ketogenesis, mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase 2 (HMGCS2). F: immunoblot for uncoupling protein-1 (UCP1) in brown adipose tissue. G: densitometry of immunoblot images of UCP1 was performed with β-actin as loading control in brown adipose tissue of four separate immunoblot membranes (n = 5–6 for regular chow-fed mice and n = 11 for HFD-fed mice). H: TNF-α and MCP-1 (Ccl2) gene expression in brown adipose tissue (n = 4–5 for regular chow-fed mice and n = 6–8 for HFD-fed mice). *P < 0.05.
The key lipolysis enzymes adipose triglyceride lipase (ATGL, or PNPLA2) and hormone-sensitive lipase (HSL, or LIPE) were expressed more highly in white adipose tissue in Muc2−/− mice fed HFD than in WT mice (Fig. 5B). Similarly, fatty acid β-oxidation was increased in Muc2−/− mice relative to WT mice fed HFD, as exemplified by the increased gene expression of palmitoyl acyl-CoA oxidase 1 (ACOX1) (Fig. 5C). More lipolysis provides more free fatty acids for β-oxidation in adipose tissue.
Because there was a more pronounced increase in body weight in HFD-fed WT mice compared with Muc2−/− mice in spite of a similar food intake and similar fecal contents, we sought other explanations that would account for the ingested calories not stored in the form of fat tissue in Muc2−/− mice. The level of physical activity was similar between HFD-fed WT and Muc2−/− mice (Fig. 5D). Since ketone synthesis in the liver is inhibited by insulin, and obese subjects show relative insulin deficiency and augmented levels of ketone bodies in the blood (14), we assessed hepatic genes involved in ketone synthesis. HFD-fed WT mice had an induced expression of genes involved in ketone body synthesis in the liver relative to chow-fed WT mice; and Muc2-deficient mice exhibited a significantly lower level of these genes than WT mice after HFD (Fig. 5E). Furthermore, the activity of uncoupling protein-1 (UCP1) in the mitochondria in mostly brown adipose tissue leads to dissipation of chemical energy as heat instead of conserving it in adenosine triphosphate (ATP) (34). The protein expression of UCP1 was higher in brown adipose tissue of Muc2-knockout mice than WT mice after 16 wk of HFD (Fig. 5, F and G). This could account for increased energy expenditure in HFD-fed Muc2-deficient mice. Because UCP1 activity is suppressed by inflammation (34), we then checked the level of inflammation in brown fat. The inflammatory markers TNF-α and Ccl2 were reduced in brown adipose tissue in Muc2-deficient mice compared with WT mice after 16 wk of HFD (Fig. 5H). Hence, the higher protein expression of UCP1 in Muc2−/− mice fed HFD might be due to the lower level of inflammation in the brown adipose tissue.
Taken together, these inflammatory and metabolic differences in white and brown adipose tissue could explain the different phenotype in features of the metabolic syndrome.
Muc2 deficiency leads to higher intestinal and systemic levels of IL-22 following HFD.
We have shown that Muc2−/− mice display elevated levels of the antimicrobials Reg3b and Reg3g in the intestine (15). IL-22 exerts an essential role in regulating the intestinal antimicrobial barrier (30, 42), inducing Reg3b, Reg3g, and mucins (30). Moreover, IL-22 directly increases expression of the lipolysis/β-oxidation genes HSL, ATGL, and ACOX1, and improves insulin sensitivity (42). IL-22 also dampens chronic inflammation in fat tissue (42), and an IL-22 receptor is also found in brown adipose tissue (43). We therefore investigated the role of IL-22 in our HFD model. IL-22 gene expression was suppressed in the small intestine of HFD-fed WT mice compared with WT mice fed regular chow (Fig. 6A), consistent with recently published data (42). Small intestinal IL-22 mRNA levels were significantly higher in Muc2-knockout mice fed HFD relative to WT mice fed HFD (Fig. 6A). Similarly, IL-22 target genes Reg3b and Reg3g were elevated in the small intestine of HFD-fed Muc2-deficient mice compared with WT mice (Fig. 6B). Muc2-knockout mice that ate the HFD demonstrated overall higher levels of inflammatory gene expression in the small (Fig. 6C) and large (Fig. 6E) intestines than HFD-fed WT mice. HFD-fed Muc2−/− mice showed signs of early inflammation as evidenced by a more pronounced submucosal edema (Fig. 6D, straight line) and inflammatory infiltrates composed primarily of neutrophils (Fig. 6D, arrow) in the ileum. Colon samples from HFD-fed Muc2−/− mice demonstrated similar early signs of inflammation, but to a lesser degree (Fig. 6F). However, increased intestinal inflammation did not result in clinical signs of diarrhea or rectal bleeding (15), and is therefore best described as subclinical intestinal inflammation.
Fig. 6.

Muc2−/− mice reveal subclinical intestinal inflammation and elevated intestinal and systemic levels of IL-22 compared with WT mice fed HFD. A–G: WT and Muc2−/− mice were fed regular chow (n = 3–5) or HFD (n = 8) for 16 wk. A: gene expression of IL-22 by qPCR in the ileum. B: gene expression of regenerating islet-derived-3 beta and gamma (Reg3b and Reg3g) by qPCR in the ileum. Gene expression of inflammatory markers IL-1β and TNF-α by qPCR in the ileum (C) and colon (E). Representative ileum (D) and colon (F) sections after hematoxylin and eosin staining. Straight line indicates the extent of the submucosa/submucosal edema, arrows indicate inflammatory infiltrates composed primarily of neutrophils, ×400 magnification. G: plasma IL-22 level. H: WT and Muc2−/− mice were fed HFD (n = 4 in each group) for 11–20 wk. Plasma levels of IL-22 at baseline and 3 h after intraperitoneal injection of flagellin. *P < 0.05.
To investigate whether increased intestinal IL-22 gene expression directly translates into higher IL-22 protein expression and secretion, plasma IL-22 levels were determined. Supporting our intestinal results, HFD-fed Muc2−/− mice showed significantly higher IL-22 plasma levels than HFD-fed WT mice (Fig. 6G). Thus increased systemic IL-22 plasma levels appear to offer an explanation for the protection of Muc2-deficient mice against features of the metabolic syndrome.
To further elucidate whether systemic hyperresponsiveness contributes to higher IL-22 plasma levels in Muc2−/− mice, we tested this possibility by administering flagellin via intraperitoneal injection. This route of delivery bypasses the gastrointestinal tract and, in particular the intestinal mucus layer, and flagellin is absorbed directly into the systemic circulation. Flagellin is a TLR5 agonist and induces IL-22 plasma levels (42). Intraperitoneal injection of flagellin resulted in higher IL-22 plasma levels in both HFD-fed WT and Muc2−/− mice. The significant difference in IL-22 plasma levels at baseline between the groups disappeared (Fig. 6H). These results suggest that the absence of the mucus layer has an important effect on the production of intestinal IL-22.
Furthermore, intestinal dysbiosis is associated with obesity and obesity-related liver disease (3, 35, 38). Correspondingly, in our experiments administration of HFD resulted in intestinal bacterial overgrowth in WT and Muc2−/− mice (Fig. 7A). Muc2−/− mice fed HFD displayed significantly higher Bacteroidetes and a trend toward a lower Firmicutes:Bacteroidetes ratio compared with WT mice; no difference in Akkermansia nor Lactobacillus was observed between these groups (Fig. 7, B and C). No difference in plasma LPS was detected between WT and Muc2-deficient mice after being fed HFD (Fig. 7D). To determine whether the phenotype of the Muc2-deficient mice depends on host-microbiome interactions, mice were fed HFD for 16 wk and then treated with antibiotics for 5 wk. WT mice fed HFD showed no significant changes in body weight (Fig. 7E), but similarly to published data (3), they exhibited significant improvements in glucose tolerance and insulin responsiveness (Fig. 7F) after an intervention therapy with antibiotics relative to their preantibiotic state. Antibiotic treatment did not change body weight, glucose tolerance, or insulin responsiveness in Muc2−/− mice compared with their preantibiotic state; their values in the GTT and ITT were also not significantly different from those of WT mice after antibiotics were administered (Fig. 7, E and F). IL-22 gene expression in the ileum was not different after antibiotic intervention in WT and Muc2-deficient mice fed HFD (Fig. 7G). Thus although antibiotic treatment results in a reduced inflammation with favorable effects on metabolic functions in WT mice fed HFD (3), the beneficial immunomodulator IL-22 was no longer elevated in the ileum of Muc2−/− mice, which could explain the vanishing metabolic advantage in the knockout mice and the similar phenotype in both WT and Muc2−/− mice given HFD and antibiotics.
Fig. 7.

HFD leads to intestinal bacterial overgrowth, bacterial translocation, and antibiotic treatment of WT mice fed HFD results in an improved glucose tolerance and insulin sensitivity. A–D: WT and Muc2−/− mice were fed regular chow (n = 4–5) or HFD (n = 5–8) for 16 wk. A: total intestinal bacteria in cecum. B: relative proportions of Firmicutes, Bacteroidetes, and Firmicutes:Bacteroidetes ratio in cecum. C: relative proportions of Akkermansia and Lactobacillus in cecum. D: LPS plasma levels. E–G: WT and Muc2−/− mice were fed HFD for 16 wk, and then an additional 5-wk HFD plus antibiotics by gavage (n = 3). E: body weights in grams. F: GTT (top) and ITT (bottom). G: gene expression by qPCR of IL-22 in ileum. *P < 0.05.
It has been reported that different HFD preparations lead to different intestinal inflammatory responses in Muc2−/− mice (22). Interestingly, male Muc2-deficient mice fed a plant-derived HFD showed a similar body weight gain, glucose tolerance, and insulin sensitivity compared with WT mice fed a plant-derived diet for 16 wk, with a similar food intake (Fig. 8, A–C). Intestinal IL-22 levels in Muc2−/− mice were not significantly different from those of WT mice fed a plant-derived HFD (Fig. 8D). Thus the protection of Muc2-deficient mice from diet-induced obesity and metabolic syndrome is largely dependent on the source of the dietary fat.
Fig. 8.

Muc2 deficiency does not protect from plant-derived HFD-induced metabolic syndrome. A–C: WT and Muc2−/− mice were fed a plant-derived HFD (n = 5–6) for 14 wk. A: body weights in grams (left) and in % relative to starting weight (right). B: GTT (left) and ITT (right). C: daily food intake of a plant-derived HFD during week 3 and week 12. D: gene expression by qPCR of IL-22 in ileum.
DISCUSSION
In this study we investigated the role of Muc2 in obesity and NAFLD in a preclinical model. Muc2-deficient mice fed HFD were protected from diet-induced weight gain, fatty liver, and insulin resistance. Additionally, they displayed less inflammation, upregulated lipolysis and β-oxidation in white adipose tissue, and increased expression of UCP1 involved in thermogenesis in brown adipose tissue after 16 wk of being fed HFD. Protection from features of the metabolic syndrome could not be explained by differences in food intake in male Muc2-deficient mice. Mice lacking Muc2 that ate HFD displayed higher intestinal and plasma levels of IL-22 and higher levels of the intestinal antimicrobials Reg3b and Reg3g in relation to WT mice fed the same diet. Increased systemic levels of IL-22 provide an explanation for the resistance against diet-induced features of the metabolic syndrome in the absence of an intestinal mucus layer.
Our results suggest that IL-22 produced and secreted by immune cells in the intestine is the driving force behind increased systemic IL-22 levels. Following intraperitoneal injection of flagellin, differences in IL-22 plasma levels between HFD-fed WT and Muc2−/− mice vanished. This suggests that the absence of the intestinal mucus layer does not lead to a generalized activation of the immune system, but the activation might be restricted to the mucosal immune system.
How can the absence of a protective barrier be beneficial for the host? The host deploys several barriers to protect the body from invading intestinal microbes and their products. Epithelial cells with their tight junctions and the mucus layer that covers the surface of intestinal epithelial cells are examples for physical barriers (35). Because Muc2 is the major secreted mucin in the intestinal tract, its absence dramatically decreases the overall mucus layer (15). If there is a breach in this mechanical barrier, as it occurs in Muc2-deficient mice, it is likely that microbial products have better access to immune cells located in the lamina propria. This could lead to the activation of immune cells such as monocytes, macrophages, and dendritic cells as a first-line, innate immune defense. Such activation primarily aims at restoration of the barrier dysfunction and initiation of a wound-healing process. Several proinflammatory mediators such as TNF-α and Ccl5 have been described to be involved. These molecules activate intracellular signaling cascades in epithelial cells that result in disruption of the tight junctions and cause further progression of disease (5, 8, 16). Components in the intestinal inflammation process such as the secretion of IL-22 are also beneficial. IL-22 is very effective at inducing antimicrobial molecules such as Reg3b, Reg3g, and mucin production (30, 42). IL-22 effectively protects from experimental colitis (48). An intestinal immune response that is characterized by predominately protective mediators will be beneficial for the host and will restore intestinal homeostasis. In Muc2 deficiency, there seems to be a very strong IL-22 response and an upregulation of IL-22 target genes following consumption of a lard-derived HFD. Currently, we can only speculate about why the absence of a mucus layer provokes this rather beneficial effect. Intestinal inflammation could be driven by dysbiosis and released products or metabolites from the dysbiotic microbiota (16). Muc2 deficiency and its associated leakiness might lead to more translocation of microbial mediators to the lamina propria that activate IL-22 expression. However, antibiotic treatment alone was not sufficient to reduce features of the metabolic syndrome in Muc2-deficient mice. It is therefore unlikely that dysbiosis alone is enhancing the mucosa-protective response in the absence of a mucin layer. Interestingly, the protective effect is lost once the source of fat is changed from animal- to plant-derived. It is therefore conceivable that food-derived metabolites drive the strong mucosal IL-22 response in the absence of mucin, which eventually will increase systemic IL-22 plasma levels. In our experiments, the plant-derived HFD had 6% fiber content, and the lard-derived HFD contained no fiber at all. Taking into account the anti-inflammatory properties of fiber (21), this could be one factor that explains the phenotype difference after Muc2−/− mice were fed a plant-derived HFD. However, the absence of the mucus layer is not beneficial in every instance. For example, intestinal pathogens such as Salmonella can penetrate and invade the host more easily (47).
IL-22 is not only good for restoring intestinal homeostasis, but it also exerts beneficial effects on a broad range of liver diseases (20, 32, 46) and the metabolic syndrome (42). A lard-derived HFD suppresses intestinal and systemic IL-22 in mice (42), which is in agreement with our results. Most interestingly, therapeutic administration of IL-22 has recently been shown to prevent and treat the metabolic syndrome in mice (42). The detailed mechanism of how IL-22 protects from diet-induced obesity is not entirely known (44). It involves suppression of inflammation, induction of lipolysis and β-oxidation in white adipose tissue (42), which we have also observed in our model. Thus increased systemic IL-22 is the mostly likely explanation why Muc2-deficient mice are protected from the metabolic syndrome. However, we cannot rule out that other mediators that might be induced in response to the intestinal barrier failure might contribute to the protection as well.
In summary, we describe a genetic model of mucin deficiency that prevents metabolic syndrome development. A strong intestinal immune response is initiated as a consequence of an intestinal barrier dysfunction. Increased levels of systemic IL-22 protect mice from diet-induced obesity and its consequences. Future studies need to determine the link between Muc2 deficiency and initiation of this beneficial restoration process in the intestinal lamina propria. Determining which metabolites cause this immune response could have tremendous clinical implications because such interventions hold promise to serve as a treatment option for patients suffering from the metabolic syndrome.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
P.H., C.T.S., M.M., K.B., S.B.H., and B.S. conception and design of research; P.H., C.T.S., M.M., A.H., L.W., and C.L. performed experiments; P.H., C.T.S., M.M., N.M.V., and B.S. analyzed data; P.H., C.T.S., M.M., N.M.V., K.B., S.B.H., and B.S. interpreted results of experiments; P.H. and C.T.S. prepared figures; P.H., C.T.S., M.M., and B.S. drafted manuscript; P.H., C.T.S., M.M., K.B., S.B.H., and B.S. edited and revised manuscript; P.H., C.T.S., M.M., L.W., C.L., K.B., S.B.H., and B.S. approved final version of manuscript.
ACKNOWLEDGMENTS
Support for this study was provided in part by Deutsche Forschungsgemeinschaft (DFG) Grant 7336/1-1 to P. Hartmann; National Institutes of Health Grants K08 DK-081830, R01 AA-020703, and U01 AA-021856 to B. Schnabl; and by Award I01BX002213 from the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development to B. Schnabl.
REFERENCES
- 1.Brenner DA, Paik YH, Schnabl B. Role of gut microbiota in liver disease. J Clin Gastroenterol 49, Suppl 1: S25–S27, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cancello R, Tordjman J, Poitou C, Guilhem G, Bouillot JL, Hugol D, Coussieu C, Basdevant A, Bar Hen A, Bedossa P, Guerre-Millo M, Clément K. Increased infiltration of macrophages in omental adipose tissue is associated with marked hepatic lesions in morbid human obesity. Diabetes 55: 1554–1561, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57: 1470–1481, 2008. [DOI] [PubMed] [Google Scholar]
- 4.Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O, Geurts L, Naslain D, Neyrinck A, Lambert DM, Muccioli GG, Delzenne NM. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 58: 1091–1103, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen P, Starkel P, Turner JR, Ho SB, Schnabl B. Dysbiosis-induced intestinal inflammation activates tumor necrosis factor receptor I and mediates alcoholic liver disease in mice. Hepatology 61: 883–894, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen P, Torralba M, Tan J, Embree M, Zengler K, Starkel P, van Pijkeren JP, DePew J, Loomba R, Ho SB, Bajaj JS, Mutlu EA, Keshavarzian A, Tsukamoto H, Nelson KE, Fouts DE, Schnabl B. Supplementation of saturated long-chain fatty acids maintains intestinal eubiosis and reduces ethanol-induced liver injury in mice. Gastroenterology 148: 203–214.e216, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collado MC, Derrien M, Isolauri E, de Vos WM, Salminen S. Intestinal integrity and Akkermansia muciniphila, a mucin-degrading member of the intestinal microbiota present in infants, adults, and the elderly. Appl Environ Microbiol 73: 7767–7770, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding S, Chi MM, Scull BP, Rigby R, Schwerbrock NM, Magness S, Jobin C, Lund PK. High-fat diet: bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS One 5: e12191, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG, Delzenne NM, de Vos WM, Cani PD. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci USA 110: 9066–9071, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fang S, Suh JM, Reilly SM, Yu E, Osborn O, Lackey D, Yoshihara E, Perino A, Jacinto S, Lukasheva Y, Atkins AR, Khvat A, Schnabl B, Yu RT, Brenner DA, Coulter S, Liddle C, Schoonjans K, Olefsky JM, Saltiel AR, Downes M, Evans RM. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat Med 21: 159–165, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ganz M, Csak T, Szabo G. High fat diet feeding results in gender specific steatohepatitis and inflammasome activation. World J Gastroenterol 20: 8525–8534, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol 29: 415–445, 2011. [DOI] [PubMed] [Google Scholar]
- 13.Guo X, Xia X, Tang R, Zhou J, Zhao H, Wang K. Development of a real-time PCR method for Firmicutes and Bacteroidetes in faeces and its application to quantify intestinal population of obese and lean pigs. Lett Appl Microbiol 47: 367–373, 2008. [DOI] [PubMed] [Google Scholar]
- 14.Hall SE, Wastney ME, Bolton TM, Braaten JT, Berman M. Ketone body kinetics in humans: the effects of insulin-dependent diabetes, obesity, and starvation. J Lipid Res 25: 1184–1194, 1984. [PubMed] [Google Scholar]
- 15.Hartmann P, Chen P, Wang HJ, Wang L, McCole DF, Brandl K, Starkel P, Belzer C, Hellerbrand C, Tsukamoto H, Ho SB, Schnabl B. Deficiency of intestinal mucin-2 ameliorates experimental alcoholic liver disease in mice. Hepatology 58: 108–119, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, Camporez JP, Shulman GI, Gordon JI, Hoffman HM, Flavell RA. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482: 179–185, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horz HP, Vianna ME, Gomes BP, Conrads G. Evaluation of universal probes and primer sets for assessing total bacterial load in clinical samples: general implications and practical use in endodontic antimicrobial therapy. J Clin Microbiol 43: 5332–5337, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest 95: 2409–2415, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259: 87–91, 1993. [DOI] [PubMed] [Google Scholar]
- 20.Ki SH, Park O, Zheng M, Morales-Ibanez O, Kolls JK, Bataller R, Gao B. Interleukin-22 treatment ameliorates alcoholic liver injury in a murine model of chronic-binge ethanol feeding: role of signal transducer and activator of transcription 3. Hepatology 52: 1291–1300, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuo SM. The interplay between fiber and the intestinal microbiome in the inflammatory response. Adv Nutr 4: 16–28, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu P, Bar-Yoseph F, Levi L, Lifshitz Y, Witte-Bouma J, de Bruijn AC, Korteland-van Male AM, van Goudoever JB, Renes IB. High beta-palmitate fat controls the intestinal inflammatory response and limits intestinal damage in mucin Muc2 deficient mice. PLoS One 8: e65878, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGuckin MA, Linden SK, Sutton P, Florin TH. Mucin dynamics and enteric pathogens. Nat Rev Microbiol 9: 265–278, 2011. [DOI] [PubMed] [Google Scholar]
- 24.McNelis JC, Olefsky JM. Macrophages, immunity, and metabolic disease. Immunity 41: 36–48, 2014. [DOI] [PubMed] [Google Scholar]
- 25.Mokdad AH, Ford ES, Bowman BA, Dietz WH, Vinicor F, Bales VS, Marks JS. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. JAMA 289: 76–79, 2003. [DOI] [PubMed] [Google Scholar]
- 26.Nakanishi Y, Murashima K, Ohara H, Suzuki T, Hayashi H, Sakamoto M, Fukasawa T, Kubota H, Hosono A, Kono T, Kaminogawa S, Benno Y. Increase in terminal restriction fragments of Bacteroidetes-derived 16S rRNA genes after administration of short-chain fructooligosaccharides. Appl Environ Microbiol 72: 6271–6276, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA 311: 806–814, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 72: 219–246, 2010. [DOI] [PubMed] [Google Scholar]
- 29.Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med 18: 363–374, 2012. [DOI] [PubMed] [Google Scholar]
- 30.Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol 29: 71–109, 2011. [DOI] [PubMed] [Google Scholar]
- 31.Pischon T, Nothlings U, Boeing H. Obesity and cancer. Proc Nutr Soc 67: 128–145, 2008. [DOI] [PubMed] [Google Scholar]
- 32.Radaeva S, Sun R, Pan HN, Hong F, Gao B. Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology 39: 1332–1342, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Rinttila T, Kassinen A, Malinen E, Krogius L, Palva A. Development of an extensive set of 16S rDNA-targeted primers for quantification of pathogenic and indigenous bacteria in faecal samples by real-time PCR. J Appl Microbiol 97: 1166–1177, 2004. [DOI] [PubMed] [Google Scholar]
- 34.Sakamoto T, Takahashi N, Sawaragi Y, Naknukool S, Yu R, Goto T, Kawada T. Inflammation induced by RAW macrophages suppresses UCP1 mRNA induction via ERK activation in 10T1/2 adipocytes. Am J Physiol Cell Physiol 304: C729–C738, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schnabl B, Brenner DA. Interactions between the intestinal microbiome and liver diseases. Gastroenterology 146: 1513–1524, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tacke F, Gabele E, Bataille F, Schwabe RF, Hellerbrand C, Klebl F, Straub RH, Luedde T, Manns MP, Trautwein C, Brenner DA, Scholmerich J, Schnabl B. Bone morphogenetic protein 7 is elevated in patients with chronic liver disease and exerts fibrogenic effects on human hepatic stellate cells. Dig Dis Sci 52: 3404–3415, 2007. [DOI] [PubMed] [Google Scholar]
- 37.Tajiri Y, Mimura K, Umeda F. High-sensitivity C-reactive protein in Japanese patients with type 2 diabetes. Obes Res 13: 1810–1816, 2005. [DOI] [PubMed] [Google Scholar]
- 38.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444: 1027–1031, 2006. [DOI] [PubMed] [Google Scholar]
- 39.Velcich A, Yang W, Heyer J, Fragale A, Nicholas C, Viani S, Kucherlapati R, Lipkin M, Yang K, Augenlicht L. Colorectal cancer in mice genetically deficient in the mucin Muc2. Science 295: 1726–1729, 2002. [DOI] [PubMed] [Google Scholar]
- 40.Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 328: 228–231, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L, Hartmann P, Haimerl M, Bathena SP, Sjowall C, Almer S, Alnouti Y, Hofmann AF, Schnabl B. Nod2 deficiency protects mice from cholestatic liver disease by increasing renal excretion of bile acids. J Hepatol 60: 1259–1267, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X, Ota N, Manzanillo P, Kates L, Zavala-Solorio J, Eidenschenk C, Zhang J, Lesch J, Lee WP, Ross J, Diehl L, van Bruggen N, Kolumam G, Ouyang W. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature 514: 237–241, 2014. [DOI] [PubMed] [Google Scholar]
- 43.Wang Z, Yang L, Jiang Y, Ling ZQ, Li Z, Cheng Y, Huang H, Wang L, Pan Y, Yan X, Chen Y. High fat diet induces formation of spontaneous liposarcoma in mouse adipose tissue with overexpression of interleukin 22. PLoS One 6: e23737, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu H, Ballantyne CM. Inflammation versus host defense in obesity. Cell Metab 20: 708–709, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wueest S, Rapold RA, Schumann DM, Rytka JM, Schildknecht A, Nov O, Chervonsky AV, Rudich A, Schoenle EJ, Donath MY, Konrad D. Deletion of Fas in adipocytes relieves adipose tissue inflammation and hepatic manifestations of obesity in mice. J Clin Invest 120: 191–202, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xing WW, Zou MJ, Liu S, Xu T, Gao J, Wang JX, Xu DG. Hepatoprotective effects of IL-22 on fulminant hepatic failure induced by d-galactosamine and lipopolysaccharide in mice. Cytokine 56: 174–179, 2011. [DOI] [PubMed] [Google Scholar]
- 47.Zarepour M, Bhullar K, Montero M, Ma C, Huang T, Velcich A, Xia L, Vallance BA. The mucin Muc2 limits pathogen burdens and epithelial barrier dysfunction during Salmonella enterica serovar Typhimurium colitis. Infect Immun 81: 3672–3683, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 29: 947–957, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]