

Abstract
The cystic fibrosis (CF) transmembrane conductance regulator (CFTR) is a chloride ion channel, the dysfunction of which directly leads to the life-shortening disease CF. Extracellular loop 1 (ECL1) of CFTR contains several residues involved in stabilizing the open state of the channel; some, including D110, are sites of disease-associated gating mutations. Structures from related proteins suggest that the position of CFTR's extracellular loops may change considerably during gating. To better understand the roles of ECL1 in CFTR function, we utilized functional cysteine cross-linking to determine the effects of modulation of D110C-CFTR and of a double mutant of D110C with K892C in extracellular loop 4 (ECL4). The reducing agent DTT elicited a large potentiation of the macroscopic conductance of D110C/K892C-CFTR, likely due to breakage of a spontaneous disulfide bond between C110 and C892. DTT-reduced D110C/K892C-CFTR was rapidly inhibited by binding cadmium ions with high affinity, suggesting that these residues frequently come in close proximity in actively gating channels. Effects of DTT and cadmium on modulation of pore gating were demonstrated at the single-channel level. Finally, disulfided D110C/K892C-CFTR channels were found to be less sensitive than wild-type or DTT-treated D110C/K892C-CFTR channels to stimulation by IBMX, suggesting an impact of this conformational restriction on channel activation by phosphorylation. The results are best explained in the context of a model of CFTR gating wherein stable channel opening requires correct positioning of functional elements structurally influenced by ECL1.
Keywords: cystic fibrosis transmembrane conductance regulator, cysteine-mediated cross-linking, chloride channel, structure-function, ATP-binding cassette transporter
cystic fibrosis (CF) is a life-shortening disease affecting ∼70,000 people worldwide. CF is directly caused by mutations imparting defects in the CF transmembrane conductance regulator (CFTR), a chloride channel expressed in several tissues and directly related to osmotic balance in epithelia (33, 35). CFTR is the only member of the ATP-binding cassette (ABC) transporter superfamily known to function as a channel (15). Similar to other mammalian ABC transporters, the protein is composed of two membrane-spanning domains (MSD1 and MSD2), each followed by cytoplasmic nucleotide-binding domains (NBD1 and NBD2). Uniquely among ABC transporters, CFTR also features a cytoplasmic regulatory R domain connecting NBD1 to MSD2, by which the channel is regulated via phosphorylation at protein kinase A (PKA) consensus sites (20, 37). It is thought that phosphorylated CFTR channels are gated via binding of ATP to the intracellular NBDs, which promotes their tight heterodimerization (28) and transmits conformational change to the transmembrane helices (TMs) forming the pore via interactions between the NBDs and intracellular loops of the MSDs (5, 18). A large body of electrophysiological evidence has indicated that the permeation pathway through CFTR is made up of significant contributions from at least three TMs (TM6, TM12, and TM1) (1, 2, 9, 16) and that changes in conformation and/or positioning of these TMs affect permeation through the pore (3, 47).
Interestingly, although the most common mutation detected in patients [the trafficking mutation F508del (7)] is located in NBD1, ∼2,000 reported mutations in CFTR, including several in the MSDs, have been reported (21). One particular area of interest is extracellular loop 1 (ECL1), where mutations at six different positions, including D110H and D110E, are associated with CF disease (40). Studies of these and other CF-causing mutations in this region, including R117H (17, 38), as well as studies using multispecies CFTR chimeras (30), have implied that ECL1 has an important role in maintaining the stability of the open pore of the channel. A recent study by our group has increased our understanding of ECL1 by characterizing charge-reversal mutants of disease-relevant residues in this region (8). Of particular interest, D110R-CFTR and E116R-CFTR displayed very brief mean burst durations (22 and 37 ms, respectively) compared with wild-type (WT)-CFTR (∼700 ms), but comparable full-conductance amplitudes. The mean burst durations of the mutants were not increased by the poorly hydrolyzable ATP analog adenylylimidodiphosphate (AMP-PNP), indicating that the defects are in “pore gating,” that is, regulation of the CFTR gate kinetically downstream of ATP binding and NBD dimerization. Charge-sparing mutations at both positions apparently rescued the mean open burst duration, at least partially, perhaps pointing to a role of negative charge at these positions in maintaining open pore stability. On the basis of charge-swapping and engineered cysteine electrophysiology experiments, E116 was suggested to interact with R104 of TM1 in the open and closed states of the channel. However, interacting open state partners with D110 were not found in that study (8). Interestingly, we also found that the reducing agent DTT significantly increased openings of D110C/K892C-CFTR in multichannel patches by apparently breaking a disulfide bond between D110C (in ECL1) and K892C [in extracellular loop 4 (ECL4)] (8). This result was intriguing in the context of the demonstrated significance of D110 in the channel's open state (17). However, important information relating to the proximity of ECL1 and ECL4 in CFTR function, such as the residue dependence, kinetics, and effects on single-channel behavior, remained to be elucidated.
In the present study, we utilized cysteine mutagenesis and electrophysiological approaches at various levels of resolution to investigate the role of ECL1 in channel function by probing in detail the effect of modifying C110 and linking it via disulfide bonding or metal coordination with C892. The data from this study strongly suggest that the positioning of the NH2-terminal end of ECL1 is important for pore gating and regulation. Interpreted in the context of other recent reports by our group and others (16), this work strengthens the hypothesis that the extracellular end of the first TM helix of CFTR is involved in regulating permeation through the channel pore and that a specific conformation in this region must be adopted for stable channel opening.
METHODS AND MATERIALS
Oocyte Preparation and Molecular Biology
Human WT-CFTR (V470M/V1470M variant) subcloned into the pGEM-HE oocyte expression vector was previously provided by D. Gadsby (Rockefeller University, New York, NY). Human WT β2-adrenergic receptor (β2-AR) subcloned into the pSP65 vector was previously provided by B. Kobilka (Stanford University, Stanford, CA). Mutations were made using the QuikChange XL mutagenesis kit (Stratagene) according to the manufacturer's instructions, and sequences were verified through the open reading frame. cRNA was transcribed using the Ambion in vitro RNA transcription kit according to the manufacturer's instructions. Xenopus laevis oocytes were injected with 1–10 ng of CFTR cRNA. For some two-electrode voltage-clamp (TEVC) experiments, cRNA encoding β2AR was premixed with CFTR cRNA at a mass-to-mass ratio of 1:20. Oocytes were incubated in modified Leibovitz's L-15 medium plus HEPES (pH 7.5), penicillin, streptomycin, and ciprofloxacin. The oocyte isolation procedure was carried out in accordance with a protocol approved by the Animal Care and Use Committee of Emory University. Recordings were typically made 48–96 h after cRNA injection.
Electrophysiology
TEVC recording.
All recordings were made at room temperature in Ringer solution (ND96) consisting of (in mM) 96 NaCl, 2 KCl, 1 MgCl2, and 5 HEPES, pH 7.5. For cadmium dose-response experiments, ND96 was made up in new plastic vessels to minimize the effect of contaminating trace metals leaching from glassware. Recordings were made using a GeneClamp 500 amplifier (Molecular Devices). Intracellular electrode resistances averaged 0.5 MΩ when 3 M KCl solution was used. Different voltage protocols were utilized depending on application. For recordings measuring conductance, the membrane voltage (Vm) was held at −20 mV (approximating the resting Vm for an expressing cell), and Vm was pulsed to −80, −60, −40, −20, and 0 mV for 200 ms every 2 s. For each CFTR variant studied, the data generated a current-voltage slope that was linear from −60 to 0 mV. Cell conductance was calculated as the slope between −40 and 0 mV. This protocol allowed recording of even highly expressing oocytes at 10-s time resolution while limiting rundown from intracellular chloride depletion. In experiments with higher desired resolution, Vm was held at −20 mV and pulsed to −60 mV for 200 ms every 2 s, and current at −60 mV was reported; altenatively, cells were held at Vm = −40 mV, and gap-free recordings were digitized at 500 Hz. In most experiments, CFTR was activated through the β2AR (via Gαs signaling to increase intracellular cAMP) with isoproterenol. Physiological activation of CFTR via β2AR has been demonstrated in lung (6), sweat glands (31), and nasal epithelia (34). This activation mode has been used successfully by our group and others in several studies to probe CFTR structure in oocytes via cysteine accessibility and modification, including studies using DTT as a reducing agent (1, 8, 27, 29). In experiments where precisely graded activation was desired, CFTR was activated by 10 μM forskolin (the adenylate kinase activator) and various concentrations of 3-isobutyl-1-methylxanthine (IBMX, the phosphodiesterase inhibitor) to reach previously described quasi-plateaus (10, 39, 45).
Single-channel recording.
Excised, inside-out patches containing several CFTR channels were recorded at room temperature, as previously described (13). Manual removal of the oocyte vitelline membrane was performed after shrinking the cell in hypertonic solution containing (in mM) 200 monopotassium aspartate, 20 KCl, 1 MgCl2, 10 EGTA, and 10 HEPES, pH 7.2 adjusted with KOH. Pipettes were pulled from borosilicate glass using a laser puller (model P-2000, Sutter). Pipette resistances averaged 10 MΩ when pipettes were filled with solution containing (in mM) 150 N-methyl-d-glutamine (NMDG) chloride, 5 MgCl2, and 10 Tris·HCl-EDTA-sucrose (TES) buffer, pH 7.5. Channels were activated and recorded in intracellular solution containing 150 mM NMDG chloride, 1.1 mM MgCl2, 2 mM Tris-EGTA, 10 mM TES buffer, 1 mM MgATP, and 127 U/ml PKA, pH 7.5. CFTR currents were measured using a patch-clamp amplifier (Axon Instruments 200B, Molecular Devices) holding in all cases at Vm = −100 mV, low-pass Bessel-filtered at 10 kHz, and recorded to digital audio tape. Traces were played back to pClamp 10.2 software, filtered at 100 Hz, and digitized at 500 Hz, and baseline was manually adjusted. Open burst durations were analyzed as described in our previous study (14), analyzing at least 100 openings pooled from at least 4 independent patches per condition. All-points amplitude histograms corresponding to each individual displayed record were fit to Gaussian distributions and shown next to the associated traces.
Generation of CFTR Model Image
Coordinates for amino acids were based on the 2.5-ns snapshot of a recently published molecular dynamics simulation of CFTR gating (32). Images were generated using PyMOL software.
Statistics
Groups of data were compared using Student's t-test. Error bars indicate standard deviation in all cases except single-channel mean burst duration, where error bars indicate standard error of the mean.
Chemicals and Reagents
Purified PKA catalytic subunit was purchased from Promega and L-15 medium from Gibco. The CFTR inhibitor Inh172 was obtained from Cayman Chemicals and diluted from frozen 20 mM stocks in DMSO. All other chemicals were purchased from Sigma Aldrich. DTT stocks (1 M) in double-distilled H2O were made fresh at the beginning of each experiment day and kept on ice. IBMX stocks (1 M) in DMSO were made fresh at the beginning of each experiment day and kept at room temperature. Cadmium solutions were made from serial dilutions of a 1 M CdCl2 stock solution in double-distilled H2O.
Limitations of Functional Cross-Linking Approaches to Elucidate Structural Details in Ion Channels
The present study uses functional cysteine linking to inform on structural details of CFTR's extracellular region. As such, the results must be interpreted in the context of two main limitations of this technique to study structural proximity. 1) Since the approach relies on a functional effect as readout, it is possible to detect only linkages that discernibly affect the function of the channel; therefore, negative results may be interpreted as lack of proximity or lack of a functional effect of that proximity. 2) As the technique requires that cysteine residues be engineered into the protein sequence, it is possible that these mutations themselves will affect channel function in a way that colors the interpretation of the effects of chemical treatments. However, in the presence of reducing agents, the single-channel behavior of both cysteine mutants for which functional effects were investigated (D110C-CFTR and D110C/K892C-CFTR) was similar to that of WT-CFTR, with the exception of moderately shortened mean open burst duration (see Figs. 6 and 7).
Fig. 6.

D110C-CFTR gating was modulated by DTT and cadmium in a manner consistent with the macroscopic results. CFTR variants were activated by 1 mM ATP and 127 U/ml PKA, and recordings were made at Vm = −100 mV. A: single-channel traces of D110C-CFTR activated by intracellular ATP and PKA and standard pipette solution (D110C-CFTR naïve), pipette solution containing 1 mM DTT (D110C-CFTR + DTT), or pipette solution containing 20 μM cadmium after preincubation of the oocyte with 1 mM DTT (D110C-CFTR + Cd2+ after DTT), all with corresponding amplitude histograms fit to Gaussian distributions. Bottom trace: expanded representation of the bracketed area in the “D110C-CFTR + Cd2+ after DTT” trace. In naïve channels, subconductance (black arrowhead) and full-conductance (gray arrowhead) openings were observed in the same patches. B: mean open burst duration of D110C-CFTR for each condition. *P < 0.05.
Fig. 7.

DTT and cadmium modulated single-channel behavior of D110C/K892C-CFTR channels. CFTR variants were activated by 1 mM ATP and 127 U/ml PKA, and recordings were made at Vm = −100 mV. A: D110C/K892C-CFTR channels recorded with standard pipette solution opened briefly and to a variable conductance level (D110C/K892C-CFTR naïve). D110C/K892C-CFTR channels in the presence of 1 mM DTT opened stably to a full conductance (D110C/K892C-CFTR + DTT). D110C/K892C-CFTR channels in the presence of 20 nM cadmium opened briefly to a range of subconductances [D110C/K892C-CFTR + 20 nM cadmium (after DTT)]. Bottom trace: expanded representation of bracketed area in the “D110C/K892C-CFTR + 20 nM cadmium (after DTT)” trace. A corresponding amplitude histogram fit to a Gaussian distribution accompanies each trace. B: mean open burst duration for each condition. *P < 0.05.
RESULTS
DTT Reduces a Putative Disulfide Bond Between D110C and K892C, Highly Potentiating Channel Activity
To probe the role of ECL1 in channel function, we initially asked whether linking a cysteine substitution of a residue in ECL1 to one in ECL4 (expected to lie across the channel pore) would stabilize the channel’s closed state in whole oocytes, using a protocol designed to minimize chloride depletion-mediated rundown (see methods and materials). When coexpressed in X. laevis oocytes with β2-AR, D110C/K892C-CFTR was activated with a saturating concentration (10 μM) of isoproterenol to reach a conductance plateau, typically within 5–10 min. Addition of 1 mM of the reducing agent DTT after maximal activation by isoproterenol elicited a large increase in conductance that failed to reach a plateau even after 5 min of continued perfusion (13.0 ± 3.2 fold increase, n = 10; Fig. 1A), presumably due to breakage of an inhibitory disulfide bond between C110 and C892. WT-CFTR channels activated in the same way were apparently not potentiated by DTT (n = 6; Fig. 1B), in agreement with reports from our laboratory and others (3, 8, 25), nor were K892C-CFTR channels potentiated by DTT (n = 4; Fig. 1C). There was no apparent effect of DTT or isoproterenol on the conductance of uninjected oocytes (n = 4; not shown). Interestingly, D110C-CFTR single-mutant channels were stably potentiated 2.87 ± 1.05 fold by DTT (n = 18; Fig. 1D), typically reaching a plateau within 5 min. D110A-CFTR channels were unaffected by DTT, directly implicating the reactive cysteine in the potentiation of D110C-CFTR by DTT (n = 4; Fig. 1E). These experiments, summarized in Fig. 1F, provide evidence that D110C and K892C form a spontaneous disulfide bond in CFTR and that this close linkage between ECL1 and ECL4 markedly perturbs channel activity.
Fig. 1.

D110C/K892C-CFTR macroscopic currents were highly potentiated by DTT. Traces show change in whole cell conductance (G) over time during activation of CFTR by stimulation of coexpressed β2-adrenergic receptor (β2-AR) using isoproterenol and subsequent exposure to DTT in the continuing presence of isoproterenol. A–E: D110C/K892C-CFTR, WT-CFTR, K892C-CFTR, D110C-CFTR, and D110A-CFTR, all activated by 10 μM isoproterenol (ISO) and then exposed to 1 mM DTT for 5 min. F: fold increase of conductance [i.e., slope between membrane potential (Vm) = −40 and 0 mV] after 5 min of DTT application for all variants tested. Dashed line indicates level of no change (i.e., “fold change of 1”). *P < 0.05.
D110C/K892C-CFTR Channels Coordinate Cadmium, Which Inhibits Channel Macroscopic Conductance
Since cysteine residues are engineered via coding into the protein of interest, it is possible that a disulfide bond could irreversibly form at any time between protein expression/folding and the time the experiment was performed, a time course of multiple days. For this reason, it has been suggested that engineered disulfide bonds can “trap” residues into rare and otherwise unnatural conformations (12). To better understand the kinetics of the C110–C892 linkage, we used acute applications of the soft metal cadmium, as successfully used to probe cysteine proximity in previous studies of CFTR (12, 24, 42) and many other types of ion channels, to perform metal-bridging experiments. To ensure that engineered cysteine residues of these channels were free to coordinate cadmium, the cells were preincubated with 1 mM DTT directly before channel activation. Cells were clamped to Vm = −40 mV and exposed to isoproterenol, and at current plateau, cadmium (20 μM) was applied for 30 s and then washed out for 30 s in the continuing presence of isoproterenol. D110C/K892C-CFTR channels were rapidly inhibited by cadmium, with negligible reversibility in the subsequent 30 s of isoproterenol alone (Fig. 2A). By contrast, D110C-CFTR channels were reversibly inhibited by 20 μM cadmium (Fig. 2B). There were no apparent significant effects of cadmium on WT-CFTR channels (Fig. 2C), in agreement with previous studies using millimolar (3) or micromolar (42) extracellular cadmium; nor was there an apparent effect on K892C-CFTR channels (Fig. 2D). The difference in the extent of recovery after removal of cadmium (quantified in Fig. 2E) between D110C-CFTR and D110C/K892C-CFTR suggests a large apparent difference in the affinity for cadmium in these mutants. Consistent with these results, dose-response experiments carried out over a seven log10 range of cadmium concentrations revealed a ∼1,000-fold higher affinity of D110C/K892C-CFTR than D110C-CFTR for cadmium (Fig. 3, A and B). A saturating cadmium concentration (20 μM) reversed a large fraction of the increase elicited in D110C/K892C-CFTR upon exposure to DTT (Fig. 3C), consistent with the idea that linkage of C110 and C892 via cadmium may inhibit channel activity in a manner similar to linkage via a disulfide bond.
Fig. 2.

Inhibition of D110C-CFTR and D110C/K892C-CFTR by cadmium demonstrated differences in reversibility. Traces show change in macroscopic current (μA) over time during activation by stimulation of coexpressed β2AR using isoproterenol. All cells were preincubated with 1 mM DTT for 5 min. A–D: D110C/K892C-CFTR, D110C-CFTR, WT-CFTR, and K892C-CFTR, all with 30-s exposures to 20 μM cadmium followed by 30 s of cadmium washout in the presence of isoproterenol. Current was reported at Vm = −40 mV; increased activation results in downward deflection (n = 4–7 per variant). E: D110C-CFTR and D110C/K892C-CFTR percent current recovery after 30 s of cadmium washout in the continuing presence of isoproterenol. *P < 0.05.
Fig. 3.

Dose-response experiments for D110C-CFTR and D110C/K892C-CFTR after DTT treatment and in the constant presence of 10 μM isoproterenol demonstrate that D110C-K892C-CFTR was ∼1,000-fold more sensitive to cadmium. A: representative records for D110C-CFTR and D110C/K892C-CFTR wherein cadmium was applied in the constant presence of 10 μM isoproterenol after 4 min of treatment with 1 mM DTT. Electrophysiological protocol was identical to that described in Fig. 1 legend. B: cadmium dose response. Kd for cadmium in the single mutant D110C-CFTR was 4.49 ± 1.65 μM; Kd in the double mutant was ∼2 nM. C: percentage of DTT-induced increase that was reversed by maximal cadmium for D110C-CFTR (2 mM cadmium) and D110C/K892C-CFTR (20 μM cadmium).
K892C in ECL4 Fails to Form Spontaneous Disulfide Bonds or Coordinate Cadmium With Two Substituted Cysteine Residues Near K892
We next asked whether cysteine linkages could be formed between K892C and other ECL1 cysteine substitutions via disulfide formation or cadmium coordination. In Fig. 4A, E115C/K892C-CFTR is shown first exposed to 1 mM DTT and then to 20 μM cadmium. There was no significant effect of either chemical on macroscopic currents (Fig. 4B). There also was no significant effect of either DTT or cadmium on D112C/K892C-CFTR (Fig. 4, B and C). Since D110C/K892C-CFTR was sensitive to DTT and cadmium but D112C/K892C-CFTR and E115C/K892C-CFTR were sensitive to neither chemical, these results suggest that ECL1 of CFTR is oriented similar to that of related ABC efflux transporters, which in crystal structures feature the NH2-terminal end positioned closer to ECL4 than the COOH-terminal end in both inward- and outward-facing conformations (Fig. 4D) (19, 23, 44).
Fig. 4.

Neither 1 mM DTT nor 20 μM cadmium significantly affected the function of other extracellular loop 1/4 (ECL1/ECL4) cysteine mutants. A: trace showing that neither 1 mM DTT nor 20 μM cadmium significantly affected E115C/K892C-CFTR. B: inhibition of current by cadmium at Vm = −60 mV after 60 s of treatment. *P < 0.05. C: potentiation of current at Vm = −60 mV by 1 mM DTT after 3 min of treatment. D: snapshot of a molecular dynamics simulation at 2.5 ns from our recent study (32), with D110, D112, E115, and K892 highlighted. Orientation of ECL1 is such that D112 and E115 are farther from K892 than is D110.
A Loss-of-Charge Mutation at D110 Affects CFTR Open Pore Stability, but Not Single-Channel Amplitude
To better understand the mechanisms of potentiation and inhibition of the DTT- and cadmium-sensitive CFTR variants, we aimed to record the behavior of single channels in the excised inside-out patch configuration. To characterize the effect of loss of the native aspartic acid at D110 on single-channel behavior, we first studied the CF-related mutant D110H-CFTR. Channels in excised patches were directly activated by bath application of 1 mM ATP and 127 U/ml PKA (without coexpression of β2-AR). D110H-CFTR opened to a full-conductance similar to that of WT-CFTR (Fig. 5A), a result consistent with that found by another group recording this mutant in lipid bilayers (17). D110H-CFTR displayed a significantly shorter mean burst duration than WT-CFTR (n = 5 and 8 patches, respectively); interestingly, the mean burst duration of this variant was very similar to that recently reported for D110E-CFTR (Fig. 5B) (8). These data, combined with our finding that D110C-CFTR macroscopic currents are unaffected by the positively charged cysteine-reactive MTS reagent [2-(trimethylammonium)ethyl]methanethiosulfonate (MTSET) (8), strongly suggest that D110 does not itself play a direct role in permeation through the CFTR channel. Since mutations at D110 significantly shortened the open burst duration, the residue is clearly involved in stabilizing the open CFTR pore; however, since a charge-sparing (D110E) and a charge-neutralizing (D110H) mutation appear to affect open burst stability similarly, there appears to be a specific role for the native aspartic acid that is not merely related to charge.
Fig. 5.

D110H-CFTR channels exhibited reduced mean burst durations but conductance levels similar to WT-CFTR. A: traces for WT-CFTR and D110H-CFTR activated by 1 mM ATP and 127 U/ml PKA. Recordings were made at Vm = −100 mV. c, Closed current level; o1, o2, and o3, open current levels for 1, 2, and 3 active channels in the patch. B: mean burst duration among D110 variants. *P < 0.05. Mean burst duration of charge-neutralized D110H-CFTR was similar to that of charge-sparing D110E-CFTR (reported in Ref. 8).
Potentiation of D110C-CFTR and D110C/K892C-CFTR by DTT and Inhibition by Cadmium Involve Modulation of Mean Burst Duration and Unitary Conductance of the Channel
To lay further groundwork for the investigation of the dramatic effects of DTT and cadmium on D110C/K892C-CFTR, we next investigated the mechanism underlying the effects of these chemicals on the single mutant D110C-CFTR via excised patch recordings with single-channel resolution. Once again, channels in excised patches were directly activated by bath application of 1 mM ATP and 127 U/ml PKA (without coexpression of β2-AR). Naïve D110C-CFTR channels demonstrated a reduced mean burst duration (154.2 ± 6.1 ms, n = 5 patches; Fig. 6, A and B) compared with WT-CFTR (P < 0.05) and a prevalent subconductance of ∼80% of full conductance alongside full-conductance openings in the same patches (Fig. 6A, black and gray arrowheads). When D110C-CFTR channels were recorded in the presence of 1 mM DTT in the pipette, we observed an increase in overall mean open burst duration (369.5 ± 16.8 ms, n = 4 patches; Fig. 6B). In addition, the prevalence of the full-conductance openings was increased by DTT, as indicated by a comparison of the all-points amplitude histograms. Since long, full-conductance openings are favored in the presence of DTT, we suggest that this single-channel behavior is indicative of D110C-CFTR channels in the reduced chemical state. We also investigated the mechanism underlying the reversible cadmium-mediated inhibition of D110C-CFTR via multichannel patch recording. Oocytes were first incubated in 1 mM DTT during shrinking in hypertonic stripping solution with periodic agitation (>10 min) and then briefly washed in intracellular solution and recorded in the presence of 20 μM cadmium in the patch pipette. As evidenced in the channel records (Fig. 6A, bottom traces) and amplitude histogram, cadmium apparently induced a range of subconductance openings in D110C-CFTR channels. Additionally, in the presence of cadmium, D110C-CFTR channels displayed a reduced mean burst duration compared with that in the presence of DTT (134.5 ± 5.8 ms, n = 5 patches; Fig. 6B). In general terms, the effects of the inhibitory cadmium on naïve D110C-CFTR channels appear to be opposite of the effects of DTT, which lengthened the burst duration and increased the prevalence of full-conductance openings.
Previously, we reported that when D110C/K892C-CFTR channels were exposed to DTT backfilled in the pipette of a multichannel patch, the number of channel openings in the presence of ATP and PKA was significantly increased upon DTT diffusing to the pipette tip and reaching the channels (8). However, the experimental design was not conducive to thorough analysis of the mechanisms controlling single-channel behavior in both conditions. In the present study, in whole oocytes, DTT treatment of D110C/K892C-CFTR channels resulted in a large potentiation in macroscopic conductance (Fig. 1). When we recorded single D110C/K892C-CFTR channels directly activated by bath application of 1 mM ATP and 127 U/ml PKA (again without coexpression of β2-AR) in the absence of DTT, the mean burst duration of untreated channels (155 ± 7.6 ms, n = 4 patches; Fig. 7, A and B) was significantly less than that of WT-CFTR (P < 0.05). The conductance of the naïve channel varied significantly: full-conductance openings, openings of the size of the dominant subconductance from D110C-CFTR channels, and subconductances of lower amplitudes (Fig. 7A, top trace). D110C/K892C-CFTR channels were also recorded with solution wherein 1 mM DTT was present throughout the pipette. These channels displayed an increased mean burst duration (440 ± 44.1 ms, n = 4 patches) compared with naïve channels and typically opened to a full conductance, as seen in the representative record (Fig. 7A, middle trace) and corresponding amplitude histogram. Not surprisingly, these data show that the behavior of DTT-treated D110C/K892C-CFTR channels was very similar to that of DTT-treated D110C-CFTR channels: both D110C/K892C-CFTR and D110C-CFTR channels opened to a reasonably stable, full-conductance state in the presence of DTT, when their cysteine residues were likely reduced. When, after 1 mM DTT exposure, D110C/K892C-CFTR channels were recorded with 20 nM cadmium in the pipette, which selectively inhibits this double mutant (Fig. 3), the channels demonstrated a decreased burst duration (122.2 ± 7.0 ms, n = 4 patches) compared with DTT-treated channels and exhibited frequent subconductance openings of variable levels (Fig. 7A, bottom traces). The burst duration results strongly suggest that both DTT and cadmium modulate the pore-gating kinetics of D110C/K892C-CFTR.
Naïve D110C/K892C-CFTR Channels Display a Reduced Sensitivity to IBMX Stimulation That Is Countered by Breakage of the Disulfide Bond
To better understand the mechanism of potentiation of D110C/K892C-CFTR by DTT, we performed TEVC experiments, wherein we activated channels with increasing concentrations of the phosphodiesterase inhibitor IBMX in the continuing presence of 10 μM forskolin (an adenylate cyclase activator), as done in several previous studies to elicit graded activation of CFTR channels expressed in oocytes (10, 39, 45, 46). Interestingly, whereas WT-CFTR was nearly maximally activated by 10 μM forskolin and 0.5 mM IBMX [comparable to previous studies (10)], naïve D110C/K892C-CFTR channels were potentiated linearly as IBMX concentration was increased to the highest concentration tested (2 mM; Fig. 8, A and B); this lack of saturation suggests a lower overall sensitivity of this channel population to the activation stimulus. D110C/K892C-CFTR channels activated by 10 μM forskolin and 2 mM IBMX were highly potentiated by 1 mM DTT, with a degree and lack of plateau after 5 min of exposure similar to activation of these channels via the β2AR in whole oocytes (10.3 ± 2.5 and 13.0 ± 3.2 fold, respectively), suggesting that at least the majority of the channel population remained in the disulfided form prior to addition of DTT. This result led us to ask whether the sensitivity of this mutant to IBMX would be modulated by DTT. We treated D110C/K892C-CFTR cells with 1 mM DTT for 10 min and then immediately activated the cells with 10 μM forskolin and IBMX, starting with 0.1 mM IBMX to minimize the amount of time between DTT reduction of the disulfide bond and channel activation. DTT-treated channels showed an increased IBMX sensitivity, as evidenced by less potentiation of macroscopic conductance with increasing concentrations of IBMX (Fig. 8, C and D). A straightforward interpretation of this somewhat normalized IBMX sensitivity relative to WT-CFTR is that the disulfided channels were somehow rendered less sensitive to a given level of phosphorylation stimulus and that breakage of the disulfide bond restored sensitivity. Additionally, the apparent change in IBMX sensitivity of D110C/K892C-CFTR upon DTT treatment is further evidence (along with the changes in single-channel behavior of this mutant) that disulfided D110C/K892C-CFTR channels are able to open (however infrequently), because if all the current we measured in naïve cells across the IBMX concentrations arose from a fraction of nondisulfided channels in the population, we would not expect their sensitivity to have been altered by DTT.
Fig. 8.

An intact inhibitory disulfide bond between ECL1 and ECL4 reduces sensitivity of D110C/K892C-CFTR to phosphorylation stimulus. CFTR currents in whole oocytes were activated by 10 μM forskolin (FSK) and increasing concentrations of IBMX, and 1 mM DTT and 20 μM Inh172 (the CFTR-specific inhibitor) were added at the indicated times. Conductance is calculated as described in Fig. 1 and 3 legends. A: conductance from naïve D110C/K892C-CFTR channels potentiated by increasing concentrations of IBMX and further potentiated by subsequent application of 1 mM DTT. B: conductance from WT-CFTR channels showing saturation at >0.5 mM IBMX and lack of potentiation at 1 mM DTT. C: conductance from DTT-treated D110C/K892C-CFTR channels with increasing concentrations of IBMX. D: potentiation of each variant normalized to 0.1 mM IBMX + 10 μM forskolin (n ≥ 3 for each variant). *P < 0.05, naïve D110C/K892C-CFTR vs. DTT-treated D110C/K892C-CFTR and WT-CFTR. #P < 0.05, naïve D110C/K892C-CFTR vs. WT-CFTR, and P = 0.087, naïve D110C/K892C-CFTR vs. DTT-treated D110C/K892-CFTR.
DISCUSSION
Several mutations in the extracellular loops of CFTR cause manifestations of CF disease. Our group and others have demonstrated that these mutations impair open pore stability (8, 17). In this study we found that when D110 in ECL1 and K892 in ECL4 were mutated to cysteine residues, the resulting CFTR variant was highly potentiated by the reducing agent DTT, presumably through breakage of a spontaneous disulfide bond that strongly stabilized a closed state of CFTR. D110C/K892C-CFTR rapidly coordinated low concentrations of the soft metal cadmium, inhibiting a large fraction of the potentiation elicited by DTT, suggesting that C110 and C892 frequently come in reasonably close proximity in actively gating channels. Neither effect could be replicated with double mutants of C892 wherein the ECL1 cysteine residue was engineered in a position COOH-terminal to D110, providing evidence that the orientation of ECL1 in CFTR is similar to that of related ABC exporters for which crystal structures have been solved (19, 23, 44). Single-channel recordings revealed that these chemical manipulations predominantly affected channel gating (open burst durations) and also had variable effects on channel subconductance behavior. Finally, disulfided D110C/K892C-CFTR channels displayed a reduced sensitivity to activation by IBMX, which was partially normalized by breakage of the disulfide bond with DTT. We will discuss these results in the context of a model of CFTR wherein channel activation and pore gating are affected by positioning of functional elements including and/or structurally influenced by this region of ECL1.
Macroscopic and Single-Channel Measures of Effects of Chemical Manipulation of D110C/K892C-CFTR Suggest an Effect of DTT on Opening Rate of the Mutant
Because most of our single-channel recordings of substantial length featured at least two channels in the patch and because these channels had brief burst durations, it was not possible to calculate reliable open channel probability values for the variants in each condition, which would allow for the comparison of steady-state opening rates. It is nonetheless possible to determine whether changes in the observed open burst behavior are sufficient to explain an observed change in macroscopic conductance without necessitating a change in channel opening rate. Specifically, the >13-fold increase of macroscopic D110C/K892C-CFTR conductance in the presence of DTT (Figs. 1 and 8) cannot be accounted for by the changes in the open channel behavior between naïve and DTT-treated channels (an ∼3-fold increase in open burst duration and a 25% increase in single-channel conductance; Fig. 7), especially when one considers that the effects of DTT on macroscopic currents were limited to that elicited by 5 min of exposure (Fig. 1). In our previous report we found that when D110C/K892C-CFTR channels were exposed to DTT backfilled in the pipette of a multichannel patch, the number of channel openings in the presence of ATP and PKA was significantly increased upon DTT reaching the patch (8). In combination, these results lead to an overall interpretation that when C110 is engaged in a disulfide bond with C892, a closed state of the channel is highly stabilized.
An Intact Disulfide Bond Linking ECL1 and ECL4 Alters Sensitivity of D110C/K892C-CFTR to Phosphorylation Stimulus
In this study we observed an apparent reduction in the sensitivity of naïve D110C/K892C-CFTR channels to IBMX relative to WT-CFTR. Interestingly, after DTT treatment, the channels were rendered apparently more sensitive to IBMX (Fig. 8C). IBMX is thought to elicit CFTR current by inhibiting phosphodiesterases and, therefore, by raising levels of cAMP in cells (10), although it also may inhibit the phosphatases that dephosphorylate CFTR (4); the effect of both of these functions of IBMX is to increase phosphorylation of CFTR. It is noteworthy that IBMX has been demonstrated to block and, possibly, directly potentiate CFTR, although neither effect alone (nor certainly the net effect of both) on macroscopic currents in phosphorylated channels is significant at ≤1 mM (36, 45), where, in this study, differences were observed between variants (Fig. 8). The difference in IBMX sensitivity, therefore, is most likely due either to a difference in propensity of the WT-CFTR, disulfided D110C/K892C-CFTR, and DTT-treated D110C/K892C-CFTR conformations to be phosphorylated or to differences in the activity of the mutants at a given level of phosphorylation. The latter interpretation would imply that disulfide-mediated restriction of ECL1 and ECL4 presents an energy barrier to opening that affects the allosteric modulation of gating that arises from phosphorylation of the R domain, as proposed in several reports by Wang et al. (41, 43) demonstrating the effect of mutations and drugs on this aspect of channel function.
Structural Significance of the Position of ECL1 With Respect to Pore Gating
Although this work has described in experimental detail the effects of chemical perturbations at position 110, from a structural standpoint, it is initially puzzling why modification of an extracellular loop, at a position not intrinsically important for permeation, is capable of inducing such significant effects on channel function and, particularly, pore gating. A clue may be found in the recent characterization of TM1 residues immediately cytoplasmic to D110. Specifically, Gao and co-workers (16) found that the conductance of A107C-CFTR was sensitive to pH, a trait normally characteristic of cysteine substitutions of residues intimately involved in chloride permeation through CFTR, such as T338C (26). If the pH sensitivity of A107C-CFTR is similarly indicative of an important role of A107 in the permeation pathway for the pore of CFTR, then a perturbation of the position of this part of TM1, perhaps as modulated by the position of the adjacent ECL1, may be expected to significantly affect the state of the pore. Therefore, it is certainly reasonable that introduction of a strong interaction between nearby C110 and another amino acid, particularly C892 across the pore, would perturb the pore in such a way as to disrupt chloride flux (Fig. 9). Furthermore, previously, we found that a spontaneous disulfide bond between R104C and E116C resulted in R104C/E116C-CFTR channels that were “locked open” until the disulfide bond was broken with DTT (8); these residues are located in TM1 and ECL1, respectively. The antagonistic effects of a disulfide bond or cadmium linkage between C110 and C892 (Fig. 9, red circle) stabilizing a closed state of CFTR vs. a disulfide bond between C104 and C116 (Fig. 9, green circle) stabilizing an open state of CFTR support a pore-gating model wherein the position of the ECLs and the extracellular end of TM1 serve as a critical determinant of permeation through the CFTR pore.
Fig. 9.
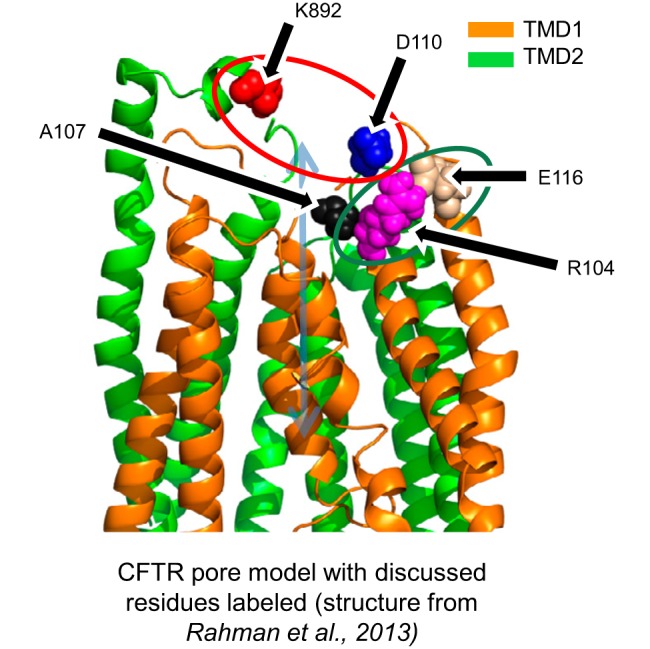
Structural interpretation of cross-linking results toward a model of CFTR gating. Structural model is built from coordinates of a 2.5-ns snapshot of a molecular dynamics simulation of CFTR previously published by our group (32). This study suggests that close proximity between cysteine-substituted residues at positions 892 and 110 (red circle) stabilizes a closed state of the pore, while we previously demonstrated that close proximity between cysteine residues at positions 104 and 116 (green circle) stabilizes an open state of the pore. Both linkages modify pore-gating behavior by impacting the position of the extracellular end of the first transmembrane helix of CFTR, possibly by altering the position of A107 (black), an amino acid predicted to lie in the permeation pathway for chloride ions.
In general, the single mutant D110C-CFTR results (mild potentiation by DTT and reversible inhibition by cadmium) also independently support the importance of the position of this region of ECL1 on CFTR pore gating and architecture, since both treatments affected the mean open burst duration and incidence of subconductances in this variant (Fig. 6). The potentiation of D110C-CFTR by DTT (Fig. 1) is not likely due to an interaction between C110 and an endogenous cysteine residue, as experimentally supported alignments fail to locate any endogenous cysteine residues near the extracellular side of the CFTR channel (1, 32). However, as we were unable to conclusively determine [among many chemical possibilities (11, 22, 25, 48)] the physical nature of the modulation of this mutant by DTT, nor in exploratory experiments were we able to identify any endogenous amino acids participating in these effects (data not shown), we limit the structural interpretation of the DTT and cadmium effects on D110C-CFTR to the suggestion that induction of an interaction between C110 and some other part of the protein destabilizes the channel open state and distorts the pore.
An Aspartic Acid at Position 110 Is Specifically Required for WT-Level Open Burst Stability in CFTR
Mutations at D110 cause CF and similarly decrease CFTR function in epithelial cells whether mutated to histidine or glutamic acid (40). Previously, we showed that charge-reversed D110R-CFTR channels display a profound pore stability defect and that D110E-CFTR channels, by comparison, opened somewhat more stably and to a WT-level conductance (8). In the present study we were able to expand on these data by recording single D110C-CFTR and D110H-CFTR channels. Interestingly, we found that D110H-CFTR and DTT-treated D110C-CFTR channels opened to a WT-like conductance with an open burst duration comparable to D110E-CFTR, despite loss of the native negative charge. In addition, Van Goor and colleagues (40) recently demonstrated that D110H-CFTR and D110E-CFTR are modulated by the US Food and Drug Administration-approved CFTR potentiator ivacaftor (VX-770, Kalydeco) with nearly identical potency and efficacy. From these data, we infer that the pore stability defect of at least the disease-related mutants at D110 may not be specifically related to charge but, instead, may require the specific orientation of charge as presented by the aspartic acid at position 110.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants R01-DK-056481 and R01-DK-075016, a grant from the Marcus Foundation (to N. A. McCarty), and National Institute of General Medical Sciences Grant T32 GM-008602-18 (to D. T. Infield).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.T.I. and N.A.M. developed the concept and designed the research; D.T.I., G.C., and C.K. performed the experiments; D.T.I. analyzed the data; D.T.I. and N.A.M. interpreted the results of the experiments; D.T.I. prepared the figures; D.T.I. drafted the manuscript; D.T.I., G.C., C.K., and N.A.M. edited and revised the manuscript; D.T.I., G.C., C.K., and N.A.M. approved the final version of the manuscript.
ACKNOWLEDGMENTS
The authors acknowledge Brandon Stauffer, Andrew Jenkins, Criss Hartzell, and Steve Traynelis (Emory University) for insightful discussion.
REFERENCES
- 1.Alexander C, Ivetac A, Liu X, Norimatsu Y, Serrano JR, Landstrom A, Sansom M, Dawson DC. Cystic fibrosis transmembrane conductance regulator: using differential reactivity toward channel-permeant and channel-impermeant thiol-reactive probes to test a molecular model for the pore. Biochemistry 48: 10078–10088, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bai Y, Li M, Hwang TC. Dual roles of the sixth transmembrane segment of the CFTR chloride channel in gating and permeation. J Gen Physiol 136: 293–309, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beck EJ, Yang Y, Yaemsiri S, Raghuram V. Conformational changes in a pore-lining helix coupled to cystic fibrosis transmembrane conductance regulator channel gating. J Biol Chem 283: 4957–4966, 2008. [DOI] [PubMed] [Google Scholar]
- 4.Becq F, Fanjul M, Merten M, Figarella C, Hollande E, Gola M. Possible regulation of CFTR-chloride channels by membrane-bound phosphatases in pancreatic duct cells. FEBS Lett 327: 337–342, 1993. [DOI] [PubMed] [Google Scholar]
- 5.Billet A, Mornon JP, Jollivet M, Lehn P, Callebaut I, Becq F. CFTR: effect of ICL2 and ICL4 amino acids in close spatial proximity on the current properties of the channel. J Cyst Fibros 12: 737–745, 2013. [DOI] [PubMed] [Google Scholar]
- 6.Bossard F, Silantieff E, Lavazais-Blancou E, Robay A, Sagan C, Rozec B, Gauthier C. β1-, β2-, and β3-adrenoceptors and Na+/H+ exchanger regulatory factor 1 expression in human bronchi and their modifications in cystic fibrosis. Am J Respir Cell Mol Biol 44: 91–98, 2011. [DOI] [PubMed] [Google Scholar]
- 7.Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O'Riordan CR, Smith AE. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63: 827–834, 1990. [DOI] [PubMed] [Google Scholar]
- 8.Cui G, Rahman KS, Infield DT, Kuang C, Prince CZ, McCarty NA. Three charged amino acids in extracellular loop 1 are involved in maintaining the outer pore architecture of CFTR. J Gen Physiol 144: 159–179, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cui G, Song B, Turki HW, McCarty NA. Differential contribution of TM6 and TM12 to the pore of CFTR identified by three sulfonylurea-based blockers. Pflügers Arch 463: 405–418, 2012. [DOI] [PubMed] [Google Scholar]
- 10.Drumm ML, Wilkinson DJ, Smit LS, Worrell RT, Strong TV, Frizzell RA, Dawson DC, Collins FS. Chloride conductance expressed by ΔF508 and other mutant CFTRs in Xenopus oocytes. Science 254: 1797–1799, 1991. [DOI] [PubMed] [Google Scholar]
- 11.Eiamphungporn W, Soonsanga S, Lee JW, Helmann JD. Oxidation of a single active site suffices for the functional inactivation of the dimeric Bacillus subtilis OhrR repressor in vitro. Nucleic Acids Res 37: 1174–1181, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El Hiani Y, Linsdell P. Metal bridges illuminate transmembrane domain movements during gating of the cystic fibrosis transmembrane conductance regulator chloride channel. J Biol Chem 289: 28149–28159, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuller MD, Thompson CH, Zhang ZR, Freeman CS, Schay E, Szakacs G, Bakos E, Sarkadi B, McMaster D, French RJ, Pohl J, Kubanek J, McCarty NA. State-dependent inhibition of cystic fibrosis transmembrane conductance regulator chloride channels by a novel peptide toxin. J Biol Chem 282: 37545–37555, 2007. [DOI] [PubMed] [Google Scholar]
- 14.Fuller MD, Zhang ZR, Cui G, McCarty NA. The block of CFTR by scorpion venom is state-dependent. Biophys J 89: 3960–3975, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gadsby DC, Vergani P, Csanady L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature 440: 477–483, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao X, Bai Y, Hwang TC. Cysteine scanning of CFTR's first transmembrane segment reveals its plausible roles in gating and permeation. Biophys J 104: 786–797, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hammerle MM, Aleksandrov AA, Riordan JR. Disease-associated mutations in the extracytoplasmic loops of cystic fibrosis transmembrane conductance regulator do not impede biosynthetic processing but impair chloride channel stability. J Biol Chem 276: 14848–14854, 2001. [DOI] [PubMed] [Google Scholar]
- 18.He L, Aleksandrov AA, Serohijos AW, Hegedus T, Aleksandrov LA, Cui L, Dokholyan NV, Riordan JR. Multiple membrane-cytoplasmic domain contacts in the cystic fibrosis transmembrane conductance regulator (CFTR) mediate regulation of channel gating. J Biol Chem 283: 26383–26390, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hohl M, Briand C, Grutter MG, Seeger MA. Crystal structure of a heterodimeric ABC transporter in its inward-facing conformation. Nat Struct Mol Biol 19: 395–402, 2012. [DOI] [PubMed] [Google Scholar]
- 20.Hunt JF, Wang C, Ford RC. Cystic fibrosis transmembrane conductance regulator (ABCC7) structure. Cold Spring Harb Perspect Med 3: a009514, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hwang TC, Kirk KL. The CFTR ion channel: gating, regulation, and anion permeation. Cold Spring Harb Perspect Med 3: a009498, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JW, Soonsanga S, Helmann JD. A complex thiolate switch regulates the Bacillus subtilis organic peroxide sensor OhrR. Proc Natl Acad Sci USA 104: 8743–8748, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J, Jaimes KF, Aller SG. Refined structures of mouse P-glycoprotein. Protein Sci 23: 34–46, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Linsdell P. Metal bridges to probe membrane ion channel structure and function. Biomol Concepts 6: 191–203, 2015. [DOI] [PubMed] [Google Scholar]
- 25.Liu X, Alexander C, Serrano J, Borg E, Dawson DC. Variable reactivity of an engineered cysteine at position 338 in cystic fibrosis transmembrane conductance regulator reflects different chemical states of the thiol. J Biol Chem 281: 8275–8285, 2006. [DOI] [PubMed] [Google Scholar]
- 26.Liu X, Zhang ZR, Fuller MD, Billingsley J, McCarty NA, Dawson DC. CFTR: a cysteine at position 338 in TM6 senses a positive electrostatic potential in the pore. Biophys J 87: 3826–3841, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCarty NA, McDonough S, Cohen BN, Riordan JR, Davidson N, Lester HA. Voltage-dependent block of the cystic fibrosis transmembrane conductance regulator Cl− channel by two closely related arylaminobenzoates. J Gen Physiol 102: 1–23, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mense M, Vergani P, White DM, Altberg G, Nairn AC, Gadsby DC. In vivo phosphorylation of CFTR promotes formation of a nucleotide-binding domain heterodimer. EMBO J 25: 4728–4739, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Norimatsu Y, Ivetac A, Alexander C, O'Donnell N, Frye L, Sansom MS, Dawson DC. Locating a plausible binding site for an open-channel blocker, GlyH-101, in the pore of the cystic fibrosis transmembrane conductance regulator. Mol Pharmacol 82: 1042–1055, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Price MP, Ishihara H, Sheppard DN, Welsh MJ. Function of Xenopus cystic fibrosis transmembrane conductance regulator (CFTR) Cl channels and use of human-Xenopus chimeras to investigate the pore properties of CFTR. J Biol Chem 271: 25184–25191, 1996. [DOI] [PubMed] [Google Scholar]
- 31.Quinton P, Molyneux L, Ip W, Dupuis A, Avolio J, Tullis E, Conrad D, Shamsuddin AK, Durie P, Gonska T. β-Adrenergic sweat secretion as a diagnostic test for cystic fibrosis. Am J Respir Crit Care Med 186: 732–739, 2012. [DOI] [PubMed] [Google Scholar]
- 32.Rahman KS, Cui G, Harvey SC, McCarty NA. Modeling the conformational changes underlying channel opening in CFTR. PLoS One 8: e74574, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, et al. . Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245: 1066–1073, 1989. [DOI] [PubMed] [Google Scholar]
- 34.Rowe SM, Clancy JP, Wilschanski M. Nasal potential difference measurements to assess CFTR ion channel activity. Methods Mol Biol 741: 69–86, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med 352: 1992–2001, 2005. [DOI] [PubMed] [Google Scholar]
- 36.Schultz BD, Frizzell RA, Bridges RJ. Rescue of dysfunctional ΔF508-CFTR chloride channel activity by IBMX. J Membr Biol 170: 51–66, 1999. [DOI] [PubMed] [Google Scholar]
- 37.Sebastian A, Rishishwar L, Wang J, Bernard KF, Conley AB, McCarty NA, Jordan IK. Origin and evolution of the cystic fibrosis transmembrane regulator protein R domain. Gene 523: 137–146, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sheppard DN, Rich DP, Ostedgaard LS, Gregory RJ, Smith AE, Welsh MJ. Mutations in CFTR associated with mild-disease-form Cl− channels with altered pore properties. Nature 362: 160–164, 1993. [DOI] [PubMed] [Google Scholar]
- 39.Smit LS, Wilkinson DJ, Mansoura MK, Collins FS, Dawson DC. Functional roles of the nucleotide-binding folds in the activation of the cystic fibrosis transmembrane conductance regulator. Proc Natl Acad Sci USA 90: 9963–9967, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Goor F, Yu H, Burton B, Hoffman BJ. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J Cyst Fibros 13: 29–36, 2014. [DOI] [PubMed] [Google Scholar]
- 41.Wang W, Li G, Clancy JP, Kirk KL. Activating cystic fibrosis transmembrane conductance regulator channels with pore blocker analogs. J Biol Chem 280: 23622–23630, 2005. [DOI] [PubMed] [Google Scholar]
- 42.Wang W, Linsdell P. Relative movements of transmembrane regions at the outer mouth of the cystic fibrosis transmembrane conductance regulator channel pore during channel gating. J Biol Chem 287: 32136–32146, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang W, Roessler BC, Kirk KL. An electrostatic interaction at the tetrahelix bundle promotes phosphorylation-dependent cystic fibrosis transmembrane conductance regulator (CFTR) channel opening. J Biol Chem 289: 30364–30378, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ward A, Reyes CL, Yu J, Roth CB, Chang G. Flexibility in the ABC transporter MsbA: alternating access with a twist. Proc Natl Acad Sci USA 104: 19005–19010, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilkinson DJ, Mansoura MK, Watson PY, Smit LS, Collins FS, Dawson DC. CFTR: the nucleotide binding folds regulate the accessibility and stability of the activated state. J Gen Physiol 107: 103–119, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilkinson DJ, Strong TV, Mansoura MK, Wood DL, Smith SS, Collins FS, Dawson DC. CFTR activation: additive effects of stimulatory and inhibitory phosphorylation sites in the R domain. Am J Physiol Lung Cell Mol Physiol 273: L127–L133, 1997. [DOI] [PubMed] [Google Scholar]
- 47.Zhang ZR, Song B, McCarty NA. State-dependent chemical reactivity of an engineered cysteine reveals conformational changes in the outer vestibule of the cystic fibrosis transmembrane conductance regulator. J Biol Chem 280: 41997–42003, 2005. [DOI] [PubMed] [Google Scholar]
- 48.Zhou P, Tian F, Lv F, Shang Z. Geometric characteristics of hydrogen bonds involving sulfur atoms in proteins. Proteins 76: 151–163, 2009. [DOI] [PubMed] [Google Scholar]