

Abstract
Ess1 is a peptidyl-prolyl cis/trans isomerase (PPIase) that binds to the carboxy-terminal domain (CTD) of RNA polymerase II. Ess1 is thought to function by inducing conformational changes in the CTD that control the assembly of cofactor complexes on the transcription unit. Ess1 (also called Pin1) is highly conserved throughout the eukaryotic kingdom and is required for growth in some species, including the human fungal pathogen Candida albicans. Here we report the crystal structure of the C. albicans Ess1 protein, determined at 1.6 Å resolution. The structure reveals two domains, the WW and the isomerase domain, that have conformations essentially identical to those of human Pin1. However, the linker region that joins the two domains is quite different. In human Pin1, this linker is short and flexible, and part of it is unstructured. In contrast, the fungal Ess1 linker is highly ordered and contains a long α-helix. This structure results in a rigid juxtaposition of the WW and isomerase domains, in an orientation that is distinct from that observed in Pin1, and that eliminates a hydrophobic pocket between the domains that was implicated as the main substrate recognition site. These differences suggest distinct modes of interaction with long substrate molecules, such as the CTD of RNA polymerase II. We also show that C. albicans ess1− mutants are attenuated for in vivo survival in mice. Together, these results suggest that CaEss1 might constitute a useful antifungal drug target, and that structural differences between the fungal and human enzymes could be exploited for drug design.
Candida albicans is an opportunistic fungal pathogen that causes life-threatening infections in immunocompromised individuals (1, 2). Candidiasis is a common hospital-acquired infection, and causes signficant mortality in patients who undergo organ transplantation or cancer chemotherapy, or are HIV-infected (3). C. albicans is polymorphic, in that cells can exist in a yeast form, or they may undergo filamentation to pseudohyphal and hyphal forms (4). This process, known as morphogenetic switching, is often associated with virulence and tissue invasiveness (5, 6) and is largely under transcriptional control (7, 8).
Therapeutic drugs for C. albicans infections typically target components of the cell membrane or cell wall biosynthetic machinery that are unique to fungi. However, many of these agents are fungistatic, inhibiting cell growth but not killing cells. Resistance to antifungal drugs is a clinical problem, and since many drugs are chemically similar, cells that show resistance to one are often resistant to others (9). Combination therapies offer a promising alternative, if they affect unrelated cellular targets (10, 11).
Peptidyl-prolyl cis/trans isomerases (PPIases)1 are one group of enzymes under study as alternative targets (12). PPIases catalyze rotation of the peptide bond preceding proline (13). The drugs cyclosporin A and FK506 target the PPIases cyclophilin A and FKBP, respectively, and have potent antifungal activity (14). One complication, however, is that these compounds, and another, rapamycin (which inhibits FKBPs), are also immunosuppressive (15). A likely strategy would be to use these drugs in combination therapies at low doses, sufficient for antifungal activity, but not for immune suppression (12), or to use non-immunosuppressive analogues (16). An alternative is to target Ess1, a member of a distinct class of PPIases called parvulins (17) that do not bind cyclosporin A, FK506, or rapamycin, and that have not been linked to immune suppression.
Ess1 was originally identified in Saccharomyces cerevisiae, where it was shown to be essential for cell growth (18), and later demonstrated to be a PPIase (19, 20). In S. cerevisiae, Ess1 (ScEss1) binds the carboxy-terminal domain (CTD) of the large subunit of RNA polymerase II (21, 22) and controls multiple stages of transcription, including initiation and elongation (23, 24). Ess1 orthologs have been identified in nearly all eukaryotic organisms that have been examined, including flies, humans, and other pathogenic fungi (25–29). In C. albicans, Ess1 is essential for growth, and deletion mutants arrest mitosis, as they do in S. cerevisiae (27). C. albicans mutants in which the dosage or activity of Ess1 has been reduced grow normally in culture but show defects in morphogenetic switching, suggesting that, in mammalian hosts, they will be attenuated for virulence. In C. albicans, Ess1 is likely to be important for proper expression of virulence-associated genes. Ess1 has also been identified in Cryptococcus neoformans, another common fungal pathogen, where it was shown to be important for virulence in a mouse model system (28). These results indicate that Ess1 may serve as a useful, broad-spectrum antifungal drug target.
The mammalian homologue of Ess1, called Pin1 (25), has been implicated in a wide range of activities, including cell cycle control, signal transduction, and regulation of transcription factors (30–32). Misregulation of human Pin1 is associated with certain cancers and neurodegenerative diseases (33), although causation has been difficult to demonstrate (32). In mice, Pin1 is not essential, although cells isolated from Pin1-deficient mice are delayed in re-entering the cell cycle after G1 arrest (34). Mammalian Pin1 is also implicated in transcriptional control (35). The crystal structure of human Pin1 has been determined and reveals two largely independent structural domains, an N-terminal WW domain and a C-terminal catalytic domain that has PPIase activity (PPIase domain) (36). The WW domain is a proline-binding module found in a large number of signaling proteins (37). In human Pin1, it is joined to the PPIase domain by a flexible linker region (38, 39) that is approximately 17 residues in length. Both domains are capable of binding proline-containing peptides, with the preferred target sites being phospho-Ser-Pro or phospho-Thr-Pro (40). Binding affinity for substrate peptides is ∼10-fold higher for the WW domain than for the PPIase domain (41). Extensive mutagenesis of the human and S. cerevisiae enzymes has shown that both domains are critical for in vivo function (reviewed in ref 42).
In this study, we determined the crystal structure of the C. albicans Ess1 apoenzyme (CaEss1) and refined it to 1.6 Å resolution. This allowed us to compare the fungal and human enzymes, to provide insights into enzyme function, and to illuminate differences that could potentially be exploited for antifungal drug design. The C. albicans and human proteins are 43% identical at the amino acid level, and as expected, the WW and PPIase domains fold to form analogous structures. However, a striking difference is revealed in the position of the two domains relative to one another. This is largely due to the presence of a novel α-helix in the so-called linker region of the fungal Ess1 that is absent in human Pin1. This helix and the interaction with nearby residues likely constrain the WW domain against the PPIase catalytic domain, and eliminate the hydrophobic pocket formed in the interdomain space of the human enzyme. These differences suggest a different mechanism by which the two domains interact to present the substrate to the active site area and may lead to strategies for drug design.
Experimental Procedures
Expression of C. albicans Ess1
The primary sequence of CaESS1 has been described (27). The CaESS1 gene was amplified by PCR and inserted between the EcoRI and NdeI sites of pUC19, resulting in plasmid pCaESS1-C. An NdeI–BamHI fragment (∼800 bp) of pCaESS1-C was recloned into the same sites of pET28a for expression in Escherichia coli strain BL21(DE3) (Novagen). The His-tagged CaEss1 fusion protein (197 residues) was purified from cells grown at 22 °C using Ni–NTA (Qiagen) affinity chromatography, and was used for enzyme assays and crystallography. The plasmid for expression of CaEss1 in C. albicans is pCaESS1, and was constructed by insertion of a HindIII–BamHI fragment from pGD-CaESS1 (27) into the same sites of pABSK2 (a gift from H. Chibana and P. T. Magee, University of Minnesota, Minneapolis, MN).
Mouse Model of Infection
Six 4–6-week-old (15–20 g) female BALB/c mice (Griffin Laboratory, Wadsworth Center) were injected with 0.1 mL of each C. albicans strain at 107 cfu/mL. For the mutant strains, three mice were sacrificed by CO2 asphyxiation on day 1 and on day 5 postinjection. For wild-type C. albicans, mice were sacrificed after 1 or 2 days, at which time they were moribund. Kidney homogenates were diluted in 1 mL of saline, and the number of colony-forming units (cfu) was determined by plating of serial dilutions onto YEPD medium containing chloramphenicol (25 μg/mL) and gentamycin (40 μg/mL) and incubating for 5–7 days at 30 °C. Presented data are average values from two or more mice.
Prolyl Isomerase Assay
A modification of a standard chromogenic assay was used (43). Briefly, 0.05–50 nmol of enzyme in 500 μL of 10 mM Tris (pH 8.0) containing 0.2 mM substrate peptide (Suc-AEPF-p-nitroanilide; Bachem) was prewarmed at 20 °C; 500 μL of prewarmed 2 mg/mL chymotrypsin in 10 mM Tris (pH 8.0) was added and rapidly mixed. Reactions were monitored at 1.1 s intervals in a Beckman DU-800 spectrophotometer, under temperature control, until the absorbance at 390 nm reached a plateau. Data analysis and calculation of turnover number were performed as described previously (44).
Purification and Crystallization of CaEss1
Recombinant CaEss1 was overexpressed as described above. Cell pellets were resuspended in a solution containing 20 mM Tris-HCl (pH 8.0) and 0.5 M NaCl. After sonication, the supernatant was applied to a Ni2+–NTA affinity column for purification. The pooled CaEss1 fractions were incubated with thrombin and dialyzed overnight at 4 °C against a digestion buffer [50 mM Tris-HCl (pH 8.0), 0.5 M NaCl, 5 mM MgCl2, and 2.5 mM CaCl2] to remove the His tag. SDS–PAGE analysis was used to monitor the completion of thrombin digestion. The sample was applied to a 2 mL benzamidine Sepharose 6B column to remove the thrombin. The flow-through was collected and concentrated, after addition of 10 mM DTT, in an Amicon stirred cell unit (Millipore) and filtered to remove precipitates. Gel filtration chromatography on a Superdex-75 column (Amersham) was used to remove trace amounts of contaminants. The peak corresponding to the correct molecular mass (∼20 kDa) was recovered, exchanged into a buffer containing 20 mM HEPES (pH 7.5), 100 mM NaCl, and 10 mM DTT, and concentrated to approximately 30 mg/mL prior to crystallization.
Crystals of CaEss1 were obtained by hanging drop vapor diffusion methods at room temperature. Drops, composed of 2 μL of the protein solution and 2 μL of the precipitation buffer, were equilibrated against a reservoir containing 1 mL of the precipitation buffer, 20–30% PEG 8000, 50 mM KH2PO4, and 0.2 M ammonium acetate. Larger crystals were obtained by microseeding the drops 24 h after the initial setup. The crystals belong to space group P21 with the following cell dimensions: a = 29.8 Å, b = 59.5 Å, c = 44.8 Å, and β = 91.1°. There is one molecule in the asymmetric unit, with a solvent content of ∼38%.
Diffraction Data Collection, Structure Determination, and Refinement
Prior to data collection, all crystals were transferred to a cryosolvent consisting of the precipitation buffer complemented with 20% (v/v) glycerol. Crystals were flash-cooled in a nitrogen gas stream at 100 K. All X-ray diffraction data were collected using an in-house R-axis IV (Rigaku) detector and processed using HKL/SCALEPACK (45). Data collection statistics are summarized in Table 1.
Table 1. Data Collection, Phasing, and Refinement Statistics.
| derivatives | ||||
|---|---|---|---|---|
|
|
||||
| native | Hg | Pt | Au | |
| data collection | ||||
| resolution (Å) | 1.6 | 2.8 | 2.3 | 1.6 |
| no. of unique reflections | 20324 | 3536 | 6615 | 18892 |
| completeness (%) | 97.1 (87.7) | 92.9 (94.9) | 95.4 (91.2) | 90.9 (70.4) |
| Rsym (%) | 4.6 (20.1) | 6.9 (32.1) | 5.7 (48) | 8.9 (24.1) |
| I/σ(I) | 29 (2.9) | 12.7 (2.7) | 22.7 (2.0) | 22 (3.7) |
| MIRAS phasing | ||||
| resolution range (Å) | 20–1.6 | |||
| no. of sites | 4 | 4 | 5 | |
| Riso (%) | 24 | 35 | 35 | |
| Rano (%) | 8.5 | 7.0 | 5.6 | |
| figure of merit | 0.37 | |||
| refinement | ||||
| no. of reflections | 20204 | |||
| Rcryst | 0.200 | |||
| Rfree | 0.242 | |||
| rmsd from ideality | ||||
| bond lengths (Å) | 0.004 | |||
| bond angles (deg) | 1.3 | |||
| dihedrals (deg) | 22.9 | |||
| average B-factor (Å2) | ||||
| protein | 27.2 | |||
| water (369) | 38.2 | |||
Attempts to determine the crystal structure of CaEss1 by molecular replacement using the coordinates of human Pin1 (36; Protein Data Bank entry 1PIN) as the search model failed. Therefore, the structure was determined by multiple isomorphous replacement/anomalous scattering (MIRAS) methods. Three heavy atom derivatives were prepared by soaking the CaEss1 crystals overnight in the crystallization buffer supplemented with 10 mM heavy atom compounds, Hg(OAc)2, K2PtCl4, and KAu(CN)2.
MIRAS phasing, using SOLVE (46), clearly defined the positions of the heavy atoms. An interpretable electron density map could be generated after density modification using RESOLVE (47). Ten fragments of apolyalanine model containing ∼60% of the CaEss1 residues could be traced automatically. Refinement of this partial model generated an interpretable electron density map for the complete molecule. The model was completed by manual rebuilding using TURBO-FRODO (48). The structure refinement was completed using CNS (49). After the rigid-body refinement, iterative cycles of simulated annealing or minimization and temperature factor refinement were carried out, interspersed with model rebuilding into σA-weighted Fo – Fc and 2Fo – Fc electron density maps using O (50). The final refinement statistics are summarized in Table 1. All but one residue (Asn11) are within the most favored (90.8%) and additionally allowed (8.5%) regions of the Ramachandran plot, as defined in PROCHECK (51).
Results And Discussion
CaEss1 Has PPIase Activity
The C. albicans Ess1 protein has been shown to complement S. cerevisiae mutants that lack Ess1 (27). However, it had not been previously demonstrated that CaEss1 has enzymatic activity. We tested this using a standard in vitro prolyl isomerase assay with synthetic peptide substrates. In this assay, chymotrypsin cleaves the substrate after it is converted from the cis to the trans form by the isomerase. Cleavage liberates a nitroaniline chromophore that absorbs at 390 nm. The results showed that CaEss1 has activity comparable to, or slightly greater than, that of the wild-type S. cerevisiae enzyme (turnover rates are 19 and 17 s−1, respectively) (Figure 1). Thus, CaEss1 is a peptidyl-prolyl isomerase.
Figure 1.

PPIase activity of C. albicans Ess1 using a standard protease-coupled assay. The substrate (Suc-AEPF-pNA) was subjected to proteolysis by chymotrypsin, which releases the p-nitroaniline (pNA) from the peptide only when the E–P bond is in the trans conformation. The release of pNA was monitored during the assay at 20 °C by its absorbance at 390 nm, and the data were linearized as described in Experimental Procedures. Shown are representative graphs of experiments carried out in triplicate. The lines indicate the least-squares fit of the data; only every 15th data point is shown for clarity. The blank indicates no isomerase enzyme added. The results indicate that CaEss1 has activity comparable to that of the control S. cerevisiae enzyme (ScEss1).
CaEss1 Is Important for the Survival of C. albicans in a Mammalian Host
We tested two CaESS1 mutant strains, previously shown to be defective in morphogenetic switching (27), for their ability to survive and proliferate in a mouse model system. In this assay, a high dose of C. albicans (106 cfu) is injected into the lateral tail vein of BALB/c mice, whereupon they establish a persistent infection, usually killing the host within 2–3 days (52, 53). Mutants attenuated for virulence should live longer and possess lower organ loads of C. albicans cells (54). In our experiments, kidneys from infected animals were evaluated after 1–5 days for the presence of C. albicans cells.
Indeed, both strains, a heterozygous mutant (ess1Δ/ESS1) and a temperature-sensitive (ts) mutant (ess1H171R/ess1Δ), were attenuated for their ability to survive in vivo. Whereas mice injected with wild-type C. albicans (SC5314) were dead or moribund after 2 days, mice injected with the mutant strains were viable until at least day 5, at which time organ load in kidney homogenates was assessed (Figure 2). The results show that organ load is reduced more than 100-fold in mice infected with ess1 mutant strains after either 1 or 5 days of injection. Thus, CaESS1 appears to be important for survival and proliferation in a host organism. For the ts strain, part of this may be due to inherent slow growth (27). However, the heterozygous strain grows normally under laboratory conditions, suggesting that the defect is more likely related to its inability to undergo morphogenetic switching. As predicted, CaESS1 infection was restored in an ess1Δ deletion strain that carried CaESS1 on an episomal plasmid. However, this rescue was lost by day 5, perhaps due to plasmid instability in vivo. Acquisition of further evidence demonstrating that CaESS1 is required for virulence will require long-term survival studies, as well as the generation of stable reconstituted lines in which CaESS1 is chromosomally integrated at the homologous locus.
Figure 2.
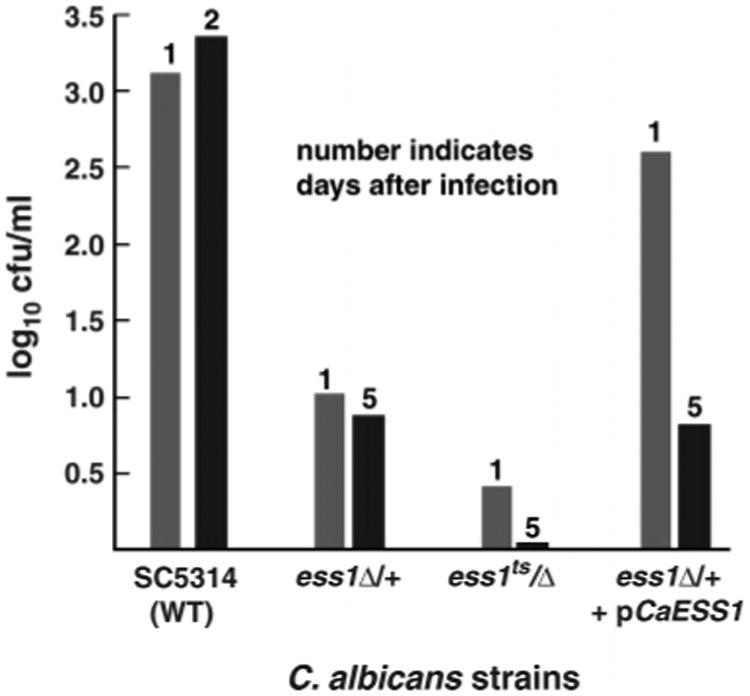
Organ load of C. albicans ess1 mutants in a mouse model of infection. The fungal burden in host animals was assessed by measuring the organ load in the kidney, which provides a sensitive and convenient method for monitoring the progression of systemic infections (54). Mice infected with different strains of C. albicans were sacrificed at the indicated number of days following initial infection (details in Experimental Procedures). The organ load of kidney homogenates is given in colony-forming units. The results show that ess1 mutants have reduced organ loads after infection for 1 day, and that mice infected with ess1 mutant strains were still viable after 5 days. The strains used were wild-type SC5314 (WT), a heterozygous mutant ess1Δ/+ (CaGD1), a ts mutant ess1H171R/Δ (CaGD3), and a “reconstituted strain”, ess1Δ/+, +pCaESS1 (CaGD2 + pABSK2-CaESS1), and have been described (27). As indicated in the figure, even a reduction of the ESS1 gene dosage by one-half (i.e., heterozygous mutant, ess1Δ/+) is sufficient to attenuate virulence nearly 100-fold in this assay.
Three-Dimensional Structure of CaEss1
To better understand the structure and function of the CaEss1 enzyme, the crystal structure was determined by MIRAS methods and refined to 1.6 Å resolution. Overall, the molecule has a flat, rectangular shape in which the three structural elements (WW domain, linker, and PPIase domain) are closely packed, with extensive interdomain contacts (Figure 3). Residues 7–37 form the WW domain, a small module consisting of a three-stranded antiparallel β-sheet, which has been shown to be important for the recognition of the phosphopeptide substrates (37, 41). The PPIase domain, residues 67–177, forms a globular domain with an α/β-fold that consists of a central four-stranded antiparallel β-sheet flanked by four α-helices. This domain harbors the active site; however, the mechanisms by which it participates in substrate recognition and by which it shares this responsibility with the WW domain remain to be established. The linker region of CaEss1, residues 38–66, is highly ordered, consisting of a type II β-turn (residues 38–41), an α-helix (residues 43–56), a short extended segment (residues 58–63), and a type I β-turn (residues 64–67). This linker interacts closely with both the WW and PPIase domains.
Figure 3.
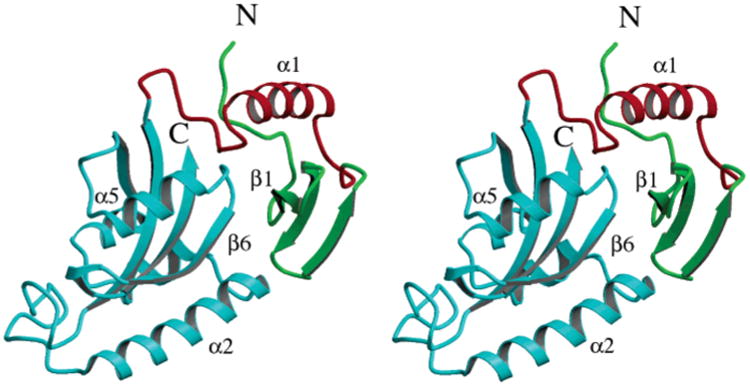
Three-dimensional structure of CaEss1. The three structural elements, the WW domain (green), the linker (red), and the PPIase domain (cyan), are tightly packed together to form a compact molecule.
Comparison with Human Pin1
The WW and PPIase domains of CaEss1 have conformations that are very similar to those of the corresponding domains of human Pin1 (Figure 4a,b) (36, 41), as had been anticipated on the basis of the high degree of sequence similarity between those segments of the proteins. However, there are major differences in the composition and conformation of the linker that joins the WW and PPIase domains, and in the relative positions of these two domains. These differences could have major implications for the mechanisms by which the two proteins interact with their substrates.
Figure 4.
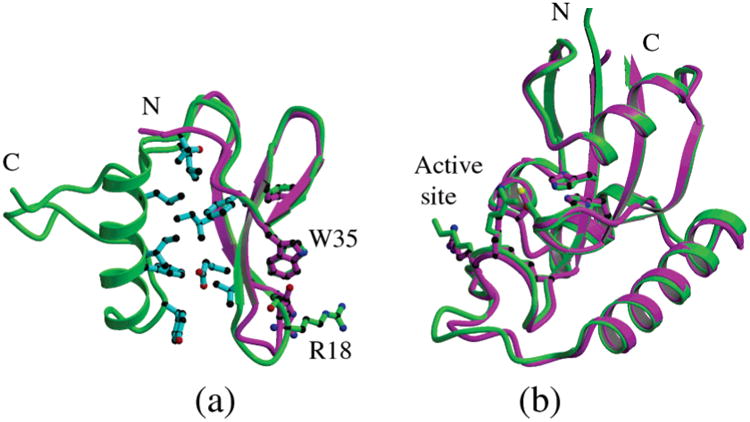
Domain structure comparison of CaEss1 with human Pin1. (a) Superposition of the WW domain of Pin1 (magenta) onto the WW domain and linker of CaEss1 (green) showing the overall similarities of the folds of the three-stranded β-sheet domains. The residues that form a hydrophobic core between the WW domain and the linker are shown in cyan. (b) Superposition of the PPIase domains of CaEss1 (green) and Pin1 (magenta).
Superposition of the WW domains of CaEss1 and Pin1 (Figure 4a) shows a highly similar conformation (rmsd of 1.25 Å for residues 8–38), with the only significant difference being a 2.0 Å shift in the position of the loop joining β-strands 1 and 2 (residues 18–21). The loop conformation seen in the CaEss1 structure appears to be more open than the one seen in the Pin1 structures (36, 41). No ligands interact with the WW domain in the CaEss1 structure, while the WW domain of Pin1 is in contact with ligands, a PEG molecule (36), or a phosphopeptide (41). Therefore, this closed conformation of the domain in the Pin1 structures may be related to ligand binding, especially since Arg18 (CaEss1 numbering) is involved in binding the phosphopeptide in the Pin1 structure. All conserved residues, and specifically residues that interact with the phosphopeptide in the Pin1 structure, have similar conformations, except for Arg18, which appears to be disordered in the CaEss1 structure. This is consistent with the idea that the flexibility of the loop containing Arg18 plays a role in substrate binding by WW domains.
The conformations of the PPIase domains of Pin1 and CaEss1 are nearly identical (Figure 4b) with an rmsd of 0.51 Å for residues 68–77 and 95–177 (CaEss1 numbering). Most importantly, all residues involved in the area of the active site are conserved and have very similar conformations. The only significant difference between the two PPIase domains is in the conformation of the loop between residues 83 and 92. This loop, which has a one-residue insertion in CaEss1 compared to Pin1, was termed the catalytic loop in the studies of Pin1, because it was observed in two different conformations in the two crystal structures of Pin1. In the complex of Pin1 with an Ala-Pro dipeptide (36), the loop had a “closed” conformation, in which it is in the proximity of residues near the active site of the PPIase domain. A sulfate ion, which is thought to substitute for a phosphate of the phosphopeptide substrate, is inserted between the loop and the active site residues. In contrast, in the structure of Pin1 complexed with a CTD phosphopeptide bound by the WW domain (41), the loop is in an open conformation, distant from the active site residues. This open conformation was attributed to the absence of ligands in the active site area. However, NMR studies of Pin1 (38, 39) have not revealed evidence of conformational flexibility in this loop, reducing the likelihood that the transition between the two conformations is significant for enzymatic activity. In the structure of CaEss1, the conformation of this loop resembles that of the closed conformation of Pin1, despite the fact that there are no ligands present in the active site. The latter fact itself is surprising since the protein was crystallized from a buffer containing 50 mM phosphate. Regardless, this result is consistent with the NMR studies for Pin1 and suggests that movement of the loop is not important for either substrate binding or catalytic activity.
Previous sequence and structural analyses of Ess1 proteins (27, 36) allowed us to delimit the extent of the CaEss1 WW and PPIase domains and an interdomain linker region that corresponds approximately to residues 38–66. This linker region, in contrast to that of Pin1, is highly structured. In fact, had it not been for extensive structural data showing that all other WW domains consist of only a three-stranded β-sheet (55, 56), it would have been more appropriate to consider the first 20 residues of the linker, including the α-helix, as an integral part of the WW domain. This is exemplified by the presence of a cluster of hydrophobic residues at the interface between the three strands of the WW domain and the helix of the linker (Figure 4a). This highly ordered structure of the linker is surprising, in view of the highly flexible nature of the linker in the Pin1 crystal structures. While the Pin1 linker is 11 residues shorter than the CaEss1 linker, electron density could not be observed for a six-residue segment of the Pin1 linker, indicative of static and/or dynamic disorder in the crystal. The residues that could be modeled adopt an extended conformation. Likewise, NMR studies of Pin1 have shown that the linker is not structured in solution (38, 39).
Domain Association of CaEss1 Contrasts with the Flexibility of Pin1
Comparisons of the crystal structures of CaEss1 and Pin1 show that the differences in the sizes and structures of the linker regions of the two proteins have a profound effect on the relative positions and mobilities of the WW and PPIase domains. In the crystal structures of Pin1, the WW domain contacts the PPIase domain through an interaction between the N-terminal end of strand β3 of the WW domain with helix R4 and strand β6 of the PPIase domain. For CaEss1, the primary interaction site involves contacts between residues of the N-terminal segment and of strand β1 of the WW domain with the loop between helix R5 (which corresponds to helix R4 of Pin1) and strand β6 of the PPIase domain (Figure 3). In addition, there are extensive interactions between the loops joining strands β2 and β3 of the WW domain with the C-terminus of helix α2, the first helix of the PPIase domain. The latter contact site is noteworthy, since both the crystallographic and NMR studies of human Pin1 do not detect interactions with this helix.
Analysis of the relative domain movements, using DYNDOM (57), shows that the WW domain of CaEss1 is rotated by approximately 108°, about an axis that runs parallel to the β-sheet of the PPIase domain, and translated by 11 Å along this axis, compared to the WW domain of Pin1 (Figure 5a). Consistent with the more highly ordered linker structure, many more close contacts exist between the two domains and between the linker and the PPIase domain in CaEss1 than in Pin1, including six hydrogen bonds (Table 2 and Figure 5b) that are distributed over a large area compared to three hydrogen bonds that are confined to a much smaller area. In fact, the total surface area of the PPIase domain rendered inaccessible by the WW domain and linker is 689 Å2 for CaEss1 compared to 587 Å2 of Pin1.
Figure 5.
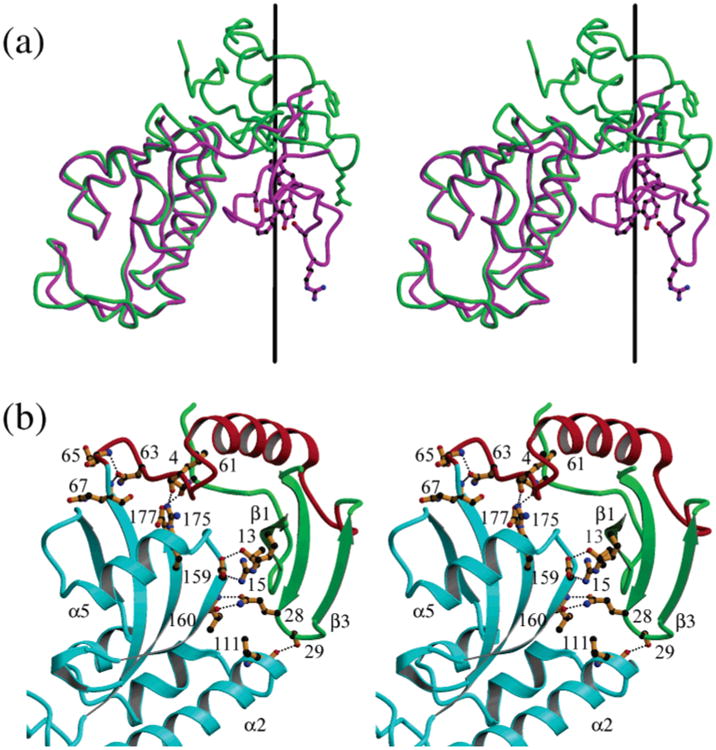
Difference in the relative positions of the WW and PPIase domains of CaEss1 (green) and Pin1 (magenta). (a) A rotation by 108° about the axis shown in black followed by a translation of 11 Å positions the WW domain of Pin1 onto that of CaEss1. (b) Interdomain contacts observed in the structure of CaEss1. Residues involved in hydrogen bonding contacts among the WW domain (green), the linker (red), and the PPIase domain (cyan) are shown in orange with black dotted lines indicating the hydrogen bonding contacts.
Table 2. Hydrogen Bonding Contacts between Residues of the WW Domain and the Linker with the PPIase Domain of CaEss1.
| WW domain atom | PPIase domain atom | distance (Å) |
|---|---|---|
| Thr4 OG1 | Gly177 O | 2.73 |
| Thr13 OG1 | Glu159 OE2 | 2.75 |
| Arg15 NH1 | Glu159 OE1 | 2.84 |
| Gln28 OE1 | Val160 N | 2.83 |
| Gln28 NE2 | Val160 O | 2.74 |
| Ser29 OG | Leu111 O | 2.68 |
|
| ||
| linker atom | PPIase domain atom | distance (Å) |
|
| ||
| Tyr49 OH | His156 ND1 | 2.69 |
| Leu61 O | Arg175 NH2 | 2.72 |
| Asn63 OD1 | Gln67 | 2.78 |
| Asp65 N | 2.79 | |
NMR studies for Pin1 have shown that, in the absence of peptide substrates, interactions between the WW and PPIase domains in solution are at best of a transient nature, and certainly less extensive than those seen in the Pin1 crystal structure (38, 39). These studies further showed that the binding of short peptides does not reduce the linker flexibility, but that it does increase the stability of the domain association. However, this change in domain association is dependent on the composition of the peptide that is used, with the most stable association resulting from binding of the optimal substrate of the enzyme (39). It was also reported that the NMR data are consistent with a model in which, upon peptide binding, the two domains interact in a fashion similar to that seen in the crystal structures. This suggests that the Pin1–phosphopeptide complex as seen in the crystal structures is indeed in the functional conformation of the molecule. For CaEss1, the highly ordered structure of the linker and the extensive contacts between the domains and the linker lead us to believe that the domain association seen in the structure of this protein is fixed in a more permanent functional conformation.
Examination of fungal and metazoan Ess1 and Pin1 sequences indicates that the novel α-helix within the linker region of CaEss1 might be fungal-specific. For example, in addition to C. albicans, linker sequences from other pathogenic fungi such as Cryptococcus neoformans, Candida glabrata, and Aspergillus nidulans are of a length similar to that of the CaEss1 linker, and secondary structure predictions suggest that they all contain an α-helix (Figure 6). The Ess1s of some generally nonpathogenic yeasts, such as Schizo-saccharomyces pombe, Kluyveromyces lactis, and S. cerevisiae, also have extended linker regions, although not as consistently high in α-helical content. In contrast, the metazoan forms of Ess1 and Pin1, ranging from Drosophila melanogaster and Xenopus laevis to mouse and human, seem either to lack these extended linker sequences or to have linkers that are not predicted to contain α-helices. These linkers are rich in Pro, a residue inconsistent with helical conformations, and Gly, a residue that promotes flexibility. Consequently, the linkers of the metazoan proteins are all expected to be unstructured. Consistent with the idea that the linker region of the fungal enzyme is important for function, a mutation in this region of S. cerevisiae Ess1, G43V (corresponding to Gly40 of CaEss1), renders the cells slow-growing and temperature-sensitive (22).
Figure 6.
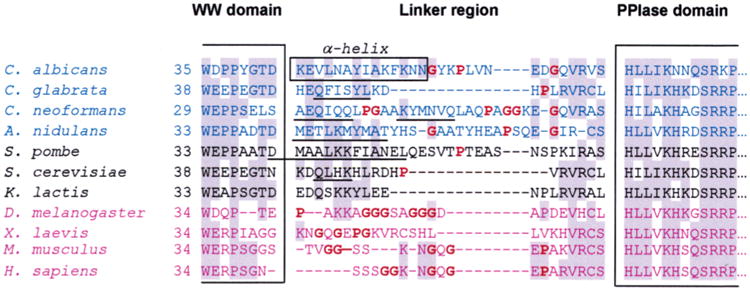
Sequence alignment of the linker region of Ess1 and Pin1 orthologs from different organisms. The sequences were aligned on the basis of highly conserved motifs within the WW and PPIase domains, some of which are shown within the large brackets. Shaded residues are the most highly conserved. The box denotes the residues in C. albicans that form a long α-helix in the crystal structure. Sequences in blue are from pathogenic fungi, and those in black are from other fungi that are not normally pathogenic. Sequences in purple are from metazoans. Bold residues (also highlighted in red) are helix-breaking residues or residues that do not promote helices (proline or glycine). Secondary structure predictions were performed for all linker regions using NNPREDICT (58). Sequences that are predicted to be α-helical are underlined. In general, fungal linker regions are predicted to include α-helices, whereas metazoan linkers probably do not.
It is tempting to suggest that the α-helix plays some fungal-specific role, for example, by interacting with other proteins that are important for virulence or other fungal-specific functions. Alternatively, the structured linker, which limits domain mobility, might serve to restrict substrate recognition. In the metazoan Pin1 proteins, flexibility in this linker may broaden the spectrum of substrates that can be recognized, and perhaps add the capacity for additional regulation. The fact that the human and Drosophila Pin1/Ess1 enzymes function in S. cerevisiae (25, 26) indicates that they recognize at least the essential fungal substrates, despite the absence of structured linkers, demonstrated for human Pin1 or predicted for Drosophila. The reverse experiment, expressing the fungal enzyme in flies or mice, has not yet been attempted.
Role of the WW Domain in Substrate Recognition
Since the WW domain has been shown to have higher substrate binding affinity than the PPIase domain (41), the relative positions and orientations of the two domains are likely to be important for the mechanism by which Ess1- and Pin1-type PPIases bind and process intact, multiple-repeat substrates, such as the CTD of RNA polymerase II. The difference in the relative orientations of the WW and PPIase domains of Pin1 and CaEss1 greatly affects the location of the phosphopeptide-binding site of the WW domain relative to the catalytic site. In the structure of the complex of Pin1 with a single repeat of the CTD peptide (YpSPTpSPS, pS = phosphoserine), the phosphopeptide is located in a cleft between the two domains (Figure 7a). However, direct interactions with the peptide are limited to residues of the WW domain. It was proposed that this cleft represents a primary substrate recognition site and that longer substrates will first be bound in this cleft and then will be positioned toward the active site. How the protein guides this extended peptide to the active site area in the PPIase domain remains to be established. The CTD of RNA polymerase II consists of multiple repeats of the heptapeptide sequence (26 in yeast and 52 in human), allowing for simultaneous interactions with the WW domain and the active site of the PPIase domain. The heptapeptide observed in the Pin1 structure is in an extended-coil conformation and is bound in a direction that would require a full 180° turn, in the extended peptide, that is at least four repeat units long, for it to reach the active site. Whether such a 180° turn can be accommodated by an extended peptide of this composition remains to be determined, although there is evidence, from the crystal structures of longer CTD peptides, 14 and 21 residues, respectively, in complexes with mRNA capping enzyme (59) and with 3′-RNA processing factors (60), that multiple copies of the repeat may generate spiral structures.
Figure 7.
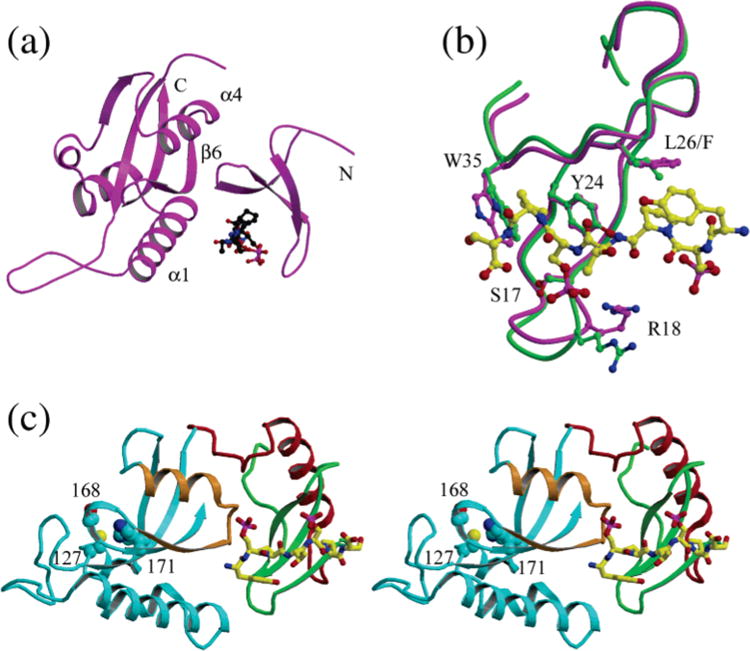
Comparison the phosphopeptide binding site of the WW domains of Pin1 and CaEss1. (a) Structure of Pin1 with the phosphopeptide bound in the cleft between the WW and PPIase domains. (b) Superposition of the WW domain of CaEss1 (green) onto that of Pin1 (magenta) with the phosphopeptide (yellow) bound, showing the similarity in the conformations of the domains and the locations of the residues that interact with the peptide. (c) Model of the location of the phosphopeptide (yellow) on the WW domain of CaEss1. The peptide binding site is found on the surface of the molecule, and an extended peptide could approach the active site area, identified by the side chains of the catalytic residues which are shown in cyan-colored CPK representation, by passing over the area of helix α5 and strand β6 of the PPIase domain (shown in brown).
In the structure of CaEss1, because of the different relative orientations of the WW and PPIase domains, there is no corresponding cleft between the two domains. While the residues of the WW domain that interact with the phospho-peptide are conserved, they are found on an exterior surface of the molecule in CaEss1 (Figure 7b). By superpositioning the WW domain of Pin1 onto that of CaEss1, we have modeled the location of the phosphopeptide on the CaEss1 structure (Figure 7c). The most important observation from this model is that the peptide binding site is exposed on the surface of the molecule and is aligned nearly parallel with the longest axis of the molecule so that an extended peptide, consisting of only three or four repeats, could interact with both the WW domain and the active site area of the PPIase domain. Access to the active site itself would require a 90° turn in the peptide conformation or a conformational change in the PPIase domain near the active site. Therefore, the differences in the observed structures of Pin1 and CaEss1 are consistent with different mechanisms by which the proteins interact with their substrate for binding to the WW domain and isomerization by the PPIase domain.
Conclusion
Here, we have presented evidence that the C. albicans homologue of Ess1 and Pin1 is a prolyl isomerase, and that it is important for survival in a mammalian host. Isomerase activity against synthetic peptide substrates is similar to that of the S. cerevisiae enzyme and of human Pin1 (61). The 1.6 Å crystal structure of CaEss1 revealed a strong similarity of the WW and PPIase domains, compared to those in human Pin1, but striking differences in the manner by which they are joined. These differences provide insight into possible functioning of parvulin-class PPIases on their natural substrates, and may suggest strategies for developing specific inhibitors for antifungal applications. Future work with CaEss1 will be aimed at understanding the role of the α-helix-containing linker region in enzyme function, and its importance in vivo.
Footnotes
Research supported in part by Grant R01 GM55108 (S.D.H.) from the National Institutes of Health. Support provided by the Molecular Genetics Core of the Wadsworth Center is gratefully acknowledged.
The atomic coordinates and observed structure factors have been deposited in the Protein Data Bank as entry 1YW5.
Abbreviations: CaEss1, C. albicans Ess1; CTD, carboxy-terminal domain; cfu, colony-forming unit; PPIase, peptidyl-prolyl isomerase; MIRAS, multiple isomorphous replacement/anomalous scattering; rmsd, root-mean-square deviation; ts, temperature-sensitive.
References
- 1.Odds FC. Candida and Candidosis: A Review and Bibliography. 2nd. Bailliere Tindall; London and Philadelphia: 1988. [Google Scholar]
- 2.Maschmeyer G, Ruhnke M. Update on antifungal treatment of invasive Candida and Aspergillus infections. Mycoses. 2004;47:263–276. doi: 10.1111/j.1439-0507.2004.01003.x. [DOI] [PubMed] [Google Scholar]
- 3.Rolston K. Overview of systemic fungal infections. Oncology. 2001;15:11–14. [PubMed] [Google Scholar]
- 4.Sudbery P, Gow N, Berman J. The distinct morphogenic states of Candida albicans. Trends Microbiol. 2004;12:317–324. doi: 10.1016/j.tim.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 5.Romani L, Bistoni F, Puccetti P. Adaptation of Candida albicans to the host environment: The role of morphogenesis in virulence and survival in mammalian hosts. Curr Opin Microbiol. 2003;6:338–343. doi: 10.1016/s1369-5274(03)00081-x. [DOI] [PubMed] [Google Scholar]
- 6.Saville SP, Lazzell AL, Monteagudo C, Lopez-Ribot JL. Engineered control of cell morphology in vivo reveals distinct roles for yeast and filamentous forms of Candida albicans during infection. Eukaryotic Cell. 2003;2:1053–1060. doi: 10.1128/EC.2.5.1053-1060.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dhillon NK, Sharma S, Khuller GK. Signaling through protein kinases and transcriptional regulators in Candida albicans. Crit Rev Microbiol. 2003;29:259–275. doi: 10.1080/713610451. [DOI] [PubMed] [Google Scholar]
- 8.Berman J, Sudbery PE. Candida albicans: A molecular revolution built on lessons from budding yeast. Nat Rev Genet. 2002;3:918–930. doi: 10.1038/nrg948. [DOI] [PubMed] [Google Scholar]
- 9.Casalinuovo IA, Di Francesco P, Garaci E. Fluconazole resistance in Candida albicans: A review of mechanisms. Eur Rev Med Pharmacol Sci. 2004;8:69–77. [PubMed] [Google Scholar]
- 10.Kontoyiannis DP, Lewis RE. Toward more effective antifungal therapy: The prospects of combination therapy. Br J Haematol. 2004;126:165–375. doi: 10.1111/j.1365-2141.2004.05007.x. [DOI] [PubMed] [Google Scholar]
- 11.Vazquez JA. Combination antifungal therapy against Candida species: The new frontier—are we there yet? Med Mycol. 2003;41:355–368. doi: 10.1080/13693780310001616528. [DOI] [PubMed] [Google Scholar]
- 12.Singh N, Heitman J. Antifungal attributes of immunosuppressive agents: New paradigms in management and elucidating the pathophysiologic basis of opportunistic mycoses in organ transplant recipients. Transplantation. 2004;27:795–800. doi: 10.1097/01.tp.0000117252.75651.d6. [DOI] [PubMed] [Google Scholar]
- 13.Schmid FX, Mayr LM, Mucke M, Schonbrunner ER. Prolyl isomerases: Role in protein folding. Adv Protein Chem. 1993;44:25–66. doi: 10.1016/s0065-3233(08)60563-x. [DOI] [PubMed] [Google Scholar]
- 14.Kunz J, Hall MN. Cyclosporin A, FK506 and rapamycin: More than just immunosuppression. Trends Biochem Sci. 1993;18:334–338. doi: 10.1016/0968-0004(93)90069-y. [DOI] [PubMed] [Google Scholar]
- 15.Blankenship JR, Steinbach WJ, Perfect JR, Heitman J. Teaching old drugs new tricks: Reincarnating immuno-suppressants as antifungal drugs. Curr Opin Invest Drugs. 2003;4:192–199. [PubMed] [Google Scholar]
- 16.Cruz MC, Goldstein AL, Blankenship J, Del Poeta M, Perfect JR, McCusker JH, Bennani YL, Cardenas ME, Heitman J. Rapamycin and less immunosuppressive analogs are toxic to Candida albicans and Cryptococcus neoformans via FKBP12-dependent inhibition of TOR. Antimicrob Agents Chemother. 2001;45:3162–3170. doi: 10.1128/AAC.45.11.3162-3170.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rahfeld JU, Schierhorn A, Mann K, Fischer G. A novel peptidyl-prolyl cis/trans isomerase from Escherichia coli. FEBS Lett. 1994;343:65–69. doi: 10.1016/0014-5793(94)80608-x. [DOI] [PubMed] [Google Scholar]
- 18.Hanes SD, Shank PR, Bostian KA. Sequence and mutational analysis of ESS1, a gene essential for growth in Saccharomyces cerevisiae. Yeast. 1989;5:55–72. doi: 10.1002/yea.320050108. [DOI] [PubMed] [Google Scholar]
- 19.Hani J, Stumpf G, Domdey H. PTF1 encodes an essential protein in Saccharomyces cerevisiae, which shows strong homology with a new putative family of PPIases. FEBS Lett. 1995;365:198–202. doi: 10.1016/0014-5793(95)00471-k. [DOI] [PubMed] [Google Scholar]
- 20.Hani J, Schelbert B, Bernhardt A, Domdey H, Fischer G, Wiebauer K, Rahfeld JU. Mutations in a peptidyl prolyl-cis/trans-isomerase gene lead to a defect in 3′-end formation of a pre-mRNA in Saccharomyces cerevisiae. J Biol Chem. 1999;274:108–116. doi: 10.1074/jbc.274.1.108. [DOI] [PubMed] [Google Scholar]
- 21.Morris DP, Phatnani HP, Greenleaf AL. Phospho-carboxyl-terminal domain binding and the role of a prolyl isomerase in pre-mRNA 3′-end formation. J Biol Chem. 1999;274:31583–31587. doi: 10.1074/jbc.274.44.31583. [DOI] [PubMed] [Google Scholar]
- 22.Wu X, Wilcox CB, Devasahayam G, Hackett RL, Arevalo-Rodriguez M, Cardenas ME, Heitman J, Hanes SD. The Ess1 prolyl isomerase is linked to chromatin remodeling complexes and the general transcription machinery. EMBO J. 2000;19:3727–3738. doi: 10.1093/emboj/19.14.3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu X, Rossettini A, Hanes SD. The ESS1 prolyl isomerase and its suppressor BYE1 interact with RNA pol II to inhibit transcription elongation in Saccharomyces cerevisiae. Genetics. 2003;165:1687–1702. doi: 10.1093/genetics/165.4.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilcox CB, Rossettini A, Hanes SD. Genetic interactions with C-terminal domain (CTD) kinases and the CTD of RNA PolII suggest a role for ESS1 in transcription and elongation in Saccharomyces cerevisiae. Genetics. 2004;167:93–105. doi: 10.1534/genetics.167.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu K, Hanes SD, Hunter T. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature. 1996;380:544–547. doi: 10.1038/380544a0. [DOI] [PubMed] [Google Scholar]
- 26.Maleszka R, Hanes SD, Hackett RL, de Couet HG, Miklos GL. The Drosophila melanogaster dodo (dod) gene, conserved in humans, is functionally interchangeable with the ESS1 cell division gene of Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1996;93:447–451. doi: 10.1073/pnas.93.1.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Devasahayam G, Chaturvedi V, Hanes SD. The Ess1 prolyl isomerase is required for growth and morphogenetic switching in Candida albicans. Genetics. 2002;160:37–48. doi: 10.1093/genetics/160.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ren P, Chaturvedi V, Hanes SD. The Ess1 prolyl isomerase is dispensable for growth but required for virulence in Crytococcus neofomans. Microbiology. 2005 doi: 10.1099/mic.0.27786-0. in press. [DOI] [PubMed] [Google Scholar]
- 29.Joseph JD, Daigle SN, Means AR. PINA is essential for growth and positively influences NIMA function in Aspergillus nidulans. J Biol Chem. 2004;279:32373–32384. doi: 10.1074/jbc.M405415200. [DOI] [PubMed] [Google Scholar]
- 30.Shaw PE. Peptidyl-prolyl isomerases: A new twist to transcription. EMBO Rep. 2002;3:521–526. doi: 10.1093/embo-reports/kvf118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou XZ, Lu PJ, Wulf G, Lu KP. Phosphorylation-dependent prolyl isomerization: A novel signaling regulatory mechanism. Cell Mol Life Sci. 1999;56:788–806. doi: 10.1007/s000180050026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Joseph JD, Yeh ES, Swenson KI, Means AR, Winkler KE. The peptidyl-prolyl isomerase Pin1. Prog Cell Cycle Res. 2003;5:477–487. [PubMed] [Google Scholar]
- 33.Lu KP. Pinning down cell signaling, cancer and Alzheimer's disease. Trends Biochem Sci. 2004;29:200–209. doi: 10.1016/j.tibs.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 34.Fujimori F, Takahashi K, Uchida C, Uchida T. Mice lacking Pin1 develop normally, but are defective in entering cell cycle from G(0) arrest. Biochem Biophys Res Commun. 1999;265:658–663. doi: 10.1006/bbrc.1999.1736. [DOI] [PubMed] [Google Scholar]
- 35.Xu YX, Manley JL. Pinning down transcription: Regulation of RNA polymerase II activity during the cell cycle. Cell Cycle. 2004;3:432–435. [PubMed] [Google Scholar]
- 36.Ranganathan R, Lu KP, Hunter T, Noel JP. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell. 1997;89:875–886. doi: 10.1016/s0092-8674(00)80273-1. [DOI] [PubMed] [Google Scholar]
- 37.Sudol M. Structure and function of the WW domain. Prog Biophys Mol Biol. 1996;65:113–132. doi: 10.1016/s0079-6107(96)00008-9. [DOI] [PubMed] [Google Scholar]
- 38.Bayer E, Goettsch S, Mueller JW, Griewel B, Guiberman E, Mayr LM, Bayer P. Structural analysis of the mitotic regulator hPin1 in solution: Insights into domain architecture and substrate binding. J Biol Chem. 2003;278:26183–26193. doi: 10.1074/jbc.M300721200. [DOI] [PubMed] [Google Scholar]
- 39.Jacobs DM, Saxena K, Vogtherr M, Bernado P, Pons M, Fiebig KM. Peptide binding induces large scale changes in inter-domain mobility in human Pin1. J Biol Chem. 2003;278:26174–26182. doi: 10.1074/jbc.M300796200. [DOI] [PubMed] [Google Scholar]
- 40.Yaffe MB, Schutkowski M, Shen M, Zhou XZ, Stukenberg PT, Rahfeld JU, Xu J, Kuang J, Kirschner MW, Fischer G, Cantley LC, Lu KP. Sequence-specific and phosphorylation-dependent proline isomerization: A potential mitotic regulatory mechanism. Science. 1997;278:1957–1960. doi: 10.1126/science.278.5345.1957. [DOI] [PubMed] [Google Scholar]
- 41.Verdecia MA, Bowman ME, Lu KP, Hunter T, Noel JP. Structural basis for phosphoserine-proline recognition by group IV WW domains. Nat Struct Biol. 2000;7:639–643. doi: 10.1038/77929. [DOI] [PubMed] [Google Scholar]
- 42.Arévalo-Rodríguez M, Wu X, Hanes SD, Heitman J. Prolyl isomerases in yeast. Front Biosci. 2004;9:2420–2446. doi: 10.2741/1405. [DOI] [PubMed] [Google Scholar]
- 43.Fischer G, Bang H, Mech C. Determination of enzymatic catalysis for the cis-trans-isomerization of peptide binding in proline-containing peptides. Biomed Biochim Acta. 1984;43:1101–1111. [PubMed] [Google Scholar]
- 44.Gemmill T, Wu X, Hanes SD. Vanishingly low levels of Ess1 prolyl-isomerase activity are sufficient for growth in Saccharomyces cerevisiae. J Biol Chem. 2005 doi: 10.1074/jbc.M412172200. [DOI] [PubMed] [Google Scholar]
- 45.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 46.Terwilliger TC, Berendzen J. Automated structure solution for MIR and MAD. Acta Crystallogr. 1999;D55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr. 2000;D56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roussel A, Cambillau C. Silicon Graphics Geometry Partners Directory. Silicon Graphics; Mountain View, CA: 1989. pp. 77–78. [Google Scholar]
- 49.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 50.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for the building of protein models in electron-density maps and the location of errors in these maps. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 51.Laskowski RA, McArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:282–291. [Google Scholar]
- 52.Diez-Orejas R, Molero G, Navarro-Garcia F, Pla J, Nombela C, Sanchez-Perez M. Reduced virulence of Candida albicans MKC1 mutants: A role for mitogen-activated protein kinase in pathogenesis. Infect Immun. 1997;65:833–837. doi: 10.1128/iai.65.2.833-837.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Navarro-Garcia F, Sanchez M, Nombela C, Pla J. Virulence genes in the pathogenic yeast Candida albicans. FEMS Microbiol Rev. 2001;25:245–268. doi: 10.1111/j.1574-6976.2001.tb00577.x. [DOI] [PubMed] [Google Scholar]
- 54.Ryley JF, McGregor S, Lister SC, Jackson KP. Kidney function in experimental systemic candidosis of mice. Mycoses. 1988;31:203–207. [PubMed] [Google Scholar]
- 55.Macias MJ, Hyvonen M, Baraldi E, Schultz J, Sudol M, Saraste M, Oschkinat H. Structure of the WW domain of a kinase-associated protein complexed with a proline-rich peptide. Nature. 1996;382:646–649. doi: 10.1038/382646a0. [DOI] [PubMed] [Google Scholar]
- 56.Kowalski JA, Liu K, Kelly JW. NMR solution structure of the isolated Apo Pin1 WW domain: Comparison to the X-ray crystal structures of Pin1. Biopolymers. 2002;63:111–121. doi: 10.1002/bip.10020. [DOI] [PubMed] [Google Scholar]
- 57.Hayward S, Berendzen HJC. Systematic analysis of domain motions in proteins from conformational change: New results on citrate synthase and T4 lysozyme. Proteins: Struct Funct Genet. 1998;30:144–154. [PubMed] [Google Scholar]
- 58.Kneller DG, Cohen FE, Langridge R. Improvements in protein secondary structure prediction by an enhanced neural network. J Mol Biol. 1990;214:171–182. doi: 10.1016/0022-2836(90)90154-E. [DOI] [PubMed] [Google Scholar]
- 59.Fabrega C, Shen V, Shurman S, Lima CD. Structure of an mRNA capping enzyme bound to the phosphorylated carboxy-terminal domain of RNA polymerase II. Mol Cell. 2003;11:1549–1561. doi: 10.1016/s1097-2765(03)00187-4. [DOI] [PubMed] [Google Scholar]
- 60.Meinhart A, Cramer P. Recognition of RNA polymerase II carboxy-terminal domain by 3′-RNA-processing factors. Nature. 2004;430:223–226. doi: 10.1038/nature02679. [DOI] [PubMed] [Google Scholar]
- 61.Zhang Y, Fussel S, Reimer U, Schutkowski M, Fischer G. Substrate-based design of reversible Pin1 inhibitors. Biochemistry. 2002;41:11868–11877. doi: 10.1021/bi0262395. [DOI] [PubMed] [Google Scholar]