

Abstract
The genus Pantoea incorporates many economically and clinically important species. The plant-associated species, Pantoea agglomerans and Pantoea vagans, are closely related and are often isolated from similar environments. Plasmids conferring certain metabolic capabilities are also shared amongst these two species. The genomes of two isolates obtained from fungus-growing termites in South Africa were sequenced, assembled and annotated. A high number of orthologous genes are conserved within and between these species. The difference in genome size between P. agglomerans MP2 (4,733,829 bp) and P. vagans MP7 (4,598,703 bp) can largely be attributed to the differences in plasmid content. The genome sequences of these isolates may shed light on the common traits that enable P. agglomerans and P. vagans to co-occur in plant- and insect-associated niches.
Keywords: Pantoea, Bacteria, Insect, Symbiosis
Introduction
The bacterial genus Pantoea contains several economically important plant pathogens, as well as strains of clinical importance [10]. Amongst the plant pathogens, Pantoea ananatis, with its broad host range (e.g. onion, eucalyptus and pineapple) and P. stewartii subsp. stewartii, the causal agent of Stewart’s wilt on maize, are the best known. The human pathogens include species such as P. septica and P. brenneri [9], although some plant-associated species have also been isolated from immuno-compromised patients [12, 17]. P. agglomerans and P. vagans are most commonly isolated from similar ecological niches, including both plant and insect hosts [41].
Three plasmids (pPag1, pPag2 and pPag3) were identified in the genome of the biocontrol strain P. vagans C9-1 [45] and it is thought that the presence of these plasmids may play a role in the physiological and ecological functioning of this strain. The plasmid, pPag1, codes for sucrose metabolism, while the plasmid, pPag2, harbours genes for an antimicrobial peptide and sorbitol utilization [33, 46]. The megaplasmid pPag3 belongs to the LPP-1 plasmids conserved among all sequenced Pantoea sppecies to date and carries genes involved in pigment production, thiamine biosynthesis and maltose metabolism [19, 46]. In contrast to P. vagans, some strains of P. agglomerans are also known to induce galls on Gypsophila spp., beet (Beta vulgaris), Douglas fir (Pseudotsuga menziesii) and Wisteria spp. [6, 37]. This ability has been linked to a genomic island that encodes a Type III secretion system and pPath plasmid genes involved in the biosynthesis of the plant hormones, indole-3-acetic acid and cytokinins [6]. P. agglomerans strains have also been shown to cause opportunistic infections in humans [15, 18].
In this study we summarize the features of a P. agglomerans (Mn107) and a P. vagans (Mn109) that were isolated from two different colonies of the fungus-growing termite Macrotermes natalensis in South Africa, and provide an overview of the draft genome sequences and annotations for these two strains. The genome sequences provide some understanding of the shared genomic features that could be linked to their survival in similar environments and the unique features that characterise the species.
Organism information
Classification and features
Both P. agglomerans MP2 (LMG 29065) and P. vagans MP7 (LMG 29064) are members of the Enterobacteriaceae in the class Gammaproteobacteria, and are thus Gram-negative, motile, non-spore-forming, rods (Fig. 1, Table 1). After incubation on Luria-Bertani agar (10 g tryptone, 5 g yeast extract, 5 g NaCl, and X g agar per litre) at 28 °C for 24 h, colonies of P. agglomerans MP2 and P. vagans MP7 are yellow, convex and round with entire margins.
Fig. 1.

Photomicrographs of source organisms. The source organisms for a P. agglomerans MP2 and of b P. vagans MP7, stained with safranin
Table 1.
Classification and general features of P. agglomerans MP2 and P. vagans MP7
| MIGS ID | Property | Pantoea agglomerans MP2 | Evidence codea | Pantoea vagans MP7 | Evidence codea |
|---|---|---|---|---|---|
| Classification | Bacteria | NAS [25] | Bacteria | NAS [25] | |
| Proteobacteria | NAS [23] | Proteobacteria | NAS [23] | ||
| Gammaproteobacteria | NAS [24, 51] | Gammaproteobacteria | NAS [24, 51] | ||
| Enterobacteriaceae | NAS [42, 44] | Enterobacteriaceae | NAS [42, 44] | ||
| Enterobacteriales | NAS [25] | Enterobacteriales | NAS [25] | ||
| Pantoea | NAS [9, 26] | Pantoea | NAS [9, 26] | ||
| Pantoea agglomerans | NAS [26, 39] | Pantoea vagans | NAS [10] | ||
| Gram stain | Negative | NAS [26] | Negative | NAS [10] | |
| Cell shape | Straight rods | NAS [26] | Short rods | NAS [10] | |
| Motility | Motile | NAS [26] | Motile | NAS [10] | |
| Sporulation | Non-sporeforming | NAS [26] | Non-sporeforming | NAS [10] | |
| Temperature range | Mesophile | NAS [26] | Mesophile | NAS [10] | |
| Optimum temperature | 30 °C | NAS [54] | 30 °C | NAS [54] | |
| pH range; Optimum | 4 - 8; 5–6 | IDA | 4 - 9; 5 -6 | IDA | |
| Carbon source | D-Glucose, L-arabinose, D-galactose, maltose, D-mannitol, D-mannose, L-rhamnose, sucrose, trehalose, D-xylose | NAS [54] | Malonic acid, L-ornithine, D-glucose, L-arabinose, D-ribose, D-galactose, sucrose, maltose | NAS [10] | |
| Energy source | Chemoorganotroph | NAS [54] | Chemoorganotroph | NAS [54] | |
| Terminal electron receptor | Not available | Not available | |||
| MIGS-6 | Habitat | Termite | IDA | Termite | IDA |
| MIGS-6.3 | Salinity | Not available | Not available | ||
| MIGS-22 | Oxygen requirement | Facultative anaerobic | NAS [54] | Facultative anaerobic | NAS [54] |
| MIGS-15 | Biotic relationship | Potential termite symbiont | Potential termite symbiont | ||
| MIGS-14 | Pathogenicity | Not available | Not available | ||
| MIGS-4 | Geographic location | Pretoria, South Africa | Mookgophong, South Africa | ||
| MIGS-5 | Sample collection | January 2010 | January 2010 | ||
| MIGS-4.1 MIGS-4.2 | Latitude – Longitude | S25 43 45.6 E28 14 09.9 | S24 40 30.5 E28 47 50.4 | ||
| MIGS-4.3 | Depth | N/A | N/A | ||
| MIGS-4.4 | Altitude | 1344 m | 1046 m |
IDA Inferred from Direct Assay, TAS Traceable Author Statement (i.e., a direct report exists in the literature), NAS Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are derived from the Gene Ontology project
aEvidence codes
The 16S rRNA gene sequences of the enteric bacteria tend to provide insufficient resolution and the phylogenetic relationships of P. agglomerans MP2 and P. vagans MP7 were therefore inferred with multi-locus sequence analysis. This analysis included closely related members in the genus Pantoea with available genome sequences, and was based on partial nucleotide sequences of four protein coding genes (i.e., atpD, carA, gyrB, infB, recA and rpoB) [57]. Our results showed that P. agglomerans and P. vagans group as sister-species (Fig. 2).
Fig. 2.
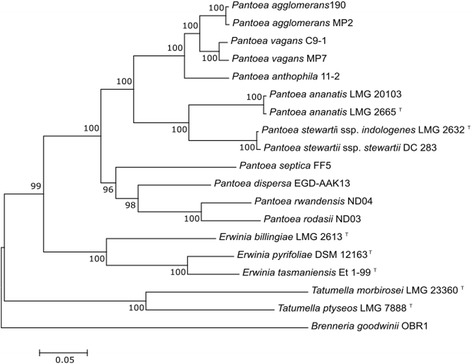
Maximum likelihood phylogenetic tree indicating the phylogenetic relationship of sequenced isolates. The maximum likelihood (ML) tree was constructed from an alignment of concatenated atpD, carA, gyrB, infB, recA and rpoB gene sequences [57]. The tree was constructed with Mega 6 [49] using the general time reversible (GTR) model [36] with the estimation of the proportion of invariable sites and gamma distribution. Bootstrap support values were calculated from 1000 bootstrap replicates. Several strains (including type strains; indicated with “T”) of Pantoea sppecies for which genome sequences are publicly available were included in the analysis [Genbank Accessions: P. agglomerans 190 [26]: GCA_000731125.1, P. vagans C9-1 [10]: GCA_000148935.1, P. anthophila 11–2 [10]: GCA_000969395.1, P. stewartii subsp. indologenes LMG 2632T [38]: GCA_000757405.1, P. stewartii subsp. stewartii DC283 [38]: GCA_000248395.2, P. ananatis LMG 2665 T [38]: GCA_000710035.1, P. ananatis LMG 20103 [38]: GCA_000025405.2, P. septica FF5 [9]: GCA_000612605.1, P. dispersa EGD-AAK13 [26]: GCA_000465555.2, P. rodasii ND03 [11]: GCA_000801085.1, P. rwandensis ND04 [11]:GCA_000759475.1]. Type strains of species of the sister genera Tatumella [Tatumella ptyseos LMG 7888 T [31, 52]: GCA_000439895.1 and Tatumella morbirosei LMG 23360 T [31]: GCA_000757425.2 (Genbank Accessions)] and Erwinia [44, 55], [Erwinia billingiae LMG 2613 T [39]: GCA_000196615.1, Erwinia pyrifoliae DSM 12163 [34]: GCA_000026985.1, Erwinia tasmaniensis Et-99: GCA_000026185.1 (Genbank Accessions)], for which genome sequences are available, were also included. Brenneria goodwinii OBR-1 [GCA_001049335.1 (Genbank Accession)] was used as outgroup
The two isolates (strain codes: MP2 = Mn109-1w1C and MP7 = Mn107-old1M) were isolated from Macrotermes natalensis termite mounds in 2010. The surface of worker termite was rinsed using phospate buffer saline and MP2 was isolated from the rinsate, which was inoculated directly onto chitin medium (4 g chitin, 0.7 g K2HPO4, 0.3 g KH2PO4, 0.5 g MgSO4.5H2O, 0.01 g FeSO4.7H20, 0.001 g ZnSO4, 0.001 g MnCl2, and 20 g of agar per litre), while MP7 was isolated from fungus comb ground in PBS and inoculated onto Carboxymethyl cellulose medium (10 g carboxymethyl cellulose and 20 g agar per litre). Isolates were streaked onto Yeast Malt Extract Agar medium (4 g yeast extract, 10 g malt extract, 4 g D-glucose and 20 g bacteriological agar per litre), and once in pure culture, they were stored in 10 % glycerol at −20 °C. The specificity and possible role of associations between fungus-growing termites and the two Pantoea isolates have not been determined, but the abundance of members of the Enterobacteriaceae in both fungus-growing termite guts [40] and fungus combs [4] suggests the possibility of a specific association.
Genome sequencing information
Genome project history
The genomes of both isolates were sequenced using the Illumina platform. Velvet [56] and Mauve [16] were employed for the assembly of the genomes and annotations were done using the Rapid Annotation using Subsystem Technology [5] and WebMGA. The genomes will remain as high quality drafts and are available from the National Center for Biotechnology Information (Tables 2 and 3). The Whole Genome Shotgun projects have been deposited at DDBJ/EMBL/GenBank under the accessions JPKQ00000000 and JPKP00000000, respectively. The versions described in this paper are version JPKQ00000000.1 and JPKP00000000.1.
Table 2.
Project information
| MIGS ID | Property | P. agglomerans MP2 | P. vagans MP7 |
|---|---|---|---|
| MIGS-31 | Finishing quality | High-quality draft | High-quality draft |
| MIGS-28 | Libraries used | 500 bp | 500 bp |
| MIGS-29 | Sequencing platforms | Illumina HiSeq mate-pair | Illumina HiSeq mate-pair |
| MIGS-31.2 | Fold coverage | 179 × | 184 × |
| MIGS-30 | Assemblers | Velvet | Velvet |
| MIGS-32 | Gene calling method | RAST | RAST |
| Genbank ID | JPKQ00000000.1 | JPKP00000000.1 | |
| Genbank Date of Release | 23/9/2014 | 23/9/2014 | |
| GOLD ID | Gp0099200 | Gp0099199 | |
| BIOPROJECT | PRJNA254768 | PRJNA254769 | |
| MIGS-13 | Source material identifier | SAMN02905153 | SAMN02905155 |
| Project relevance | Potential termite symbiont | Potential termite symbiont |
Table 3.
Summary of the genomes
| Label | Size (Mb) | Topology | INSDC identifier | RefSeq ID | |
|---|---|---|---|---|---|
| Pantoea agglomerans MP2 | Chromosome 1 | 3988.2 | circular | JPKQ0100001-13 | NZ_JPKQ01000001.1-13.1 |
| Plasmid 1 | 184.9 | circular | JPKQ01000014 | NZ_JPKQ01000014.1 | |
| Plasmid 2 | 292.9 | circular | JPKQ01000015 | NZ_JPKQ01000015.1 | |
| Plasmid 3 | 531.5 | circular | JPKQ01000016 | NZ_JPKQ01000016.1 | |
| Pantoea vagans MP7 | Chromosome 1 | 3913.1 | circular | JPKP01000001-6 | NZ_JPKP01000001.1-6.1 |
| Plasmid 1 | 176.9 | circular | JPKP01000007 | NZ_JPKP01000007.1 | |
| Plasmid 2 | 508.6 | circular | JPKP01000008 | NZ_JPKP01000008.1 |
Growth conditions and genomic DNA preparation
Pure cultures of the MP2 and MP7 isolates that were initially grown at 28 °C on YMEA plates was then cultured in Luria-Bertani broth (10 g tryptone, 5 g yeast extract, and 5 g NaCl per litre). DNA was subsequently extracted from the cultures using the Qiagen DNeasy blood and tissue kit (Qiagen, CA). DNA quality was assessed using a NanoDrop™ spectrophotometer.
Genome sequencing and assembly
The genomes of the two isolates were sequenced using mate-paired Illumina sequencing using the HiSeq Platform at the Beijing Genomics Institute. Libraries with an insert size of 500 bp were generated and sequence lengths of 90 bp in both directions were obtained. After filtering out reads with >10 % Ns and/or 25–35 bases of low quality (≤Q20), and removing adapter and duplication contamination as well as trimming read ends, approximately 850 Mb of sequence data remained per isolate. The sequence reads were assembled using Velvet [56] and the sequencing and assembly metrics are given in Table 2. Contigs generated in this way were further assembled into contiguous scaffolds by alignment against the closest complete genomes, based on BLAST, of P. vagans C9-1 [45] and the draft genome of Pantoea sp. SL1-M5 [1] using the progressive Mauve algorithm in Mauve 2.3.1 [16]. The final genomes had coverage of ca. 180 ×, where that of MP2 consisted of 16 contigs and that of MP7 consisted of 8 contigs (Figs. 3 and 4).
Fig. 3.
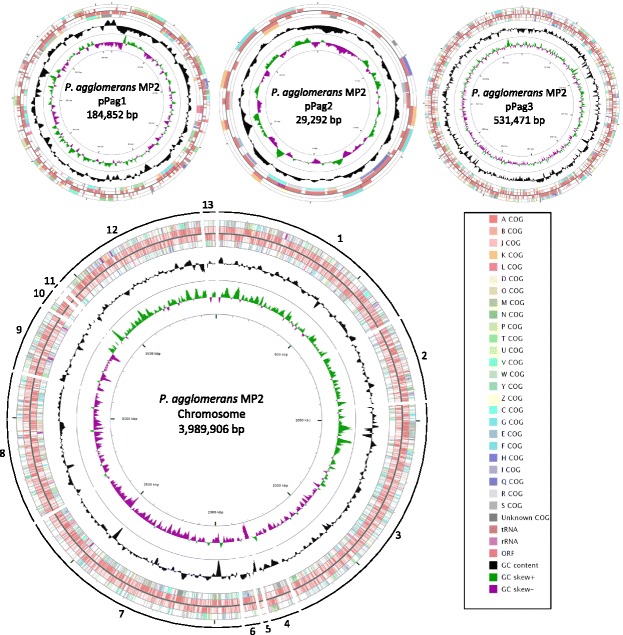
The genome structure of P. agglomerans MP2. The genome consists of 1 chromosome and 3 plasmids. The order of the contigs was based on the publicly available complete genome sequence of P. vagans C9-1 [45]. The sizes of the contigs varied significantly with the smallest being just below 5 kbp (contig 5) and the largest being just less than 800 kbp (contig 3). The open-reading frames (ORFs) for the forward and reverse strands are indicated in the inner tracks, flanked by the COG classes associated with the respective ORFs. The GC content across the genome is indicated in black, with the GC skew (calculated as [G-C/G + C]) indicated in green and purple, respectively [48]
Fig. 4.
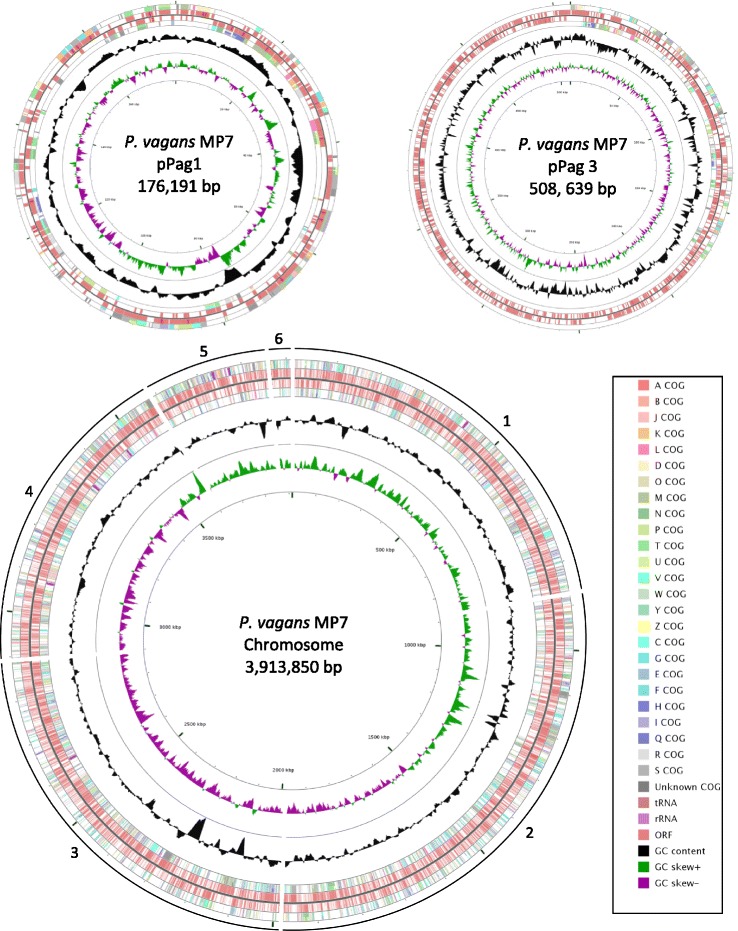
The genome structure of P. vagans MP7. The genome consists of 1 chromosome and 2 plasmids. The order of the contigs was based on the complete genome sequence of P. vagans C9-1 which is publicly available [45]. The contigs varied in size with the largest (contig 2) being approximately 1,010 kbp and the smallest (contig 6) being just below 50 kbp. The predicted ORFs are indicated in the inner tracks and are flanked with the COG classes associated with each of the ORFs. The GC content of the various regions within the genome is indicated in black, with the GC skew indicated in green and purple [48]
Genome annotation
The genomes were annotated using the RAST pipeline [5]. RAST initiated the annotation by predicting RNA molecules, followed by an initial gene prediction and placing of the genome into phylogenetic context. The most closely related genomes were used to assess protein families using FIGfams (i.e., sets of protein sequences that are similar along their full length and that likely represent isofunctional homologs). The remaining genes were then assessed against the FIGfam database [5], followed by metabolic reconstruction. The number of protein-coding genes with functional predictions was thus based on the subsystem technology of RAST.
Both genomes were also subjected to analysis on WebMGA, where comparisons to the Clusters of Orthologous Genes [50] and Protein family (pfam) databases [7] were performed with rpsblast [2]. Signal peptide prediction and transmembrane helix prediction for the protein-coding genes in the genomes were performed using Phobius [32]. CRISPR repeats were detected using the CRISPRs database [29] (Table 4).
Table 4.
Nucleotide content and gene count levels of the genomes
| Attribute | Pantoea agglomerans MP2 (total) | Pantoea vagans MP7 (total) | ||
|---|---|---|---|---|
| Value | % of totala | Value | % of totala | |
| Genome size (bp) | 4,733,829 | 100 % | 4,598,703 | 100 % |
| DNA coding (bp) | 4,043,819 | 85.4 % | 3,948,783 | 85.9 % |
| DNA G + C (bp) | 2,614,812 | 55.2 % | 2,541,699 | 55.3 % |
| DNA scaffolds | 16 | - | 8 | - |
| Total genesb | 4449 | - | 4277 | - |
| Protein coding genes | 4355 | 100 % | 4181 | 100 % |
| RNA genes | 94 | 2.2 % | 91 | 2.2 % |
| Pseudo genes | - | - | 2 | 0.1 % |
| Genes in internal clusters | - | - | - | - |
| Genes with function prediction | 3470 | 79.7 % | 3351 | 80.1 % |
| Genes assigned to COGs | 3686 | 84.6 % | 3608 | 86.3 % |
| Genes with Pfam domains | 2124 | 48.8 % | 2064 | 49.4 % |
| Genes with signal peptides | 810 | 18.6 % | 768 | 18.4 % |
| Genes with transmembrane helices | 927 | 21.3 % | 906 | 21.7 % |
| CRISPR repeats | 4 | 0.09 % | 3 | 0.07 % |
aThe percentage of total is based on either the size of the genome in base pairs or the total number of protein coding genes in the annotated genome
bAlso includes pseudogenes and other genes
Genome properties
The total genomes of P. agglomerans MP2 and P. vagans MP7 were 4,733,829 bp and 4,598,703 bp in size, respectively (Table 4; Figs. 3 and 4). The P. agglomerans MP2 genome includes three closed plasmids which show high sequence similarity and synteny to pPag1, pPag2 and pPag3 of P. vagans C9-1. The genome of P. vagans MP7 on the other hand incorporates only copies of pPag1 and pPag3. The pPag2-harbored herbicolin biosynthetic locus of P. vagans C9-1 is absent from the genomes of both MP2 and MP7 [33], while the pPATH pathogenicity island [37] is likewise absent from both strains. For P. agglomerans MP2, 85.4 % (4,043,819 bp) of the genome coded for 4,449 genes. Of these, 4,355 genes were protein-coding. For P. vagans MP7, 85.9 % (3,948,783 bp) of the genome coded for 4181 protein-coding genes. The majority of protein-coding genes had functional predictions using both RAST annotations and the COG database (Table 4). A high number of genes code for proteins that are involved in metabolism (COG codes C, G, E, F, H, I, P and Q) with fewer genes involved in all other classes (Table 5).
Table 5.
Number and proportion of genes associated with 25 COG functional categories
| P. agglomerans MP2 | P. vagans MP7 | ||||
|---|---|---|---|---|---|
| Code | Value | % of totala | Value | % of totala | Description |
| J | 196 | 4.50 % | 194 | 4.54 % | Translation |
| A | 1 | 0.02 % | 2 | 0.05 % | RNA processing and modification |
| K | 358 | 8.22 % | 331 | 7.74 % | Transcription |
| L | 147 | 3.38 % | 137 | 3.20 % | Replication, recombination and repair |
| B | - | - | - | - | Chromatin structure and dynamics |
| D | 42 | 0.96 % | 42 | 1.00 % | Cell cycle control, Cell division, chromosome partitioning |
| Y | - | - | - | - | Nuclear structure |
| V | 48 | 1.10 % | 50 | 1.17 % | Defence mechanisms |
| T | 228 | 5.24 % | 225 | 5.26 % | Signal transduction mechanisms |
| M | 239 | 5.49 % | 242 | 5.66 % | Cell wall/membrane biogenesis |
| N | 90 | 2.07 % | 92 | 2.15 % | Cell motility |
| Z | - | - | - | - | Cytoskeleton |
| W | - | - | - | - | Extracellular structures |
| U | 78 | 1.79 % | 82 | 1.92 % | Intracellular trafficking and secretion |
| O | 137 | 3.15 % | 133 | 3.11 % | Posttranslational modification, protein turnover, chaperones |
| C | 209 | 4.80 % | 206 | 4.82 % | Energy production and conversion |
| G | 395 | 9.07 % | 378 | 8.84 % | Carbohydrate transport and metabolism |
| E | 405 | 9.30 % | 405 | 9.47 % | Amino acid transport and metabolism |
| F | 96 | 2.20 % | 100 | 2.34 % | Nucleotide transport and metabolism |
| H | 164 | 3.77 % | 165 | 3.86 % | Coenzyme transport and metabolism |
| I | 117 | 2.69 % | 106 | 2.48 % | Lipid transport and metabolism |
| P | 244 | 5.60 % | 248 | 5.80 % | Inorganic ion transport and metabolism |
| Q | 77 | 1.77 % | 69 | 1.61 % | Secondary metabolites biosynthesis, transport and catabolism |
| R | 450 | 10.33 % | 430 | 10.05 % | General function prediction only |
| S | 393 | 9.02 % | 387 | 9.05 % | Function unknown |
| - | 669 | 15.36 % | 669 | 15.64 % | Not in COGs |
aThe total is based on the total number of predicted protein coding genes in the annotated genomes
Insights from the genome sequences
The genomes of the sequenced isolates were compared to the publicly available genomes of P. agglomerans 190 and P. vagans C9-1 [45] to determine the average nucleotide identity [28, 43] values between the isolates (Table 6). The ANI calculations were done with JSpecies [43] using the BLAST function, which is based on fragmenting the genomic sequence into pieces of 1,020 nucleotides long and performing similarity searches to determine homology between the genomic fragments.
Table 6.
Average nucleotide identity (ANI) values for the sequenced isolates and additional strains representative of the lineages of Pantoea
| P. agglomerans 190 | P. agglomerans MP2 | P. vagans C9-1 | P. vagans MP7 | P. anthophila 11-2 | P. ananatis LMG 2665 | P. stewartii sp. stewartii DC283 | P. stewartii sp. indologenes LMG2632 | P. dispersa EGD-AAK13 | P. rwandensis ND04 | |
|---|---|---|---|---|---|---|---|---|---|---|
| P. agglomerans 190 | --- | 98.06 | 90.66 | 90.83 | 87.96 | 78.79 | 78.87 | 78.73 | 78.83 | 78.05 |
| P. agglomerans MP2 | 98.75 | --- | 91.88 | 91.81 | 89.08 | 79.89 | 79.72 | 79.64 | 79.89 | 78.95 |
| P. vagans C9-1 | 90.66 | 91.12 | --- | 96.62 | 87.56 | 78.79 | 78.81 | 78.75 | 78.75 | 78.1 |
| P. vagans MP7 | 90.87 | 91.17 | 96.71 | --- | 87.57 | 78.9 | 78.84 | 78.69 | 78.6 | 78.11 |
| P. anthophila 11-2 | 88.03 | 88.49 | 87.65 | 87.59 | --- | 78.97 | 78.9 | 78.72 | 78.92 | 77.93 |
| P. ananatis LMG 2665 | 78.65 | 79.28 | 78.71 | 78.77 | 78.81 | --- | 83.77 | 83.62 | 77.19 | 76.69 |
| P. stewartii subsp. stewartii DC283 | 79.01 | 79.48 | 78.99 | 78.98 | 79.05 | 83.87 | --- | 98.99 | 77.54 | 76.92 |
| P. stewartii subsp. indologenes LMG2632 | 78.58 | 79.2 | 78.59 | 78.6 | 78.57 | 83.6 | 98.72 | --- | 77.13 | 76.61 |
| P. dispersa EGD-AAK13 | 78.68 | 79.35 | 78.69 | 78.64 | 78.85 | 77.3 | 77.37 | 77.27 | --- | 82.97 |
| P. rwandensis ND04 | 78.03 | 78.44 | 78.02 | 78.01 | 77.97 | 76.81 | 76.78 | 76.73 | 83.02 | --- |
The number of shared genes within and between species ranged from 3,400 to 3,500. Based on the ANI values, the isolates grouped with representatives of the designated species, as species cut-off values are suggested at 95 % for ANI [28].
Conclusion
The two bacteria described in this report were phylogenetically and genomically very closely related, but clearly belonged to different species. The ANI values supported the identification of isolates MP2 and MP7 as P. agglomerans and P. vagans, respectively.
Their similarity in genomic content may allow P. agglomerans and P. vagans to occupy the same or overlapping niches and perform the same or similar functional roles. This is consistent with what has been observed before where isolates of P. agglomerans and P. vagans occur in similar environmental niches and may even co-occur in the same environment [40]. Although recombination among micro-organisms occupying the same niche is common [3, 27], our data indicated that P. agglomerans and P. vagans have remained sufficiently distinct to identify them as separate species. This suggests that their ability to occupy the same niche is likely a function of their shared genes [13, 30, 35], but that the integrity of their individual genomic complements is protected by barriers that limit genetic exchange or gene flow between these species [14, 47].
Members of the genus Pantoea are often considered generalists that are isolated from a wide variety of environments [10, 19, 26]. Large metabolic repertoires (unpublished data, Marike Palmer) may allow species of this genus to form opportunistic associations with many potential hosts including insects [8, 53]. These associations, as with the biocontrol isolates [41], may be based on the Pantoea isolates outcompeting potentially harmful bacteria in the respective environments as microbial antagonists. This is likely also true for P. agglomerans and P. vagans and their association with termites, however recent evidence (unpublished data, Michael Poulsen) suggest that the bacterial species may provide nitrogen fixation capabilities to the termites. It is possible that the antimicrobial [21, 22, 41] and metabolic capabilities (especially pectinolytic and other carbohydrate degrading enzymes) [8] of these bacteria allow them to outcompete other, potentially harmful micro-organisms, while also providing carbohydrates and other compounds for the termites to utilize [20].
Acknowledgements
The authors would like to acknowledge funding from the Danish Council for Independent Research, Natural Sciences (STENO grant: Michael Poulsen), the National Research Foundation (NRF) (RCA Fellowship: Pieter De Maayer) and the NRF/Dept. of Science and Technology Centre of Excellence in Tree Health Biotechnology (CTHB), South Africa. We would like to thank Wai-Yin Chan for assistance with the annotation of the genomes.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MPa performed annotations, constructed genome maps, calculated the genome metrics and drafted the manuscript. PDM constructed the genome assemblies, assisted with the submission of sequences, provided guidance for the annotations and revised the manuscript. MPo performed collections and isolations of the isolates and provided support with drafting and revising the manuscript. EVZ provided organism information, performed culturing of the organisms and assisted with submission of isolates and revision of the manuscript. ETS, TAC and SNV participated in the coordination of the study, provided support with interpretation of the data and helped draft the manuscript. All authors read and approved the final manuscript.
References
- 1.Adams AS, Jordan MS, Adams SM, Suen G, Goodwin LA, Davenport KW, Currie CR and Raffa KF. Cellulose-degrading bacteria associated with the invasive woodwasp Sirex noctilio. ISME J. 2011;5:1323–31. [DOI] [PMC free article] [PubMed]
- 2.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, and Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. [DOI] [PMC free article] [PubMed]
- 3.Andam CP, Gogarten JP. Biased gene transfer in microbial evolution. Nat Rev Microbiol. 2011;9:543–55. doi: 10.1038/nrmicro2593. [DOI] [PubMed] [Google Scholar]
- 4.Aylward FO, Suen G, Biedermann PHW, Adams AS, Scott JJ, Malfatti SA, Glavina del Rio T, Tringe SG, Poulsen M, Raffa KF, Klepzig KD, Currie CR. Convergent bacterial microbiotas in the fungal agricultural systems of insects. mBio. 2014;5. doi:10.1128/mBio.02077-14. [DOI] [PMC free article] [PubMed]
- 5.Aziz R, Bartels D, Best A, DeJongh M, Disz T, Edwards R, Formsma K, Gerdes S, Glass E, Kubal M, Meyer F, Olsen G, Olson R, Osterman A, Overbeek R, McNeil L, Paarmann D, Paczian T, Parrello B, Pusch G, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A and Zagnitko O. The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genomics. 2008;9:75. [DOI] [PMC free article] [PubMed]
- 6.Barash I, Manulis-Sasson S. Recent evolution of bacterial pathogens: the gall-forming Pantoea agglomerans case. Annual Review of Phytopathology. 2009;47:133–52. doi: 10.1146/annurev-phyto-080508-081803. [DOI] [PubMed] [Google Scholar]
- 7.Bateman A, Birney E, Durbin R, Eddy SR, Howe KL, Sonnhammer ELL. The Pfam protein families database. Nucleic Acids Res. 2000;28:263–6. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Behar A, Jurkevitch E, Yuval B. Bringing back the fruit into fruit fly–bacteria interactions. Mol Ecol. 2008;17:1375–86. doi: 10.1111/j.1365-294X.2008.03674.x. [DOI] [PubMed] [Google Scholar]
- 9.Brady CL, Cleenwerck I, Venter SN, Engelbeen K, De Vos P and Coutinho TA: Emended description of the genus Pantoea, description of four species from human clinical samples, Pantoea septica sp. nov., Pantoea eucrina sp. nov., Pantoea brenneri sp. nov. and Pantoea conspicua sp. nov., and transfer of Pectobacterium cypripedii (Hori Brenner et al. 1973 emend. Hauben et al. 1998 to the genus as Pantoea cypripedii comb. nov. Int J Syst Evol Microbiol. 1911;2010(60):2430–40. doi: 10.1099/ijs.0.017301-0. [DOI] [PubMed] [Google Scholar]
- 10.Brady CL, Venter SN, Cleenwerck I, Engelbeen K, Vancanneyt M, Swings J, and Coutinho TA. Pantoea vagans sp. nov., Pantoea eucalypti sp. nov., Pantoea deleyi sp. nov. and Pantoea anthophila sp. nov. Int J Syst Evol Microbiol. 2009;59:2339–45. [DOI] [PubMed]
- 11.Brady CL, Cleenwerck I, Van der Westhuizen L, Venter SN, Coutinho TA, De Vos P. Pantoea rodasii sp. nov., Pantoea rwandensis sp. nov. and Pantoea wallisii sp. nov., isolated from Eucalyptus. International journal of systematic and evolutionary microbiology. 2012;62:1457–64. doi: 10.1099/ijs.0.032615-0. [DOI] [PubMed] [Google Scholar]
- 12.Cataño JC, Echeverri LM, Szela C. Bacterial contamination of clothes and environmental items in a third-level hospital in Colombia. Interdiscip Perspect Infect Dis. 2012 doi: 10.1155/2012/507640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coenye T, Gevers D, Van de Peer Y, Vandamme P, Swings J. Towards a prokaryotic genomic taxonomy. FEMS Microbiol Rev. 2005;29:147–67. doi: 10.1016/j.fmrre.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 14.Cohan FM. What are bacterial species? Annu Rev Microbiol. 2002;56:457–87. doi: 10.1146/annurev.micro.56.012302.160634. [DOI] [PubMed] [Google Scholar]
- 15.Cruz AT, Cazacu AC, Allen CH. Pantoea agglomerans – a plant pathogen causing human disease. J Clin Microbiol. 2007;45(6):1989–1992. [DOI] [PMC free article] [PubMed]
- 16.Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Baere T, Verhelst R, Labit C, Verschraegen G, Wauters G, Claeys G, and Vaneechoutte M. Bacteremic infection with Pantoea ananatis. J Clin Microbiol. 2004;42:4393–5. [DOI] [PMC free article] [PubMed]
- 18.De Champs C, Le Seaux S, Dubost JJ, Boisgard S, Sauvezie B, Sirot J. Isolation of Pantoea agglomerans in two cases of septic monoarthritis after plant thorn and wood sliver injuries. J Clin Microbiol. 2000;38:460–1. doi: 10.1128/jcm.38.1.460-461.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Maayer P, Chan W-Y, Blom J, Venter S, Duffy B, Smits T, and Coutinho TA. The large universal Pantoea plasmid LPP-1 plays a major role in biological and ecological diversification. BMC Genomics. 2012;13:625. [DOI] [PMC free article] [PubMed]
- 20.De Vries EJ, Jacobs G, Sabelis MW, Menken SB, Breeuwer JA. Diet–dependent effects of gut bacteria on their insect host: the symbiosis of Erwinia sp. and western flower thrips. Proc R Soc Lond Ser B-Biol Sci. 2004;271:2171–8. doi: 10.1098/rspb.2004.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dillon RJ, Charnley AK. Chemical barriers to gut infection in the desert locust: in vivo production of antimicrobial phenols associated with the bacterium Pantoea agglomerans. J Invertebr Pathol. 1995;66:72–5. doi: 10.1006/jipa.1995.1063. [DOI] [Google Scholar]
- 22.Dillon RJ, Dillon VM. The gut bacteria of insects: nonpathogenic interactions. Annu Rev Entomol. 2004;49:71–92. doi: 10.1146/annurev.ento.49.061802.123416. [DOI] [PubMed] [Google Scholar]
- 23.Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey’s manual of systematic bacteriology, second edition, volume 2, part B. New York: Springer; 2005. p. 1. [Google Scholar]
- 24.Garrity GM, Bell JA, Lilburn T. Class III. Gammaproteobacteria class. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey’s manual of systematic bacteriology, second edition, volume 2, part B. New York: Springer; 2005. p. 1. [Google Scholar]
- 25.Garrity GM, Holt JG. Taxonomic outline of the Archaea and Bacteria. In: Garrity GM, Boone DR, Castenholz RW, editors. Bergey’s manual of systematic bacteriology, second edition, volume 2, part B. 2nd edition. New York: Springer; 2001. [Google Scholar]
- 26.Gavini F, Mergaert J, Beji A, Mielcarek C, Izard D, Kersters K, and De Ley J. Transfer of Enterobacter agglomerans (Beijerinck 1888) Ewing and Fife 1972 to Pantoea gen. nov. as Pantoea agglomerans comb. nov. and description of Pantoea dispersa sp. nov. Int J Syst Bacteriol. 1989;39:337–45.
- 27.Gogarten JP, Doolittle WF, Lawrence JG. Prokaryotic evolution in light of gene transfer. Mol Biol Evol. 2002;19:2226–38. doi: 10.1093/oxfordjournals.molbev.a004046. [DOI] [PubMed] [Google Scholar]
- 28.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57:81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 29.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–7. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hacker J, Carniel E. Ecological fitness, genomic islands and bacterial pathogenicity. EMBO reports. 2001;2:376–81. doi: 10.1093/embo-reports/kve097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hollis DG, Hickman FW, Fanning GR, Farmer JJ, Weaver RE, Brenner DJ. Tatumella ptyseos gen. nov., sp. nov., a member of the family Enterobacteriaceae found in clinical specimens. J Clin Microbiol. 1981;14:79–88. doi: 10.1128/jcm.14.1.79-88.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Käll L, Krogh A, Sonnhammer ELL. Advantages of combined transmembrane topology and signal peptide prediction: the Phobius web server. Nucleic Acids Res. 2007;35:W429–32. doi: 10.1093/nar/gkm256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kamber T, Smits TM, Rezzonico F, Duffy B. Genomics and current genetic understanding of Erwinia amylovora and the fire blight antagonist Pantoea vagans. Trees. 2012;26:227–38. doi: 10.1007/s00468-011-0619-x. [DOI] [Google Scholar]
- 34.Kim WS, Gardan L, Rhim SL, Geider K. Erwinia pyrifoliae sp. nov., a novel pathogen that affects Asian pear trees (Pyrus pyrifolia Nakai) International Journal of Systematic Bacteriology. 1999;49(2):899–906. doi: 10.1099/00207713-49-2-899. [DOI] [PubMed] [Google Scholar]
- 35.Lan R, Reeves PR. Intraspecies variation in bacterial genomes: the need for a species genome concept. Trends Microbiol. 2000;8:396–401. doi: 10.1016/S0966-842X(00)01791-1. [DOI] [PubMed] [Google Scholar]
- 36.Lanave C, Preparata G, Sacone C, Serio G. A new method for calculating evolutionary substitution rates. J Mol Evol. 1984;20:86–93. doi: 10.1007/BF02101990. [DOI] [PubMed] [Google Scholar]
- 37.Manulis S, Barash I. Pantoea agglomerans pvs. gypsophilae and betae, recently evolved pathogens? Mol Plant Pathol. 2003;4:307–14. doi: 10.1046/j.1364-3703.2003.00178.x. [DOI] [PubMed] [Google Scholar]
- 38.Mergaert J, Verdonck L, Kersters K. Transfer of Erwinia ananas, synonym, Erwinia uredovora and Erwinia stewartii to the genus Pantoea emend. as Pantoea ananas, Serrano 1928 comb. nov. and Pantoea stewartii, Smith 1898 comb. nov., respectively, and description of Pantoea stewartii subsp. indologenes subsp. nov. Int J Syst Bacteriol. 1993;43:162–73. doi: 10.1099/00207713-43-1-162. [DOI] [Google Scholar]
- 39.Mergaert J, Hauben L, Cnockaert MC, Swings J. Reclassification of non-pigmented Erwinia herbicola strains from trees as Erwinia billingiae sp. nov. Int J Syst Bacteriol. 1999;49:377–83. doi: 10.1099/00207713-49-2-377. [DOI] [PubMed] [Google Scholar]
- 40.Otani S, Mikaelyan A, Nobre T, Hansen LH, Koné NA, Sørensen SJ, Aanen DK, Boomsma JJ, Brune A and Poulsen M. Identifying the core microbial community in the gut of fungus-growing termites. Mol Ecol. 2014;23:4631–44. [DOI] [PubMed]
- 41.Pusey PL. Biological control agents for fire blight of apple compared under conditions limiting natural dispersal. Plant Dis. 2002;86:639–44. doi: 10.1094/PDIS.2002.86.6.639. [DOI] [PubMed] [Google Scholar]
- 42.Rahn O. New principles for the classification of bacteria. Zentralblatt für Bakteriologie, Parasitenkunde, Infektionskrankheiten und Hygiene. Abteilung II. 1937;96:273–86. [Google Scholar]
- 43.Richter M, Rosselló-Móra R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci. 2009;106:19126–31. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Skerman VBD, McGowan V, Sneath PHA. Approved lists of bacterial names. Int J Syst Bacteriol. 1980;30:225–420. doi: 10.1099/00207713-30-1-225. [DOI] [PubMed] [Google Scholar]
- 45.Smits THM, Rezzonico F, Kamber T, Goesmann A, Ishimaru CA, Stockwell VO, Frey JE and Duffy B. Genome sequence of the biocontrol agent Pantoea vagans strain C9-1. J Bacteriol. 2010;192:6486–7. [DOI] [PMC free article] [PubMed]
- 46.Smits THM, Rezzonico F, Pelludat C, Goesmann A, Frey JE, Duffy B. Genomic and phenotypic characterization of a nonpigmented variant of Pantoea vagans biocontrol strain C9-1 lacking the 530-kb megaplasmid pPag3. FEMS Microbiol Letters. 2010;308:48–54. doi: 10.1111/j.1574-6968.2010.01994.x. [DOI] [PubMed] [Google Scholar]
- 47.Sorek R, Zhu Y, Creevey CJ, Francino MP, Bork P, Rubin EM. Genome-wide experimental determination of barriers to horizontal gene transfer. Science. 2007;318:1449–52. doi: 10.1126/science.1147112. [DOI] [PubMed] [Google Scholar]
- 48.Stothard P, Wishart DS. Circular genome visualization and exploration using CGView. Bioinformatics. 2004;21:537–9. doi: 10.1093/bioinformatics/bti054. [DOI] [PubMed] [Google Scholar]
- 49.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol Biol Evol. 2013;30:2725–9. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–6. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Validation of publication of new names and new combinations previously effectively published outside the IJSEM. List no. 106. Int J Syst Evol Microbiol. 2005;55:2235–38. [DOI] [PubMed]
- 52.Validation of the publication of new names and new combinations previously effectively published outside the IJSB. List No. 8. Int J Syst Bacteriol. 1982;32:266–68.
- 53.Vorwerk S, Martinez-Torres D, Forneck A. Pantoea agglomerans-associated bacteria in grape phylloxera (Daktulosphaira vitifoliae, Fitch) Agric For Entomol. 2007;9:57–64. doi: 10.1111/j.1461-9563.2006.000319.x. [DOI] [Google Scholar]
- 54.Whitman WB, Goodfellow M, Kämpfer P, Busse HJ, Trujillo ME, Suzuki K, and Ludwig W. Bergey’s manual® of systematic bacteriology (vol 5). New York: Springer; 2012.
- 55.Winslow CEA, Broadhurst J, Buchanan RE, Krumwiede C, Rogers LA, Smith GH. The families and genera of the bacteria: final report of the committee of the society of American bacteriologists on characterization and classification of bacterial types. J Bacteriol. 1920;5:191–229. doi: 10.1128/jb.5.3.191-229.1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Y, Qiu S. Examining phylogenetic relationships of Erwinia and Pantoea species using whole genome sequence data. Antonie van Leeuwenhoek. 2015;108:1037–46. doi: 10.1007/s10482-015-0556-6. [DOI] [PubMed] [Google Scholar]