

Abstract
Background
Drug discovery and design are important research fields in bioinformatics. Enumeration of chemical compounds is essential not only for the purpose, but also for analysis of chemical space and structure elucidation. In our previous study, we developed enumeration methods BfsSimEnum and BfsMulEnum for tree-like chemical compounds using a tree-structure to represent a chemical compound, which is limited to acyclic chemical compounds only.
Results
In this paper, we extend the methods, and develop BfsBenNaphEnum that can enumerate tree-like chemical compounds containing benzene rings and naphthalene rings, which include benzene isomers and naphthalene isomers such as ortho, meta, and para, by treating a benzene ring as an atom with valence six, instead of a ring of six carbon atoms, and treating a naphthalene ring as two benzene rings having a special bond. We compare our method with MOLGEN 5.0, which is a well-known general purpose structure generator, to enumerate chemical structures from a set of chemical formulas in terms of the number of enumerated structures and the computational time. The result suggests that our proposed method can reduce the computational time efficiently.
Conclusions
We propose the enumeration method BfsBenNaphEnum for tree-like chemical compounds containing benzene rings and naphthalene rings as cyclic structures. BfsBenNaphEnum was from 50 times to 5,000,000 times faster than MOLGEN 5.0 for instances with 8 to 14 carbon atoms in our experiments.
Keywords: Benzene ring, Naphthalene ring, Enumeration, Breadth-first search
Background
Enumeration of chemical compounds is important in bioinformatics, and has been adapted to several applications such as drug discovery and design [1–3], structure elucidation [4–6], and analyses of chemical spaces [7–13]. It is defined as a problem of generating all non-redundant chemical structures satisfying some constraints. For example, a chemical formula, which consists of the number of each atom included in the compound, is given as an input. There are several algorithms for enumerating chemical compounds from a chemical formula and most of them use a molecular graph to represent a chemical compound, where the nodes and edges of the graph refer to atoms and bonds of the chemical compound, respectively. Some of those algorithms are claimed to be able to enumerate various chemical structures without restriction of the structure, such as MOLGEN [14] and Open Molecule Generator (OMG) [15]. It was reported that OMG is able to deal with different valences for a kind of atom, and was not efficient for several instances compared with MOLGEN. While the remaining ones, such as EnuMol [16, 17] as well as BfsSimEnum and BfsMulEnum [18], have a limitation of the structure of enumerated compounds, such as acyclic compounds for BfsSimEnum and BfsMulEnum and compounds with no cycle except for benzene rings for EnuMol, the methods consume significantly less computational time. There are also related application softwares, e.g. SmiLib [19] and CLEVER [20], that generate chemical compounds from given fragments. The limitation of these tools is that they require a library of desired chemical fragments, which can be generated by the enumeration tool.
Our previous methods, BfsSimEnum and BfsMulEnum, use a tree structure, instead of a general graph, to represent a chemical compound and call it a molecular tree so they can generate only tree-like chemical compounds. In this work, we develop BfsBenNaphEnum, which aims to reduce the limitation of previous methods by extending them such that they can enumerate chemical compounds containing only benzene rings and naphthalene rings as cyclic structures, which are six carbon atoms cyclic structures and ten carbon atoms bicyclic structures, respectively. Pólya proposed a group-theoretic method for isomer counting of single cyclic structures such as a benzene ring, a naphthalene ring, and an anthracene ring using the cycle index, from which many studies followed [21]. However, structures enumerated by these methods are restricted to certain types. Indeed, Meringer wrote that up to now the only way to calculate the number of isomers belonging to an arbitrary molecular formula is to use structure generators [22]. Suzuki et al. considered the problem of enumerating structures having monocyclic graph structures, each of which has exactly one cycle [23]. An enumeration method for tree-like chemical compounds containing only benzene rings as cyclic structures has been implemented on EnuMol web server (http://sunflower.kuicr.kyoto-u.ac.jp/tools/enumol/). On the other hand, our method can enumerate compounds containing naphthalene rings in addition to benzene rings. Moreover, the proposed algorithm can calculate the number of benzene rings and naphthalene rings from chemical formula, while users have to specify the number of benzene rings in EnuMol.
Chemical structures considered in this study can be represented by a molecular tree, where a benzene ring is converted to a node with valence six and a naphthalene ring is considered as two benzene nodes having a special bond. We name that special bond as a merge bond. Since a merge bond merges two carbon atoms of two benzene rings together, it reduces the number of carbon atoms with free valence electron of two benzene rings by two so we represent a merge bond by a double-edge. Moreover, benzene nodes cannot have double bonds with other nodes because they bond with other non-benzene atoms by a single bond [24]. This means that a double-edge represents a double bond if it connects two non-benzene nodes, while it represents a merge bond if it connects two benzene nodes. Therefore, bonds in a benzene ring and a naphthalene ring are considered as the same bond and Kekulé representation is not included in this work. Besides, this work uses a two-dimensional molecular tree to represent a chemical structure so it cannot deal with stereoisomers. For tautomeric, this work considers two structures in a pair of tautomeric as non- redundant compounds and generates both of them.
BfsSimEnum and BfsMulEnum are modified to return a set of molecular trees as the output, given a chemical formula, the number of benzene rings, and the number of naphthalene rings. After that, an attribute called carbon position list is added into benzene nodes in a molecular tree to represent the way that benzene nodes bond with their adjacent nodes. This attribute is important because bonding with different carbon atoms in a benzene ring may result in different chemical structures. Finally, for each molecular tree from BfsSimEnum and BfsMulEnum, we generate a set of molecular trees whose nodes adjacent to benzene nodes are labeled with a carbon position such that all chemical structures are enumerated without redundancy based on normal form rule.
For evaluating our proposed method, we perform computational experiments for several instances, and compare the execution time by our method with that by MOLGEN. We show that our proposed method is efficient for enumerating chemical compounds containing benzene rings and naphthalene rings, and is from 50 times to 5,000,000 times faster than MOLGEN for several instances in our experiments.
Preliminaries
Enumeration problem
Let Σ be a finite set of labels of atoms, for example, Σ={C,N,O,H }, where ‘C’, ‘N’, ‘O’, and ‘H’ denote carbon, nitrogen, oxygen, and hydrogen atoms, respectively. A molecular graph is defined as a multi-graph G(V, E), where V is a set of nodes and E is a set of multi-edges, also denoted by V(G) and E(G), respectively. Each node is labeled with an atom-label in Σ, while each edge represents the bond between two atoms and the multiplicity of edge represents the bond type. The degree of each node is equal to the valence of its atom. Let deg(v) and l(v) be the degree and the label of node v, respectively. Let val(li) be the valence of the atom represented by label li in Σ. It should be noted that there exist different valences for a kind of atom, for example, carbon atoms of CO2 and CO. For this case, it is sufficient to put two distinct labels C and C(2) in Σ, and to define val(C)=4 and val(C(2))=2. Let num(G,li) be the total number of nodes labeled with label li in molecular graph G. Then, the enumeration problem is defined as follows.
Problem1.
Given the numbers of atoms for all labels li∈Σ, the number nb of benzene rings, and the number nn of naphthalene rings, enumerate all non-redundant molecular graphs G such that for all li∈Σ, deg(v)=val(l(v)) for all nodes v∈V(G), and G includes exactly nb benzene rings, nn naphthalene rings, and no other cyclic structures. It must be noted that nb and nn can be zero.
In the case that the input chemical formula contains five or less carbon atoms, BfsStructEnum can enumerate only tree-like chemical compounds by specifying the number of benzene rings and the number of naphthalene rings to be zero. Because we enumerate molecular trees such that degree of each node equals to valence of atom label of that node, charged molecules cannot be enumerated automatically. However, they can still be enumerated by specifying a charged atom as a new kind of atom type with appropriate valence value.
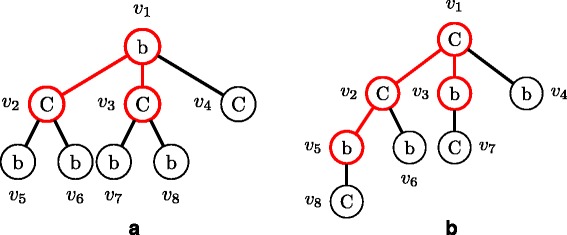
Since our enumeration methods deal with a chemical compound as a node-labeled rooted ordered tree for efficient enumeration, we contract cyclic structures appearing in a molecular graph to single nodes. Concretely, we contract a benzene ring to a node, called benzene node, labeled with a special label ‘b’, and contract a naphthalene ring to two benzene nodes connected by a special bond, called merge bond, represented by a double edge (see Fig. 1). Since six carbon atoms contained in a benzene ring are contracted into a benzene node, we need to remember which carbon atom in the benzene ring connects to its adjacent node in a molecular graph. Hence, we add an attribute called carbon position list to each benzene node. Figure 1b shows examples of carbon position lists using numbers assigned to carbon atoms in benzene rings in Fig. 1a. We call such a node-labeled rooted ordered tree whose benzene nodes are attributed with carbon position lists a carbon position-assigned molecular tree. We enumerate carbon position-assigned molecular trees instead of molecular graphs.
Fig. 1.
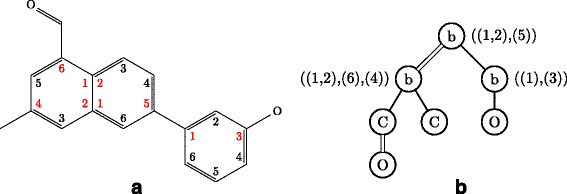
Example of a molecular graph including benzene rings and naphthalene rings. a A molecular graph including one benzene ring and one naphthalene ring. b A rooted tree contracted from the left graph. It is noted that hydrogen atoms are omitted
Center-rooted and left-heavy
In our previous work, we defined the normal form for molecular trees without any cyclic structures using center-rooted and left-heavy to avoid its redundant generation. In this work, we also utilize center-rooted and left-heavy for carbon position-assigned molecular trees, of which properties do not depend on carbon position lists.
A molecular tree T is called center-rooted if its root is the center node (see Fig. 2a) or one endpoint of the center edge of the longest path in T (see Fig. 2b). The center can be either a node or an edge depending on the length of the longest path.
Fig. 2.
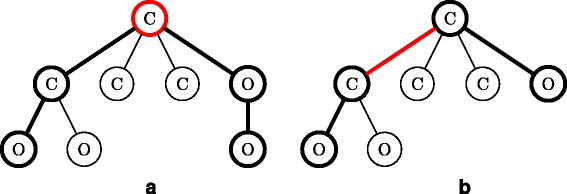
Illustration of center-rooted molecular trees. a Center of the longest path is a node. b Center of the longest path is an edge. The thick lines indicate one of the longest paths and the center node/edge is shown in red
In order to define a left-heavy tree, atom-labels must be ordered so that they can be compared with each other, for example, b >C>N>O>H for Σ={b,C,N,O,H }, where ‘b’ denotes a special atom representing a benzene ring. Let T(u) be the ordered subtree rooted at u in T. Let u and v be two nodes in a molecular tree T, (u1,u2,…,uh) and (v1,v2,…,vk) be lists of child nodes of u and v, respectively. It is defined that T(u)>sT(v) if l(u)>l(v) (Fig. 3a) or there exists an integer i such that T(uj)=sT(vj) for all j<i and (T(ui)>sT(vi) (Fig. 3b) or i=k+1≤h (Fig. 3c)). If T(u)>sT(v) or T(v)>sT(u) does not hold, it is said that T(u)=sT(v).
Fig. 3.
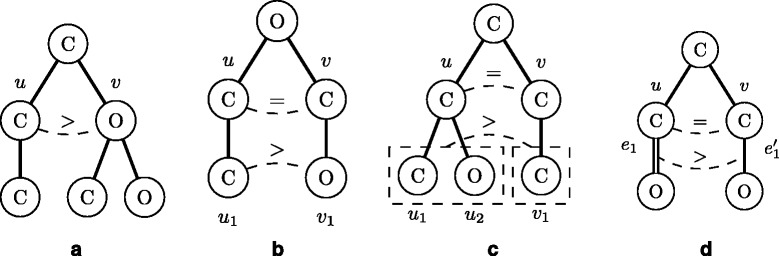
Illustration of three molecular trees such that T(u)>s T(v) or T(u)>m T(v). a l(u)>l(v). b l(u)=l(v), T(u 1)>s T(v 1). c l(u)=l(v), T(u 1)=s T(v 1), h=2>1=k. d T(u)=s T(v), m u l(e 1)>m u l(e1′)
Let mul(e) and mul(u,v) be the multiplicity of edge e=(u,v). Let (e1,e2,…,em) and () be two lists of edges in T(u) and T(v) in breadth-first search (BFS) order (see Fig. 4), respectively. T(u)>mT(v) if T(u)>sT(v), or if T(u)=sT(v) and there exists an integer i such that mul(ej)=mul(ej′) for all j<i, and mul(ei)>mul(ei′) (Fig. 3d). If T(u)>mT(v) or T(v)>mT(u) does not hold, it is said that T(u)=mT(v).
Fig. 4.
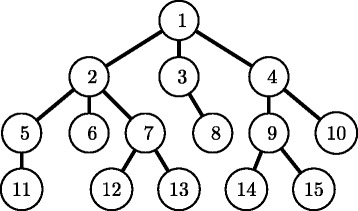
Illustration of breadth-first search (BFS) order. Numbers indicate BFS order for this example
Let child(v)=(v1,v2,…) be a list of all child nodes of node v in BFS order. It is defined that a molecular tree T is left-heavy if T(vi)≥mT(vi+1) holds for all nodes v in T and all i=1,…,|child(v)|−1.
It should be noted that center-rooted and left-heavy are different from centroid-rooted and left-heavy defined by Fujiwara et al. [16], for example, the molecular tree in Fig. 1b is center-rooted and is not centroid-rooted because the number of nodes in the left subtree by removing the root, 4, is more than (total number of nodes −1)/2=(7−1)/2=3. In addition, their left-heavy is defined using depth-first search order, not our breadth-first search order.
Carbon position list
Let s=(v1,v2,…,vn) be a list of nodes, |s| and s[ i] denote the size and the i-th element of s, respectively. Let Tsub(v1,v2) be the left-heavy tree rooted at v1 that consists of the connected component including v1 when the edge (v1,v2) is deleted from T (see Fig. 5). Tsub(v1,v2)=mT(v1) if v1 is a child of v2 in T. Let index(v,T) be the order of v∈V(T) by traversing a center-rooted left-heavy molecular tree T with BFS order, which is also denoted by index(v) if T is clear.
Fig. 5.
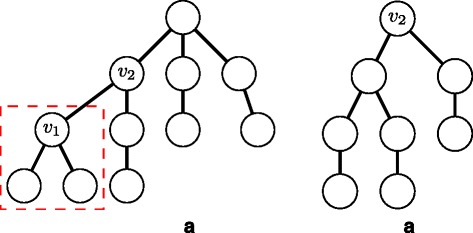
Illustration of subtree T sub(v 1,v 2). a A molecular tree T and T sub(v 1,v 2), which is surrounded by a red rectangle. b T sub(v 2,v 1)
Proposition1.
For a node v that has the parent node vp and a child node vc in a center-rooted molecular tree T, Tsub(vp,v)≠mTsub(vc,v).
Proof.
The height of Tsub(vp,v) is larger than that of Tsub(vc,v) because T is center-rooted. Hence, Tsub(vp,v) is always different from Tsub(vc,v).
We define an equality T1=CT2 for two rooted carbon-position assigned trees T1 and T2 if T1=mT2, and Cv1T1=Cv2T2 for all benzene nodes v1∈V(T1), where v2∈V(T2) satisfies index(v1,T1)=index(v2,T2), and is a list of lists, called a carbon position list explained later, for a benzene node v in T. For convenience, we define another equality by removing the condition that Cr1T1=Cr2T2 for the roots r1 and r2 of T1 and T2, respectively, from the conditions of T1=CT2, if r1 and r2 are benzene nodes.
For a node v having the parent vp and a child vc, Tsub(vp,v)≠CTsub(vc,v) if Tsub(vp,v)≠mTsub(vc,v). Hence, only carbon position lists of descendent benzene nodes are needed to determine whether or not for child nodes and of v.
Definition1.
An adjacent node list of a benzene node v in a carbon position-assigned molecular tree T is defined as a list of lists of nodes adjacent to v using carbon position lists of descendent benzene nodes such that
for all i,
if ,
for all i,j,
if (v, v′) is a merge bond for some i,
if (v, v′) is not a merge bond, and .
Figure 6 shows examples of carbon position-assigned molecular trees, where benzene node v1 in each tree has adjacent nodes v2,v3,v4,v5. Then, and index(v4)<index(v5), so we have . Also for T2, . For T3, because (v2,v1) is a merge bond. If (v2,v1) is not a merge bond and Cv2T3=Cv3T3, then .
Fig. 6.
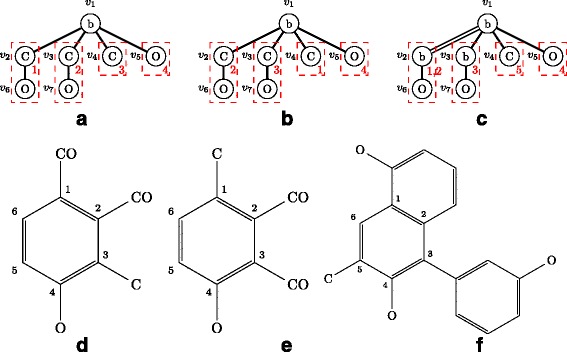
Examples of adjacent node lists and carbon position lists. a T 1. b T 2. c T 3. d Molecular graph of T 1. e Molecular graph of T 2. f Molecular graph of T 3. Red numbers represent carbon positions of node v 1
Proposition2.
For a benzene node v that has the parent node vp in a center-rooted molecular tree T, .
Proof.
If v has no child, it is clear because the adjacent node of v is only vp. We assume that v has a child vc. From Proposition 1 and index(vp)<index(vc), always holds.
A carbon position list of a benzene node v in T is a list of lists, where is a list of carbon positions of the nodes in . It is sufficient to enumerate in ascending order because each node in has the same subtree. If is a merge bond, has two carbon positions instead of one as usual. It should be noted that and two carbon positions are assigned for a merge bond because a naphthalene ring shares two carbon atoms between two benzene rings. In the examples of Fig. 6, for , for , for .
Definition2.
An adjacent node list for a naphthalene ring with two benzene nodes v1, v2, where (v1,v2) is a merge bond, is defined as a list of lists of nodes adjacent to v1 or v2 except v1 and v2 such that
for all i,
if ,
for all i,j,
if , where bn(v) is v1 or v2 that is adjacent to v.
For a benzene node v2 that is connected by a merge bond with the parent node v1, we suppose that the carbon atoms having positions 1,2 in v2 are connected with the carbon atoms having positions in v1, respectively, where x takes an integer between 1 and 6, and (see Fig. 7a). Here, consider the case that v1 has the parent node vp. If T is in normal form (Definition 6), position 1 is assigned to the carbon atom connected with vp (Proposition 5). Then, from Proposition 1, Tsub(vp,v1)≠CTsub(vc,v2) for any child node vc of v2, Tsub(vp,v1)≠CTsub(vc,v1) for any child node vc of v1 except v2, and the naphthalene ring is not symmetric. Consider the case that v1 does not have a parent node, that is, v1 is the root. If , the naphthalene ring can be symmetric only with respect to the axis denoted by the dashed red line in Fig. 7a. Then, it is not needed to consider the other symmetry for the naphthalene ring.
Fig. 7.
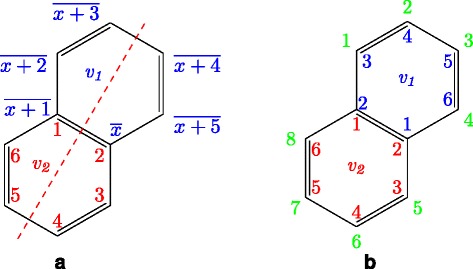
Correspondence between carbon positions in a naphthalene ring. a Correspondence between carbon positions involved with a merge bond in two benzene rings. b Correspondence between carbon positions of a naphthalene ring and two benzene rings in the case of . The upper benzene ring v 1 is the parent of the lower benzene ring v 2. denotes (x mod 6)+1. Blue, red, and green numbers are positions of , , and , respectively. The dashed red line denotes the symmetric axis of ϕ ref
Consider the case that . We can prove that x=1 if T is in normal form (see Proposition 4). Then, a carbon position list of a naphthalene ring consisting of two benzene nodes v1, v2 is a list of lists determined from and according to the following rule, where is a list of carbon positions of nodes in in ascending order.
Definition3.
Carbon positions in a naphthalene ring correspond to carbon positions in two benzene nodes v1,v2, where v1 is the parent node of v2, if , as follows (see Fig. 7b).
For the benzene ring of v1, positions 1,2 are assigned to carbons of the merge bond in . Position i (i=3,…,6) in corresponds to i−2 in .
For the benzene ring of v2, positions 1,2 are assigned to carbons of the merge bond in . Position i (i=3,…,6) in corresponds to i+2 in .
Figure 8 shows examples of carbon position lists for a naphthalene ring, where is T4 with and , is T4 with and . Then, , , and .
Fig. 8.
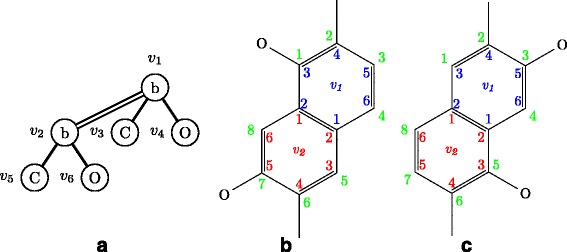
Example of carbon position lists for a naphthalene ring. a T 4. b Molecular graph of , which is T 4 with , . c Molecular graph of , which is T 4 with , and
Definition4.
For carbon position lists , , where AvT1=AvT2, it is defined that CvT1<CvT2 if there exist two integers i and j such that
CvT1[ i′][j′]=CvT2[ i′][j′] for all i′<i and all ,
CvT1[ i][j′]=CvT2[ i][j′] for all j′<j,
CvT1[ i][ j]<CvT2[ i][ j].
This definition is applied to comparison of and for a naphthalene ring with v1 and v2 in the same way.
In the example of Fig. 6, T1 and T2 have the same tree structure, and Cv1T2=((1),(4),(2,3))<((3),(4),(1,2))=Cv1T1 because Cv1T2[ 1][ 1]=1<3=Cv1T1[ 1][ 1].
Let Autb and Autn be the automorphism groups of a benzene ring and a naphthalene ring, respectively (see Fig. 9). Autb is generated from rotation of π/3 radians and reflection. For ϕb∈Autb, v1 is adjacent to v2 in a benzene ring if and only if ϕb(v1) is adjacent to ϕb(v2) in a benzene ring. Autn is generated from rotation of π radians and reflection. We suppose that a list of carbon positions for a map ϕ and is in ascending order by sorting elements of the list because all nodes in have the same subtree. For example, ϕb(Cv1T1)=((6),(5),(1,2)) for and the reflection map ϕb by the perpendicular bisector between carbon atoms of 1 and 2.
Fig. 9.
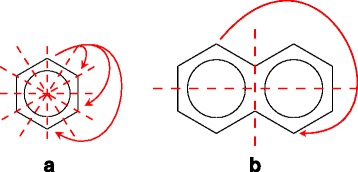
Illustration of automorphism of a benzene ring and a naphthalene ring. a A benzene ring. b A naphthalene ring. Dashed lines indicate reflections, curves indicate rotations, where all automorphisms are not shown
Normal form of a carbon position-assigned molecular tree
In order to prevent generating redundant molecular trees in enumeration, we define a normal form of a carbon position-assigned molecular tree.
Definition5.
Let P be a path in T consisting of n nodes (v1,v2,…,vn)(n≥2). P is called a symmetric path if the following conditions are satisfied.
,
for all , where ⌊x⌋ is the largest integer less than or equal to x,
for all benzene nodes , where satisfies , and v∈V1∖V2 means that v∈V1 and v∉V2.
Proposition3.
For a center-rooted molecular tree, either of and is the root if the length of a symmetric path (v1,⋯,vn) is even. Otherwise, the depth of is less than that of any node in the path.
Proof.
For a path (v1,⋯,vn), vi+1 and vn−i must be the parent nodes of vi and vn−i+1, respectively, for if n is odd and for if n is even due to the center rooted property. Therefore, if the length of path is odd, is the parent node of both and , which means that the depth of is less than that of any node in the path.
In the case that n is even, either or has the least depth among all nodes in the path and another node is the child node of that node. Assume that between these two nodes the parent node is va and the child node is vb. va cannot have a parent node because the height of Tsub(vp,va), where vp is the parent node of va, cannot be equal to the height of Tsub(vc,vb) for any nodes vc that are adjacent to vb due the center-rooted condition, which means that Tsub(va,vb)=mTsub(vb,va) cannot be hold and the first condition of symmetric path is violated. In other words, va, which is either or , is the root node of the tree if n is even.
We say that v1 is left of vn for a symmetric path (v1,…,vn) when is the root, or index(v1)<index(vn).
Figure 10 shows examples of symmetric paths, (v2,v1,v3) in T5 and (v5,v2,v1,v3) in T6, where , , and Cv4T6=Cv6T6.
Fig. 10.
Examples of symmetric paths. The red lines denote symmetric paths. a T 5, where (v 2,v 1,v 3) is a symmetric path, and . b T 6, where (v 5,v 2,v 1,v 3) is a symmetric path, and C v 4 T 6=C v 6 T 6
We define an inequality T1>CT2 for carbon position-assigned molecular trees T1 and T2 if T1>mT2, or T1=mT2, and there exists an integer i such that vi is a benzene node, CviT1>Cvi′T2, and CvjT1=Cvj′T2 for all benzene nodes vj with j>i, where index(vk,T1)=index(vk′,T2) for all k=1,…,|V(T1)|.
Definition6.
Let ϕref be the reflection map with the symmetric axis shown in Fig. 7a. A carbon position-assigned molecular tree T that contains a carbon position list for each benzene node v is in normal form if the following conditions are satisfied.
T is center-rooted and left-heavy.
T(v)≥mTsub(r,v) if the center of the longest path in T with the root r is the edge (r,v).
Positions in each sublist of for each benzene node v are in ascending order.
for all benzene nodes v that is not connected by a merge bond with the parent node and all ϕb∈Autb.
- For benzene nodes v1,v2 connected by a merge bond such that v1 is the root of T,
- for all ϕn∈Autn if , where is related with and by Definition 3.
- if and .
Tsub(v1,v2)≥CTsub(vn,vn−1) for all pairs v1,vn of nodes such that the path (v1,…,vn) is a symmetric path, v1 and vn(=v2) are not connected by a merge bond, and v1 is left of vn.
We call a tree in normal form a normal tree.
Figure 8 also shows molecular trees in normal form and not in normal form. For condition 4 of the definition, , . and satisfy conditions 1, 2, 3, and 4. For condition 5, , whereas for rotation ϕrot of π radians, and violates the condition. It is noted that is rotated by π radians from . For condition 6, v1 and v2 are connected by a merge bond. Thus, is a normal tree, and is not a normal tree.
Proposition4.
For a normal tree T with a benzene node v1 that is connected by a merge bond with its child node v2 and satisfies , positions 1,2 are assigned to the merge bond in the benzene ring of v1. Furthermore, if for all ϕn∈Autn, then for all ϕb∈Autb.
Proof.
We assume that there exists a node vl as a left sibling of v2, and vl is the leftmost child of v1. Since T is left-heavy, T(vl)≥mT(v2), and l(vl)=l(v2)=‘b’ is needed. However, T(vl)=CT(vc), where vc is the leftmost child of v2, because . Hence, T(vl)<mT(v2). It contradicts the assumption, and v2 is the leftmost child of v1. Therefore, . From condition 4 of Definition 6, , and positions 1,2 are assigned to the merge bond, that is x=1 in Fig. 7a.
For a map ϕb∈Autb other than the identity and reflection map ϕref for a benzene ring, because each of ϕb(1) and ϕb(2) is at least 2. From and the correspondence between and , . Therefore, for all ϕb∈Autb.
Proposition5.
For a benzene node v of a normal tree T, is always equal to 1.
Proof.
If v is not connected by a merge bond with the parent node, from condition 4, must be the least possible carbon position list. Hence, . Otherwise, from Definition 3, .
Lemma1.
Given a molecular graph G without cyclic structures except benzene rings and naphthalene rings, G can be represented by a normal tree.
Proof.
We can assign numbers to carbons in benzene rings and naphthalene rings of G such that the conditions of Definition 6 are satisfied.
Lemma2.
Given two different molecular graphs G1 and G2, they cannot be represented by the same normal tree.
Proof.
We can unambiguously obtain a molecular graph from a normal tree by replacing all benzene nodes with benzene rings according to its carbon position lists.
Proposition6.
For a normal tree T with a path (v1,…,vn), G′ is the molecular graph obtained from the tree T′ by removing Tsub(v1,v2) and Tsub(vn,vn−1) except v1 and vn from T, where v1 is left of vn. If there is a non-identity map ϕ of the automorphism group of G′ satisfying ϕ(vi)=vn−i+1 for all i=1,…,n, then Tsub(v1,v2)≥CTsub(vn,vn−1), where ϕ in G′ is naturally extended to T.
Proof.
If , then Tsub(v1,v2)>mTsub(vn,vn−1), and Tsub(v1,v2)>CTsub(vn,vn−1). We assume . If the path (v1,…,vn) is a symmetric path, Tsub(v1,v2)≥CTsub(vn,vn−1) from condition 6. We assume that (vi+1,…,vn−i) is a symmetric path for some i, and index(vi,Tsub(vi+1,vi+2))>index(vn−i+1,Tsub(vn−i,vn−i−1)) (see Fig. 11). Then,
| (1) |
Fig. 11.
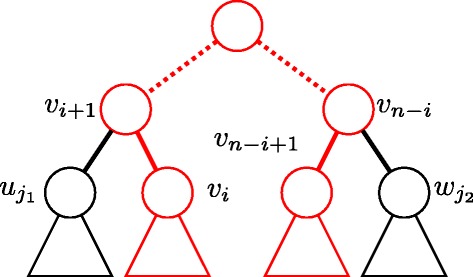
Illustration of an automorphism ϕ in the proof. The red path indicates (v 1,…,v n), where ϕ(v i)=v n−i+1 for all i=1,…,n
Let uj and wj be child nodes of vi+1 and vn−i, respectively. Then, and , where j1=index(vn−i+1,Tsub(vn−i,vn−i−1)) and j2=index(vi,Tsub(vi+1,vi+2)). If vi+1 and vn−i are benzene nodes, , , and T(vi)=CT(vn−i+1) because and ϕ(vi)=vn−i+1.
We assume that vi+1 and vn−i are not benzene nodes. For child nodes uj of vi+1, T(uj)≥CT(uj+1) because (uj,vi+1,uj+1) is a symmetric path. Also for child nodes wj of vn−i, T(wj)≥CT(wj+1). From the definition of ϕ, T(uj)=CT(ϕ(uj)) for all uj≠vi. If index(ϕ(uj+l))<index(ϕ(uj)) for uj,uj+l≠vi and l>0, T(uj)≥CT(uj+l)=CT(ϕ(uj+l))≥CT(ϕ(uj))=CT(uj). It means T(uj)=CT(uj+l). We assume that index(ϕ(uj))<index(ϕ(uj+l)) for all uj≠vi, that is, ϕ(uj)=wj+1 for all j=j1,…,j2−1. Then,
| (2) |
If Tsub(vi+1,vi+2)>CTsub(vn−i,vn−i−1), then there is an integer j (j1≤j≤j2) such that T(uj)>CT(wj), and it contradicts Eq. (2). Therefore, Tsub(vi+1,vi+2)=CTsub(vn−i,vn−i−1), and T(vi)=CT(vn−i+1). Also for the case that (vi+1,…,vn−i) is a symmetric path for some i and index(vi,Tsub(vi+1,vi+2))<index(vn−i+1,Tsub(vn−i,vn−i−1)), then T(vi)=CT(vn−i+1). Thus, Tsub(v1,v2)≥CTsub(vn,vn−1).
Lemma3.
Given two different normal trees T1 and T2, T1 does not represent the same molecular graph as T2.
Proof.
We assume that T1 represents the same molecular graph as T2. Let G1 and G2 be molecular graphs transformed from T1 and T2, respectively, where each carbon in benzene rings and naphthalene rings is connected with adjacent atoms according to carbon position lists of T1 and T2. From the assumption, there is an isomorphism ψ from G1 to G2. It means that l(v1)=l(ψ(v1)) for all v1∈V(G1), (ψ(v1),ψ(v2))∈E(G2) if and only if (v1,v2)∈E(G1), and mul(ψ(v1),ψ(v2))=mul(v1,v2).
Consider the case that the automorphism group Aut(G1) of G1 has only elements ϕ such that ϕ(v1)≠v2 for v1 and v2 belonging to distinct benzene rings. Let T(G) be the molecular tree without carbon position lists, obtained from G by contracting benzene rings and naphthalene rings to benzene nodes, and satisfying conditions 1, 2 of Definition 6. We suppose that maps ψ and ϕ in G1 are naturally extended to T(G1). Since T1 is different from T2, there is a benzene node v1∈V(T1) such that
| (3) |
If v1 is not connected by a merge bond with the parent node, there is a non-identity map ϕb∈Autb such that because T1 and T2 represent the same molecular graph. It contradicts condition 4 of Definition 6. Suppose that v1 is connected by a merge bond with the parent node vp and CvpT1=Cψ(vp)T2. If , then vp is the root, and there is a non-identity map ϕn∈Autn such that because T1 and T2 represent the same molecular graph. It contradicts condition 5a. Otherwise, . If vp is not the root, then T1 does not represent the same molecular graph as T2 because Tsub(va,vp), where va is the parent of vp, is different from other subtrees connected to the naphthalene ring. It contradicts the assumption. If vp is the root, and because T1 and T2 represent the same molecular graph. It contradicts condition 5b.
Consider the case that there is an element ϕ∈Aut(G1) such that ϕ(v1)=v2 for v1 and v2 belonging to distinct benzene rings. Since T1 is different from T2, there is a benzene node v1∈V(T1) such that
| (4) |
Here, we suppose that conditions 3, 4, 5 are satisfied for all benzene nodes in T1 and T2. Then, there is a path from v1 to ϕ(v1)=vn, (v1,…,vn), in T1. Since T1 and T2 represent the same molecular graph,
| (5) |
Here, we can assume that v1 is left of vn and ψ(v1) is left of ψ(vn) without loss of generality. Then, from Proposition 6, for paths of (v1,…,vn) and (ψ(v1),…,ψ(vn)),
| (6) |
because T1 and T2 are normal trees. There is no carbon position lists that satisfy Eqs. (4), (6) and (7).
Therefore, T1 does not represent the same molecular graph as T2.
Methods
We propose an algorithm BfsBenNaphEnum for enumerating chemical compounds containing benzene rings and naphthalene rings as cyclic structures. BfsBenNaphEnum utilizes our previously developed algorithms BfsSimEnum, BfsMulEnum [18], and assigns carbon position lists.
Modification of BfsSimEnum and BfsMulEnum
Suppose that the numbers of atoms with label li for all li∈Σ, the numbers nb, nn of benzene rings and naphthalene rings are given. BfsBenNaphEnum introduces a special label ‘b’ representing a benzene node to Σ with b>li∈Σ and val(b)=6, and executes BfsSimEnum to generate all non-redundant molecular trees T such that for li∈Σ except li=b,C and num(T,b)=nb+2nn, num(T,C)=nC−6nb−10nn. At this time, all edges of enumerated trees are single because BfsSimEnum generates only simple trees. Then, we modify BfsMulEnum to assign nn merge bonds to edges between benzene nodes in each tree enumerated by BfsSimEnum in addition to adding bonds to edges between usual nodes. It should be noted that multiple bonds cannot be assigned to edges connected to benzene nodes since a carbon atom in benzene rings and naphthalene rings is connected with another adjacent atom by a single bond.
Assignment of carbon positions for molecular trees
In this algorithm, we traverse along the tree T from the rightmost deepest benzene node to the root in reverse BFS order because an adjacent node list depends on carbon position lists of descendant nodes. For each benzene node v we found, we assign a carbon position list not to violate the conditions of normal form.
The pseudocode of assignment part in BfsBenNaphEnum is given in Algorithms 1 and 2. We always assign carbon position 1 to the first node in (line 20 in ASSIGN function) due to Proposition 5, which is the parent node of v if v is not the root (Proposition 2). If v is the root and , we assign carbon position lists in Table 1 (see also Fig. 12) to v immediately for the sake of efficiency. Carbon position lists in Table 1 satisfy condition 4 of the normal form, and all the cases are included in the table.
Table 1.
Carbon position lists for , where v is the root, and
| 3 | 0 | ((1,2,3)), ((1,2,4)), ((1,3,5)) |
| 3 | 3 | ((1,2,3),(4,5,6)), ((1,2,4),(3,5,6)), ((1,3,5),(2,4,6)) |
| 4 | 0 | ((1,2,3,4)), ((1,2,3,5)), ((1,2,4,5)) |
| 5 | 0 | ((1,2,3,4,5)) |
| 6 | 0 | ((1,2,3,4,5,6)) |
Fig. 12.
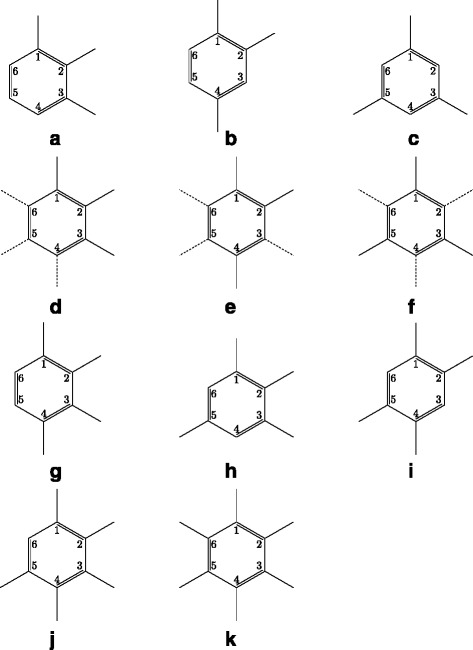
Illustration of benzene rings having each carbon position list in Table 1. a ((1,2,3)). b ((1,2,4)). c ((1,3,5)). d ((1,2,3),(4,5,6)). e ((1,2,4),(3,5,6)). f ((1,3,5),(2,4,6)). g ((1,2,3,4)). h ((1,2,3,5)). i ((1,2,4,5)). j ((1,2,3,4,5)). k ((1,2,3,4,5,6)). Solid and dashed lines correspond to and , respectively


For other carbon positions from 2 to 6, we use ASSIGN_CHILD to assign such positions to the remaining adjacent nodes. For example, let T1 in Fig. 6 be output without any carbon position list by BfsMulEnum. T1 has a benzene node v1, and . First, carbon position 1 is assigned to , that is, . Since v1 is the root and , Table 1 is not used, and the other nodes v5,v2,v3 are assigned by ASSIGN_CHILD. For v5, each carbon position from 2 to 6 is examined (line 26 in ASSIGN_CHILD). For v2, each position from 2 to 6 except the position assigned to v5 is examined (line 27). For v3, each position from 2 to 6 that is more than the position assigned to v2 except the position assigned to v5 is examined (line 27) because v2 and v3 have the same subtree and condition 3 must be satisfied. Thus, are examined, where ((1),(6),(2,3)),((1),(6),(2,4)),((1),(5),(2,3)) and so on are discarded in the next step.
For each benzene node v, after assignment of a carbon position list to , whether or not violates conditions 4, 5 of the normal form is confirmed (lines 5, 11, 14 in ASSIGN_CHILD). After carbon position lists are assigned to all benzene nodes, condition 6 is confirmed (line 4 in ASSIGN).
Since an input of this part, that is, an output of BfsMulEnum, satisfies conditions 1, 2 of the normal form, BfsBenNaphEnum always outputs normal trees. In ASSIGN_CHILD, a distinct carbon position list is always assigned, and all patterns are assigned (line 28). Hence, BfsBenNaphEnum outputs all distinct normal trees.
Theorem1.
BfsBenNaphEnum outputs all non-redundant molecular graphs that are solutions of Problem 1.
Figure 13 shows another example T7 of molecular trees. T7 includes four benzene nodes v5, v4, v3, v2 in reverse BFS order, and edges (v2,v4), (v3,v5) are merge bonds. First, our algorithm assigns carbon position lists for as . In a similar way, for , . For , are examined. In line 5 of ASSIGN_CHILD, ((1),(4,5)) and ((1),(5,6)) are discarded because ϕb(((1),(4,5)))=((1),(3,4)), ϕb(((1),(5,6)))=((1),(2,3)) for the reflection map ϕb with respect to the axis through positions 1 and 4, and these violate condition 4. In a similar way, for , are assigned. After carbon position lists are assigned to all benzene nodes, condition 6 is confirmed in line 4 of ASSIGN. If Cv2T7≠Cv3T7, then there is one symmetric path, , and T7(v2)≥CT7(v3) must be satisfied. It means that Cv4T7=Cv5T7=((1,2),(3)),((1,2),(4)),((1,2),(5)),((1,2),(6)) and Cv2T7=((1),(3,4))>Cv3T7=((1),(2,3)), or Cv4T7>Cv5T7 and Cv2T7≠Cv3T7. Hence, there are structures. If Cv2T7=Cv3T7=((1),(2,3)) (or Cv2T7=Cv3T7=((1),(3,4))), then , and both of T7(v2)≥CT7(v3) and T7(v4)≥CT7(v5), that is, Cv4T7≥Cv5T7, must be satisfied. Hence, there are 4+3+2+1=10 structures. In total, 16+10·2=36 structures are generated by BfsBenNaphEnum for T7.
Fig. 13.
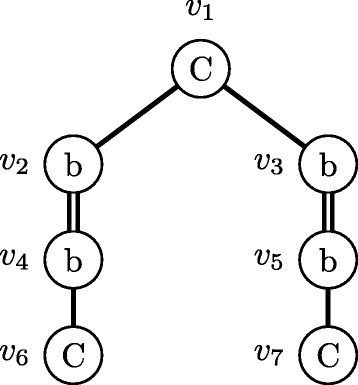
Example of a molecular tree T 7
Results
In this section, we show that our proposed method can enumerate chemical compounds with benzene rings and naphthalene rings correctly and efficiently. For the evaluation, although MOLGEN 3.5 is more suitable than MOLGEN 5.0 to enumerate tree-like compounds because MOLGEN 3.5 offered the possibility to define substructures like benzene or naphthalene as macro atoms but MOLGEN 3.5 cannot handle all the cases provided in Table 2, we compared proposed tool with MOLGEN 5.0. Thereby, we implemented it and installed another well-known general purpose structure generator, MOLGEN 5.0, on a computer with 3.47 GHz intel Xeon CPU and 23.5 GiB memory, and compared their computational time. The implementation of BfsBenNaphEnum is available on our supplementary web site, http://sunflower.kuicr.kyoto-u.ac.jp/jira/bfsenum/.
Table 2.
Results on execution time (sec), the number of enumerated structures by BfsBenNaphEnum and MOLGEN, and the number of chemical compounds exist in PubChem database for several instances
| Chemical formula | #atoms | #all compounds in PubChem | #enumerated structures | Computational time (sec) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | b | C | N | O | H | BfsBenNaphEnum | MOLGEN | |||
| C 7 O 2 H 8 | 0 | 1 | 1 | 0 | 2 | 8 | 728 | 19 | 0.001 | 0.053 |
| C 8 O 3 H 10 | 0 | 1 | 2 | 0 | 3 | 10 | 1602 | 307 | 0.002 | 0.124 |
| C 9 O 4 H 10 | 0 | 1 | 3 | 0 | 4 | 10 | 1469 | 6406 | 0.010 | 1.699 |
| C 10 N 2 O 4 H 10 | 0 | 1 | 4 | 2 | 4 | 10 | 1592 | 8,333,991 | 12.260 | 957.53 |
| 1 | 0 | 0 | 2 | 4 | 10 | 7980 | 0.031 | 69.51 | ||
| C 11 N 2 H 10 | 0 | 1 | 5 | 2 | 0 | 10 | 790 | 9012 | 0.021 | 630.44 |
| 1 | 0 | 1 | 2 | 0 | 10 | 56 | 0.005 | 24.061 | ||
| C 12 N 1 O 1 H 11 | 0 | 1 | 6 | 1 | 1 | 11 | 1582 | 80,883 | 0.155 | 2,611.57 |
| 0 | 2 | 0 | 1 | 1 | 11 | 33 | 0.001 | 98.99 | ||
| 1 | 0 | 2 | 1 | 1 | 11 | 888 | 0.009 | 560.98 | ||
| C 13 O 2 H 12 | 0 | 1 | 7 | 0 | 2 | 12 | 1239 | 162,122 | 0.289 | 6,497.55 |
| 0 | 2 | 1 | 0 | 2 | 12 | 190 | 0.002 | 2,069.3 | ||
| 1 | 0 | 3 | 0 | 2 | 12 | 2458 | 0.013 | 1,731.92 | ||
| C 14 O 4 H 12 | 0 | 1 | 8 | 0 | 4 | 12 | 1 397 | 19,514,480 | 35.655 | 197,264.54 |
| 0 | 2 | 2 | 0 | 4 | 12 | 15,581 | 0.021 | 107,509.42 | ||
| 1 | 0 | 4 | 0 | 4 | 12 | 337,178 | 1.061 | 97,326.71 | ||
Since MOLGEN can enumerate chemical compounds without restriction on the structure, we must specify a benzene ring and a naphthalene ring as a substructure so that the enumerated structures contain only benzene rings and naphthalene rings as cyclic structures. As can be seen from Table 2, where ‘n’ and ‘b’ denote a naphthalene ring and a benzene ring, respectively, BfsBenNaphEnum enumerated chemical compounds much faster than MOLGEN while giving the same number of enumerated structures. BfsBenNaphEnum was from 50 times to 5,000,000 times faster than MOLGEN for instances with 8 to 14 carbon atoms. Table 2 also compares the number of discovered compounds in PubChem, which are not limited to tree-like chemical compounds, with the number of compounds enumerated by the proposed algorithm for several chemical formulas. When the number of carbon atoms is large (greater than 8 in this case), the number of discovered compounds is much less than the number of enumerated compounds. This implies that there are still a numerous number of unknown compounds to be discovered, which possibly include some essential compounds. In this study, we examined chemical formulas including up to two benzene rings and one naphthalene ring because MOLGEN was not able to output results in practical time for chemical formulas including more benzene rings and naphthalene rings.
We plotted the relation between the number of enumerated structures and the computational time for both methods in Fig. 14, where both x-axis and y-axis are in a log scale. It is seen from the figure that the execution time of BfsBenNaphEnum is much smaller than that of MOLGEN.
Fig. 14.
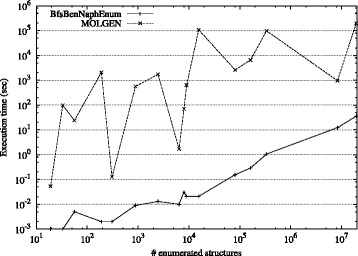
Relation between the number of enumerated structures and the computational time (sec)
Discussion
Our algorithm is limited to tree-like chemical structures without any cyclic structures except benzene rings and naphthalene rings while MOLGEN does not have such limitation. Therefore, in the future, we would like to extend the algorithm such that it can enumerate more complex cyclic structures, such as polycyclic aromatic compounds and nucleotides. Besides, in order to make enumeration tools practical, we need to rank enumerated structures because a large number of structures are usually enumerated. For that purpose, it might be useful to employ drug likeness filters such as Lipinski RO5, and QED score. Incorporation of such filters into our system is also important future work.
Conclusions
We proposed a way to represent a benzene ring in a molecular tree by regarding it as a new defined atom with valence six and introducing a new attribute named carbon position list to benzene nodes. Carbon position of an atom specifies which carbon in a benzene ring that the corresponding atom bonds with. We also proposed a new kind of bond called merge bond that merges two benzene rings together to form a naphthalene ring. With merge bond a molecular tree can represent a structure containing naphthalene rings without defining new kind of atom. Moreover, since a benzene ring and a naphthalene ring are symmetric structures, we defined a rule to assign carbon position lists such that no redundant structures due to the symmetry of a benzene ring and a naphthalene ring are enumerated.
The algorithm of this work consists of two main steps. Given the number of benzene rings, the number of naphthalene rings as well as a chemical formula, BfsSimEnum and BfsMulEnum are applied such that they can enumerate molecular trees with benzene nodes. Next, the new extension BfsBenNaphEnum assigns carbon position lists to benzene nodes in normal molecular trees.
To show the performance of our algorithm, all non-redundant chemical structures were enumerated for several chemical formulas by BfsBenNaphEnum and MOLGEN 5.0, a well-known general purpose structure generator. It is shown that our algorithm is reliable since it generated the same number of structures as MOLGEN, while expended much less computational time. BfsBenNaphEnum was from 50 times to 5,000,000 times faster than MOLGEN for instances with 8 to 14 carbon atoms in our experiments. This is mainly because the number of nodes decreases from six to one for each benzene ring and from ten to two for each naphthalene ring in a chemical structure and because we enumerate chemical structures in the form of trees instead of graphs.
Acknowledgements
This work was partially supported by Grants-in-Aid #26240034, #24500361, and #25-2920 from MEXT, Japan.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JJ and MH developed, implemented the methods, and drafted the manuscript. YZ and TA participated in the discussions during the development of the methods and helped draft the manuscript. All authors read and approved the final manuscript.
Contributor Information
Morihiro Hayashida, Email: morihiro@kuicr.kyoto-u.ac.jp.
Tatsuya Akutsu, Email: takutsu@kuicr.kyoto-u.ac.jp.
References
- 1.Ward RA, Kettle JG. Systematic enumeration of heteroaromatic ring systems as reagents for use in medicinal chemistry. J Med Chem. 2011;54(13):4670–7. doi: 10.1021/jm200338a. [DOI] [PubMed] [Google Scholar]
- 2.Blum LC, Reymond JL. 970 million druglike small molecules for virtual screening in the chemical universe database gdb-13. J Am Chem Soc. 2009;131(25):8732–3. doi: 10.1021/ja902302h. [DOI] [PubMed] [Google Scholar]
- 3.Mishima K, Kaneko H, Funatsu K. Development of a new de novo design algorithm for exploring chemical space. Mol Inform. 2014;33(11-12):779–89. doi: 10.1002/minf.201400056. [DOI] [PubMed] [Google Scholar]
- 4.Funatsu K, Sasaki S. Recent advances in the automated structure elucidation system, chemics. utilization of two-dimensional NMR spectral information and development of peripheral functions for examination of candidates. J Chem Inform Comput Sci. 1996;36(2):190–204. doi: 10.1021/ci950152r. [DOI] [Google Scholar]
- 5.Meringer M, Schymanski EL. Small molecule identification with MOLGEN and mass spectrometry. Metabolites. 2013;3:440–62. doi: 10.3390/metabo3020440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koichi S, Arisaka M, Koshino H, Aoki A, Iwata S, Uno T, Satoh H. Chemical structure elucidation from 13C NMR chemical shifts: Efficient data processing using bipartite matching and maximal clique algorithms. J Chem Inform Model. 2014;54:1027–35. doi: 10.1021/ci400601c. [DOI] [PubMed] [Google Scholar]
- 7.Bytautas L, Klein DJ, Schmalz TG. All acyclic hydrocarbons: Formula periodic table and property overlap plots via chemical combinatorics. New J Chem. 2000;24(5):329–36. doi: 10.1039/a906939i. [DOI] [Google Scholar]
- 8.Faulon J, Visco DP, Roe D. Enumerating molecules. Rev Comput Chem. 2005;21:209. [Google Scholar]
- 9.Koch MA, Schuffenhauer A, Scheck M, Wetzel S, Casaulta M, Odermatt A, Ertl P, Waldmann H. Charting biologically relevant chemical space: A structural classification of natural products (sconp) Proc Natl Acad Sci U S A. 2005;102(48):17272–7. doi: 10.1073/pnas.0503647102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mauser H, Stahl M. Chemical fragment spaces for de novo design. J Chem Inf Model. 2007;47(2):318–24. doi: 10.1021/ci6003652. [DOI] [PubMed] [Google Scholar]
- 11.Andricopulo AD, Guido RV, Oliva G. Virtual screening and its integration with modern drug design technologies. Curr Med Chem. 2008;15(1):37–46. doi: 10.2174/092986708783330683. [DOI] [PubMed] [Google Scholar]
- 12.Reymond JL, van Deursen R, Blum LC, Ruddigkeit L. Chemical space as a source for new drugs. MedChemComm. 2010;1(1):30–8. doi: 10.1039/c0md00020e. [DOI] [Google Scholar]
- 13.Bürgi JJ, Awale M, Boss SD, Schaer T, Marger F, Viveros-Paredes JM, Bertrand S, Gertsch J, Bertrand D, Reymond JL. Discovery of potent positive allosteric modulators of the α3β2 nicotinic acetylcholine receptor by a chemical space walk in chembl. ACS Chem Neurosci. 2014;5(5):346–59. doi: 10.1021/cn4002297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gugisch R, Kerber A, Kohnert A, Laue R, Meringer M, Rücker C, Wassermann A. MOLGEN 5.0, a molecular structure generator. Sharjah, United Arab Emirates: Bentham Science Publishers Ltd.; 2012. [Google Scholar]
- 15.Peironcely JE, Rojas-Chertó M, Fichera D, Reijmers T, Coulier L, Faulon JL, Hankemeier T. OMG: Open Molecule Generator. J Cheminformatics. 2012;4:21. doi: 10.1186/1758-2946-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fujiwara H, Wang J, Zhao L, Nagamochi H, Akutsu T. Enumerating treelike chemical graphs with given path frequency. J Chem Inf Model. 2008;48(7):1345–57. doi: 10.1021/ci700385a. [DOI] [PubMed] [Google Scholar]
- 17.Shimizu M, Nagamochi H, Akutsu T. Enumerating tree-like chemical graphs with given upper and lower bounds on path frequencies. BMC Bioinformatics. 2011;12:14–3. doi: 10.1186/1471-2105-12-S14-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao Y, Hayashida M, Jindalertudomdee J, Akutsu T. Breadth-first search approach to enumeration of tree-like chemical compounds. J Bioinformatics Comput Biol. 2013;11:1343007. doi: 10.1142/S0219720013430075. [DOI] [PubMed] [Google Scholar]
- 19.Schüller A, Hähnke V, Schneider G. SmiLib v2.0: A Java-based tool for rapid combinatorial library enumeration. QSAR Comb Sci. 2007;26(3):407–10. doi: 10.1002/qsar.200630101. [DOI] [Google Scholar]
- 20.Song CM, Bernardo PH, Chai CLL, Tong JC. CLEVER: Pipeline for designing in silico chemical libraries. J Mol Graph Model. 2009;27(5):578–83. doi: 10.1016/j.jmgm.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 21.Trinajstić N. Chemical Graph Theory. Boca Raton, Florida: CRC Press; 1992. [Google Scholar]
- 22.Meringer M. Handbook of Chemoinformatics Algorithms. Boca Raton, Florida: CRC Press; 2010. [Google Scholar]
- 23.Suzuki M, Nagamochi H, Akutsu T. Efficient enumeration of monocyclic chemical graphs with given path frequencies. J Cheminformatics. 2014;6:31. doi: 10.1186/1758-2946-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hardinger SA, University of California LADoC . Biochemistry: Chemistry 14D: Organic Reactions and Pharmaceuticals : Course Thinkbook, Lecture Supplements, Concept Focus Questions, OWLS Problems, Practice Problems. Plymouth, MI 48170: Hayden-McNeil Pub; 2008. [Google Scholar]