

Abstract
A substantial portion of metabolism involves transformation of phosphate esters, including pathways leading to nucleotides and oligonucleotides, carbohydrates, isoprenoids and steroids, and phosphorylated proteins. Because the natural substrates bear one or more negative charges, drugs that target these enzymes generally must be charged as well but small charged molecules can have difficulty traversing the cell membrane other than by endocytosis. The resulting dichotomy has stimulated abundant effort to develop effective prodrugs, compounds that carry little or no charge to enable them to transit biological membranes but then able to release the parent drug once inside the target cell. This chapter will present recent studies on advances in prodrug forms, along with representative examples of their application to marketed and developmental drugs.
Keywords: Prodrugs, Phosphate, Phosphonate, Bisphosphonate, Nucleotide, Isoprenoid
1 Introduction
Metabolism employs phosphorus compounds in myriad ways. Phosphate esters are central to information storage, serve as the currency for energy exchange, and contribute to membrane fluidity. They are important intermediates in carbohydrate metabolism, in formation of nucleotides and their assembly into RNA and DNA, and in steroid fabrication and protein lipidation through the isoprenoid biosynthesis pathways. They hold key roles in cell signaling processes, including G-protein coupled receptors (GPCR), second messengers such as cAMP or phosphatidylinositol, and enzyme activation/deactivation through protein phosphorylation. Within the cell, the phosphate group commonly serves as a tunable leaving group. A phosphate mono- or diester is relatively stable when free in metabolic surroundings because of its negative charge at physiological pH [1], but is readily activated upon complexation to various counterions in an enzyme’s active site. Thus phosphorus compounds provide both abundant opportunities for drug design and intrinsic challenges. Chief among the challenges is the inherent conflict between a molecule that must be anionic to bind to the active site of an enzyme but first must penetrate the membrane to access that enzyme, as well as the necessity to mimic a potential leaving group with a substructure sufficiently stable to survive delivery.
A longstanding and still common strategy for preparation of more stable analogues of phosphate esters (1) is based upon formal replacement of the ester oxygen with carbon to afford the corresponding phosphonate (2) [2]. Although phosphonates are known from a number of natural sources, implicating the existence of enzymes capable of C-P bond cleavage [3], it is widely recognized that phosphonates have greater metabolic stability. Replacement of the phosphate ester oxygen with a simple –CH2– group may preserve much of the size and shape of the original substrate but it also impacts the second pKa value, with the phosphonic acid less acidic by 0.5 to 1.5 pKa units [4]. Incorporation of this carbon also eliminates the possibility of enzymatic interaction with oxygen lone pairs at this position. Both of these concerns can be addressed by addition of fluorine or oxygen substituents on the alpha carbon, although this necessarily has on impact on steric issues and can introduce a stereogenic center as well [5]. Nevertheless, numerous phosphonate analogues of biologically active phosphates have been prepared.
Phosphate monoesters are charged at physiological pH, as are the corresponding phosphonates, and those with small and/or hydrophilic substituents may have difficulty in diffusion across biological membranes and often require endocytosis for cell entry [6]. While this limitation may be ameliorated in the case of compounds with larger, more lipophilic substituents [7], it is always a concern that must be considered in drug design. Thus efforts to prepare biologically active organophosphorus compounds often have been followed by studies to delineate strategies that temporarily mask any negative charges at physiological pH. Potential drugs based upon this approach may offer a number of advantages over their non-protected counterparts. In particular, addition of cell-cleavable protecting/masking groups (i.e. the prodrug approach) can: 1) increase oral bioavailability; 2) enhance cell penetration; 3) improve the specificity of tissue delivery; and 4) avoid or minimize degradation in serum via cellular sequestration. In addition, once the prodrug moieties are cleaved within the cell, the same factors that would normally limit entry of the non-protected drug into the cell now can restrict the free drug from leaving the cell. Thus, prodrugs can effectively allow the drug to achieve elevated concentrations within cells, further enhancing efficacy [8].
Study of phosphonate prodrugs may have a long history, but pro-drug strategies to protect and deliver phosphate monoesters also have been of great interest. Efficient delivery of a phosphate monoester into the cell can afford important metabolic advantages. For example, nucleoside analogues such as arabinofuranosyl cytidine (AraC) and gemcitabine (GemC) undergo activation after cell entry by conversion to the corresponding mono-, di- and ultimately triphosphates [9], and use of a protected phosphate may allow intersection with natural metabolic processes at a later stage [10]. This strategy may be particularly important with nucleotide prodrugs where it can allow the agent to bypass the rate-limiting initial phosphorylation [11]. Furthermore, phosphate prodrugs may confer stability to serum phosphatases, and therefore support more effective dosing.
However, use of prodrugs is not without substantial challenges. In particular, choosing the best protecting group is difficult for several reasons. Most notably, cellular cleavage of the protecting groups can often generate products which are viewed as disadvantageous or even toxic. In addition, the protecting groups must strike a balance between allowing absorption in the intestines and allowing cleavage in the blood or target cell.
Given the tremendous importance of phosphonate and phosphate prodrugs, research in this area has grown dramatically and has been the subject of periodic reviews [12–14]. Many reviews have focused exclusively on nucleotide prodrugs [15–18] or hepatitis C (HCV) [19–21]. However, it has been more than six years since a comprehensive phosphorous prodrug review [22]. Therefore, we have attempted herein to compare and contrast recent investigations of phosphonate and phosphate pro-drugs, and to consider the present applications and future potential of such agents. After a brief consideration of the structural factors involved, representative examples of these strategies will be presented.
2 General considerations
The majority of prodrug applications of phosphates and phosphonates seek to facilitate passive diffusion through the cell membrane by masking negative charge until the compound is within the cell. This requires attention to two primary considerations: 1) what type of derivative will be employed; and 2) what process or processes will be entrusted to remove the protecting groups once the prodrug is within the cell.
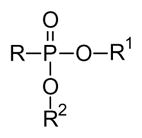
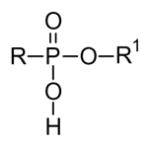
For applications in synthetic chemistry, a wide variety of protecting groups for phosphonates and phosphates is known [23]. Those employed to provide prodrugs fall into two general categories, esters (3–6) and amides (7–10). Yet even after that initial selection is made, there are a number of other concerns to address and an equally large array of solutions has been developed. When two charges must be masked, will two of the same groups be employed in a diester (e.g. 3 or 4) or will two different groups be used (e.g. 5 or 6)? Use of two different groups necessarily results in formation of a stereogenic center at phosphorus, but may allow more flexibility in terms of the cleavage step(s). For either variation, use of two alcohols allows formation of an acyclic derivative while use of a single diol affords a cyclic derivative. Parallel considerations underlie the selection of amine-derived prodrugs, where again use of two different amines or an unsymmetrical diamine (e.g. 9 or 10) generates a stereogenic phosphorus. Finally, mixed ester-amides (e.g. 11 or 12) are also well known, and can offer some advantages in terms of both the cleavage step and organ or tissue targeting.
Equally complex is determination of the strategy that will be employed for regeneration of the drug once it is within the cell. In most cases, this is assumed to involve a specific enzyme or enzymes, but those can include an esterase, an amidase, a phosphatase, or even a redox process. Furthermore, some prodrugs have been reported that rely upon thermal cleavage, and the possibility of photochemical cleavage may be available through photodynamic therapy approaches.
A dramatically different prodrug application has been to employ phosphates as anions to make a specific drug more water soluble. This strategy is particularly valuable where the drug itself is an alcohol (e.g. metronidazole, shown as the phosphate derivative 13) [24] or a phenol (e.g. tamoxifen, shown as the nucleotide derivative 14). In the case of metronidazole, addition of the phosphate enhances water solubility 50-fold and the prodrug is cleaved rapidly in human serum [25]. Further derivatization of metronidazole through conjugation with a nucleoside 3′-phosphate has been explored as a strategy to enhance oral bioavailability [26]. In the case of 4-hydroxytamoxifen, incorporation of the nucleotide element was desired to make this prodrug a substrate for a ribonuclease [27], a strategy designed to release a natural metabolite upon generation of the drug [26]. Variations of this approach have been employed with a number of other drugs [28–31] through functionalization of phenol substructures such as in prodrugs 15–17 [32], the nitrogen of phenyl hydantoins like derivatives 18 and 19, and the relatively acidic carbon of β-dicarbonyl compounds [33]. While this appears to be an area of growing interest, in light of the recent review [32] it will not be considered in greater depth here.
3 Ester prodrugs
A host of different alcohols can be employed to prepare phosphorus-based esters, and many examples have been reported. Some of the more useful approaches are listed in Table 1. Representative cases will be described in the following sections.
Table 1.
Prodrug variations based upon ester linkages
| Symmetrical diesters | ||
| R1 = R2 | ||
| Alkyl | -CH3 |
|
| Benzyl | -CH2C6H4X (X = H, OAc, OCH3) | |
| Aryl | -C6H5 | |
| acyloxyalkyl (POM) | -CH2OC(O)C(CH3)3 | |
| alkoxycarbonyloxy alkyl (POC) | -CH2OC(O)OCH(CH3)2 | |
| S-acylthioalkyl (SATE) | -CH2CH2SC(O)R | |
| Unsymmetrical diesters | ||
| R1 ≠ R2 |
|
|
| cycloSal | ||
| HepDirect |
|
|
| Monoesters | ||
| R1 = |
|
|
| steroidal | cholesteryl | |
| glycerol-fatty alcohol | -CH2OCH2(CH2)14CH3 | |
| Internal monoesters and mixed esters | ||
| R1 = |
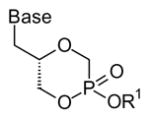
|
|
| Cidofovir and HPMPA | -H | |
| glycerol-fatty alcohol | -CH2OCH2(CH2)14CH3 | |
3.1 Symmetrical diesters
Conceptually, the simplest derivative of a phosphonic acid that would be neutral at physiological pH may be a diester derived from a small alcohol such as methanol or ethanol. Dimethyl esters are readily prepared, and do not introduce stereochemical issues at phosphorus. However, despite the emergence of organophosphorus hydrolases in various bacteria [34], perhaps driven by the widespread use of phosphorus-based insecticides, simple dialkyl esters of phosphonates appear to be surprisingly stable in mammalian systems (cf. bisphosphonate examples [35–37], nucleoside phosphonate examples [38–40], and a squalene synthase inhibitor [41]). Some phosphonate dibenzyl esters also have been examined and show more promise. In one study, the parent benzyl group was converted to the free drug too slowly to be of value, but more substituted systems can have greater utility as prodrugs [42,43]. For example, the p-acetoxybenzyl system can be activated by esterase-mediated hydrolysis of the acetate [38] and the p-methoxybenzyl esters undergo cytochrome P mediated activation [42]. While the later process was suggested to involve cleavage of the methyl ether, it should be noted that chemical cleavage of p-methoxybenzyl ethers involves oxidation of the benzylic position which could also facilitate drug regeneration within the cell [23,44]. Whatever the mechanism of drug release, the bis[(p-methoxy)benzyl] prodrug showed enhanced plasma stability which allowed intercellular drug release as well enhanced metabolism in the liver.
Aryl esters would be expected to be more chemically reactive because of the greater acidity of phenol vis-à-vis methanol. In some cases diaryl phosphonates have proven to be of utility as prodrugs [45]. This early study also confirmed that simple alkyl esters were generally incapable of functioning as prodrugs, while demonstrating that diphenyl esters performed well, releasing the free acid in vivo at high concentrations as well as exhibiting physiochemical properties that were more conducive to pharmaceutical formulation. More recently it has been suggested that the ability to tune the reactivity of diaryl esters through choice of their substituents should make discovery of useful prodrugs in this family feasible [46,47].
Phosphonate diesters derived from more complex natural alcohols, for example glucose, might be more amenable to metabolic hydrolysis. Unfortunately, most carbohydrate-based alcohols also would add significant chemical complexity in terms of their stereogenic centers as well as potential issues of regiochemical control during their formation. Furthermore, the enzymes that metabolize carbohydrate phosphates, such as glucose-6-phosphatase, have natural substrates that are dianions or anions, and may be unlikely to hydrolyze neutral compounds. Thus the early studies on phosphonate prodrugs often relied upon enzymatic cleavage mediated by enzymes other than phosphatases. Particularly prominent among these strategies were those that relied upon nonspecific esterases for an initial hydrolysis, followed by a chemical decomposition that liberated the desired drug [14].
Much of the early interest in phosphonate prodrugs can be traced to key studies during the quest to develop drugs against the human immunodeficiency virus (HIV), hepatitis, and other viral diseases. After De Clercq, Holy, Rosenberg and their colleagues reported strong antiviral activity of acyclic nucleoside phosphonates such as 9-[2-(phosphonomethoxy)ethyl]adenine (PMEA) and (S)-9-(3-hydroxy-2-phosphonyl-methoxypropyl) adenine ((S)-HPMPA [48] (Reviewed in [49]), the activity of these compounds stimulated great interest in development of prodrug forms to enhance the membrane permeability of these drugs. An early focus was pivaloyloxymethyl (POM) modified phosphonates, a prodrug format that first had been advanced for use with phosphate monoesters [50,51]. However this approach was readily adapted to phosphonates such as foscarnet esters (20) [52] and a phosphonate inhibitor of insulin receptor tyrosine kinase [53]. Ultimately, the POM moieties were applied to PMEA leading, after extensive studies, to the approval of adefovir dipivoxil (21, Hepsera) by Gilead for treatment of Hepatitis B (HBV) (Reviewed by Lee and Martin [54]).
The POM group also has been applied to a variety of other phosphonates including bisphosphonates such as compound 22 [55]. This pro-drug of an inositol monophosphatase inhibitor exhibits an increase of more than 2500-fold in activity relative to the parent drug. Such a dramatic impact, together with the clinical utility of drugs such as adefovir dipivoxil, has fostered continued interest in the use of POM groups in phosphonate and phosphate prodrugs to the point where this is often the first prodrug form examined for any new phosphorus-containing drug [22,56]. At the same time, concerns are periodically expressed about the metabolic byproducts of POM cleavage, including the toxicity [42,43] and carcinogenicity [45] of formaldehyde as well as the burden resulting from elimination of pivalic acid as a carnitine derivative. Thus while the POM group is a valued addition to the line-up of potential prodrug forms, research has continued into other variations.
A closely related strategy for preparation of phosphonate prodrugs is based on use of a carbonate ester such as isopropyloxycarbonyloxymethyl derivatives (POC). Esterase-mediated cleavage of this prodrug results in loss of carbonate and 2-propanol, rather than pivalic acid [57]. Therefore, in contrast to POM prodrugs, the POC prodrugs should not impact carnitine levels. An early study [58] was the basis for selecting the POC derivatives of acyclic nucleosides for development, because it showed significantly increased potency towards HIV-1 in a cell-based assay as well as a longer serum half-life than the corresponding POM compound. Ultimately these studies have led to the clinical use of tenofovir disoproxil (23) for treatment of viral infections [59]. While the POC compounds may have lower levels of chemical stability than their POM analogues [60], this can be used to advantage in preparation of the parent phosphonates from POC-protected intermediates [61].
Other ester prodrugs have been based on derivatives of 2-mercapto ethanol. These prodrugs are reminiscent of the POM prodrugs in that cellular cleavage to the free drug is mediated by esterases. For example, S-acylthioalkyl ester (SATE) derivatives undergo enzymatic hydrolysis of the ester, thus liberating a thiol which in turn can attack carbon to afford episulfide and the free drug [62]. This strategy avoids formation of formaldehyde but instead yields episulfide and pivalic acid which may interfere with carnitine metabolism as noted above. However, an advantage of this group is that it can be applied to both phosphonates (e.g. compound 24, [63]) and phosphates (e.g. compound 25, [64])
The POM (26), SATE (27), and POC (28) esters all undergo cleavage through an initial ester hydrolysis, and there is some similarity in the non-drug products formed. As noted above, hydrolysis of a POM compound (26) releases both pivalic acid (29) and, after decomposition of the resulting intermediate 30, formaldehyde (31) and the parent drug (32) (Scheme 1). Hydrolysis of a common SATE ester (27) also affords pivalic acid (29) and a thiol intermediate (33) that decomposes to give episulfide (34), which may be as problematic as formaldehyde. Hydrolysis of a POC compound (28) affords isopropyl alcohol (35) and ultimately carbon dioxide (36), which may be the least worrisome byproducts in this series, but issues such as the stability of the prodrug also must be considered.
Scheme 1.

The products of POM, POC, and SATE prodrug cleavage
3.2 Unsymmetrical diesters
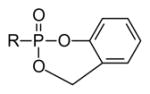
In contrast to symmetrical phosphonate diesters, which do not result in a stereogenic center at the phosphorus atom, unsymmetrical phosphonate diesters (or phosphate triesters) introduce asymmetry at this position. However, unsymmetrical diesters also may allow deployment of different strategies for deprotection, or even distribution of a second drug to the cell [65], while symmetrical diesters may be limited to a single strategy for unmasking the parent drug. For example, since the introduction of the cycloSal approach [66] several substituted salicyl alcohol derivatives have been employed to prepare different prodrug forms of nucleotides and carbohydrate phosphates and phosphonates [67]. Even the early studies on cycloSal, which employed derivatives designed to be removed by chemical means once within the cell, showed that this prodrug form can afford additional efficacy. For example, 100–600 fold increases in activity relative to their respective non-phosphorylated controls have been observed [68]. However, while comparisons of cycloSal groups in phosphate triesters with the activity of the parent phosphate ester would be more attractive, it also can be more difficult to determine if, for example, the phosphate has limited stability in the culture medium.
Studies performed on acyclic nucleoside phosphonates prepared as cycloSal prodrugs revealed a limited impact on cell permeability and release of the prodrug 37 relative to the parent phosphonate PMEA [69]. Here the cycloSal approach imparted only 1–2 fold improvement in cell activity relative to the free phosphonic acids, which was less than cell-cleavable approaches such as the bis-POM strategy. To the extent that removal of the cycloSal group is dependent upon chemical hydrolysis, with no additional increase in hydrolysis seen in the presence of plasma, once such a prodrug enters the cell it also may diffuse out rather than be unmasked which would result in limited increases in potency. Realizing this limitation, the Meier group has prepared newer cycloSal derivatives that impart a cell cleavable component prior to the chemical hydrolysis step [70,71]. These papers describe second and third generation cycloSal technology as well as some dramatic improvements in efficacy.
Studies on cycloSal prodrugs have continued for many years, and they have been well-rewarded. For example, classical resolution of stereo-isomers at phosphorus potentially is possible whenever the cycloSal pro-drug is a derivative of a nucleoside or nucleoside analogue that contains stereogenic centers in itself [72]. An early observation of an 11-fold difference in the biological activity of the phosphorus stereoisomers [72] has encouraged development of methods to allow stereocontrolled synthesis. Recently it has proven feasible to use valine-derived auxiliaries (e.g. 38) to control formation of the phosphorus stereocenter via Pv chemistry [73]. Through controlled synthesis of the phosphorus stereocenter, and judicious placement of a methyl substituent on the aromatic ring, it was possible to observe 7- to 20-fold differences in the antiviral activity of d4TMP pro-drugs (e.g. 39) [74]. The biochemical and/or physiological basis for such significant differences in activity is/are not yet clear, but they clearly validate the effort devoted to diastereoselective syntheses and justify additional research into the cycloSal prodrugs. Applications of the cycloSal group as a leaving group in synthesis of 1,6-diglycopyranosyl-phosphates only further enhances the value of this group [75,76].
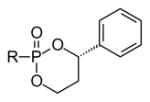
A different approach based on unsymmetrical diesters deliberately places a stereogenic center in a diol esterified to the phosphorus moiety. Although this introduces a stereogenic phosphorus atom, it also can allow tissue-targeted cleavage. Erion and colleagues have pioneered the use of this type of cyclic phosphonate prodrug, e.g. compound 40 (reviewed by [77]). Such compounds exhibit high stability in plasma but undergo rapid oxidative cleavage mediated by a cytochrome P450 enzyme expressed in the liver (CYP3A4) [78] and thus are known as HepDirect prodrugs. When compared to the bis-POM prodrug of adefovir, it was found that the HepDirect approach causes higher drug accumulation in the liver and lower accumulation in the kidney and intestine in rats after oral delivery [79]. This strategy is effective with phosphonates and phosphates, given that similar distribution was seen with a HepDirect modified AraC [80]. In this study a cyclic 1-(aryl)-1,3-propanyl prodrug of AraC was evaluated for its ability to target this compound to the liver. There was a substantial increase in mice, compared to the non-phosphorylated AraC as a control. The HepDirect technology also was applied to a 2′,3′-carbonate to make an orally available compound (39%) with high liver potency [81,82].
The HepDirect strategy has been applied to a variety of compounds other than nucleotides or nucleotide analogues. For example, compound 41 was prepared as a prodrug for a thyroid hormone receptor agonist, and was shown to lower cholesterol and triglyceride levels with decreased impact on other tissues [83,84]. A variety of compounds in this vein has been prepared, and the various stereoisomers were separable by column chromatography and HPLC on a nonracemic column. Unfortunately, the simple aryl compounds had limited aqueous solubility and functioned as inhibitors of CYP3A4. Through use of a pyridyl or substituted pyridyl ring system (e.g. 42), inhibition of CYP3A4 was diminished, water solubility was increased, and more effective inhibition of glucose production was observed, especially in the 2R,4S isomer [85]. The general strategy also has been applied to prepare prodrugs of a phenol (43) [86] and an alcohol (44) [87].
The mechanism of drug release from the prodrug 45 is believed to involve enzymatic oxidation of the benzylic position to afford the hemiketal 46 [80]. Because this oxidation takes place primarily in the liver, the prodrug effectively targets this organ. After ring opening (47), a β-elimination results in liberation of the drug (49) and an enone (48) derived from the masking group. It has not yet been identified whether or not generation of the enone, a potential Michael acceptor, is problematic. Regardless, the authors suggest that the mechanism of HepDirect prodrug release results in gradual delivery of the original drug, therefore indirectly delaying its elimination [87].
3.3 Monoesters
While small lipophilic diesters can neutralize two negative charges in phosphates (or phosphonates) and thus facilitate passive diffusion across biological membranes, it has long been recognized that monoesters derived from larger lipophilic groups also may offer advantages. Among those advantages, monoesters avoid the complexities of phosphorus stereochemistry in the salt form (e.g. the anion of compound 50). In some very early work this was exploited with cholesterol esters. For example, the cholesteryl ester of 5-fluoro-2′-deoxyuridine was prepared via a DCC coupling of the nucleotide with cholesterol [88], and subsequently tested for activity in Ehrlich ascites cells. It displayed modest activity even though it is almost insoluble in water [89]. Much more recent studies have provided more facile synthetic routes [90] to the cholesteryl esters, but further studies on the efficacy of this prodrug strategy have yet to appear.
Studies with other monoesters have been both more extensive and more successful. Perhaps most notable is work with lysophospholipids (or their analogues) as phosphonate masking groups [91,92]. Because lysophospholipids are metabolized by phospholipase A2 cleavage of dietary phospholipids and readily absorbed in the GI tract, it was hypothesized that conjugation with nucleotide analogs would enhance the oral bioavailability of the associated drug. An early study by Hostetler demonstrated the feasibility of the phospholipid conjugation approach through preparation of acyclovir diphosphate dimyristoylglycerol, which can be viewed as an analog of the naturally occurring CDP-DAG. This compound was very effective at inhibiting replication of herpes-simplex virus (HSV) in cells deficient of thymidine kinase [93]. Likewise, several alkyl glycerol-3-phosphate derivatives of acyclovir and azidothymidine with extended alkyl chains increase intracellular levels of the monophosphate by over ten-fold, with strong in vitro anti-viral activity [94]. These compounds also had an elevated and sustained oral bioavailability relative to the non-phosphorylated drug. This approach reduces the levels of the drug in the kidney and increases its levels in the liver. Differences also have been noted in the lung accumulation with varied lipid moieties [95].
Some of the strongest effects of this strategy were observed with lipid esters of the acyclic nucleoside phosphonates [91]. Across evaluation with a number of viruses, there were routinely several log unit increases in activity associated with use of the lipid esters. Of special significance was a >10,000-fold increase in in vitro potency of hexadecyloxypropyl (HDP)-(S)-HPMPA against HIV-1 [96], a >5,000-fold increase in potency of the HDP-HPMPA against orf virus [97], and a >2,000-fold increase in potency of HDP-CDV against Epstein–Barr virus (EBV) [98] relative to the respective free acyclic nucleoside phosphonates. These increases may be a consequence of their association with membranes leading to increased cellular half-lives. One indication of the effectiveness of this strategy is that two lipid nucleoside conjugates presently are in clinical trials. Brincidofovir (51, CMX001) is an HDP-conjugated cidofovir in trials for CMV [99], while the related prodrug CMX157 (52) is an HDP-conjugated tenofovir in trials for HIV. This progress certainly encourages further investigation of phosphonate monoesters [100,101].
3.4 Cyclic monoesters and mixed diesters
Specific examples of acyclic nucleoside phosphonates offer unique opportunities for prodrugs based upon an intramolecular cyclization. For example, cidofovir (53) can be converted to a cyclic monoester (cyclic cidofovir, 54) which in turn can be further modified at the remaining acid function (reviewed by Krylov [102]). As a prodrug, the cyclic cidofovir 54 offers advantages over the parent compound in that only one ionizable function remains, and hydrolysis to the parent drug does not release any additional products. At the same time, upon ionization of compound 54 there is no longer a stereogenic center at phosphorus, so the only stereochemistry is that inherent to cidofovir itself. The cyclic form of cidofovir also was reported to have 10- to 40-fold lower nephrotoxicity than the parent compound [103]. However, formation of derivatives may further increase uptake and allow targeting [103].
One cidofovir derivative in this vein was based upon salicylate esters (e.g. 55). For example, formation of the aryl ester from n-butyl salicylate gave a mixture of the axial and equatorial phosphorus esters, but these diastereomers were separated. The axial isomer showed greater chemical stability, and thus was assayed as an individual enantiomer. Based on the biological results, including stability in plasma and in liver and intestinal homogenates, it was hypothesized that breakdown occurs via an esterase-mediated hydrolysis of the carboxylate ester of the salicylate, followed by complete release of the drug [104]. The use of long lipid esters also has been investigated [105]. For example, the efficacy of phospholipid esters of cyclic cidofovir (e.g. 56), cidofovir, and HPMPA has been investigated, providing valuable comparisons [106,107].
An intriguing approach to cyclic cidofovir prodrugs has employed ester linkages to serine derivatives (57) [108,109]. In addition to reduction of the phosphonate charge, this approach may impart the ability for the prodrug to act as a substrate for peptide/amino acid transporters in the GI tract. However, while uptake was increased, especially with a Val-Ser derivative, it was not due to hPEPT1 transport as these compounds bind to hPEPT1 but do not act as substrates [110]. Use of amino acid esters also has been applied to prepare prodrugs of other nucleoside analogues including HPMPA, and at times improved oral availability was observed [111].
More traditional masking groups also have been used in combination with an internal ester. The acyclic nucleotide analogue based on 2,6-diaminopurine has been further modified as its POM (58) and HDP derivatives [112], suggesting that virtually any ester (vide supra) or amide (vide infra) might be employed to obtain derivatives of the cyclic esters available from nucleotide analogues such as cidofovir.
Finally, internal esters are not limited to acyclic nucleoside phosphonates or even to phosphonates in general. For example, the cyclic nucleotide 59 has been reported to show very promising activity against the hepatitis C virus [113,114]. In this case the issue of phosphorus stereochemistry is ameliorated by selective formation of the desired stereoisomer, which has been accomplished on a kilo scale including purification by crystallization [115]. A variety of different phosphate esters of this general structure has been reported, and studies have described a mechanism of drug release that may include cytochrome P activation [114]. Again in this case, the reported studies strongly suggest that even more varied phosphate esters could be employed.
4 Amidate prodrugs
While many have employed phosphorus esters to prepare prodrugs, others have developed phosphoramidate derivatives (reviewed by Mehellou [116]). As early as 1990, Devine and McGuigan had hypothesized that nucleotides containing at least one phosphoramidate linkage would exhibit cellular conversion to the free phosphate, potentially enhanced by activity of HIV protease [117]. Extensive studies in the intervening years have demonstrated amply the validity of that hypothesis with both diamidates and aryloxy phosphoramidates (reviewed in [118]), and each class offers advantages. Examples will be described in the following sections.
4.1 Symmetrical diamidates
As described above for diesters, it can be argued that from a conceptual standpoint the simplest phosphordiamidates are those where symmetry avoids the issues inherent to introduction of phosphorus stereochemistry. Recently, McGuigan and colleagues have claimed just such advantages for a diamidate strategy [119,120]. When the amide is derived from an amino acid, achieving a neutral species requires esterification of the carboxyl group as well (e.g. 60). After examination of a series of twenty-five diamidates for activity against HCV [120] and HIV replication, and anti-proliferative effects in several cell lines, it was found that this strategy is sufficient to afford good cellular activity with several different nucleoside analogues [119]. Furthermore, preparation of symmetrical bisamidates from the symmetrical diesters has been reported [121].
Symmetrical bisamidates also have been used to prepare prodrugs of phosphonates. For example, compound 61 was prepared as an inhibitor of fructose 1,6-bisphosphatase [122]. The phosphonodiamidate prodrug was far more potent in human hepatocytes than the free acid and, after comparison with a broad range of other prodrugs, the bisamidate was judged superior and selected for clinical trials [123]. While Phase II trials with compound 61 ultimately proved disappointing, studies with second generation analogues continue [124].
4.2 Amidate esters
Some of the earliest work on azidothymidine (AZT) monophosphates reported that simple alkyl diesters were inactive in a cellular model of HIV but that the combination of an alkyl monoester with a phosphoramidate led to surprisingly good cellular potency [117]. In an impressive series of studies that continue to this day [125] McGuigan and colleagues evaluated lactate-derived systems and diaryl phosphates, before identifying aryloxy phosphoramidates as a highly efficient prodrug approach (reviewed in [118]). Work applying the aryl phosphoramidate technology to nucleoside phosphonates has led to the clinical trials of several such agents.
Most recent work on aryl phosphoramidates has employed protected L-alanine esters for the amine moiety [126]. In several cases where the D-alanine analogues were prepared, they were found to display significantly lower activity [127–129]. One comparison of PMPA prodrugs reported that a phenyl phosphoramidate displayed 10-fold greater activity than the corresponding POC prodrug.
While the amide component of phosphoramidates appears to be centering on L-alanine derivatives, considerable variations in the aryl ester component are still reported. Simple phenyl esters may well be the most extensively employed substructure [130,131], especially in nucleotide analogues such of the general structure 62 [116,127,132–136]. However in cases such as the hydroxamic acid 63, an inhibitor of 6-phosphogluconate dehydrogenase for use in trypanosomiasis, addition of simple substituents such as methyl groups to the phenyl ring has resulted in more than a 10-fold increase in potency [137]. In some cases, deployment of a naphthyl ester (e.g. 64) has accompanied preparation of the phenyl ester [116,132,133] while other studies have focused on the naphthyl esters [138–141].
While aryl phosphoramidates based on amino acids are by far the most common examples of this class, further variations in the ester and amine components are certainly known [142]. For example, in place of an aryl ester, a 3-acyloxymethoxypropyl group has been studied (e.g. 65), and hydrolysis of the ester was found to be ~20-fold faster than the corresponding phenyl phosphoramidate [131]. In this work, small differences in the reactivity of the RP and SP isomers were noted, but it was not possible to identify which one was the more reactive.
An interesting alternative to amines derived from amino acids is based on the 4-chlorobutyl system as reflected in compound 66. After reduction and loss of the nitrofuryl group, cyclization of the butyl group delivers the active nucleotide, as shown by the preservation of activity in cells deficient in the kinase that generates FdUMP [143] or AraC monophosphate from the corresponding nucleosides [144].
Finally, a bidentate ligand also has been employed to prepare cyclic phosphoramidate esters such as compound 67 [145]. The (N-3-hydroxypropyl)-amino esters could be prepared from commercially available amino acids, ultimately allowing preparation of both the SP and RP isomers, and the phosphorus stereochemistry was assigned based on NMR data. Small differences in activity were found between the phosphorus stereoisomers, and the most active compounds in the series were comparable or slightly greater in potency when compared to the POM analogue.
4.3 Monoamidates
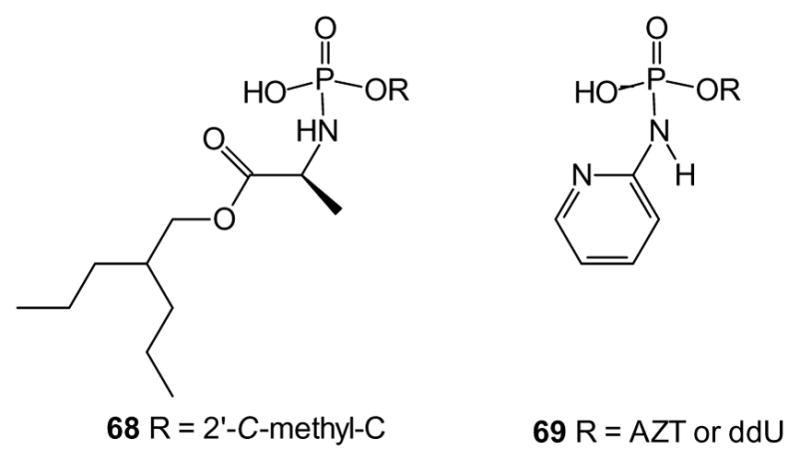
Symmetrical diamidates do not introduce a stereocenter at phosphorus, but in cases such as aryl (or alkyl) phosphoramidates where two different protecting groups are employed, introduction of a stereogenic center at phosphorus can become a concern. Formation of a stereocenter can be avoided if but one amidate is used, as long as the single negative charge in the resulting salt is resonance delocalized, and an additional benefit is that drug release does not generate phenol. One recent study of monoamidates prepared compounds such as 68, where the longer chain alanyl ester was intended to regain some of the lipophilicity sacrificed by removal of the phenyl ester [146]. The resulting compound proved very effective at generating high levels of the nucleoside triphosphate in human hepatocytes.
A similar strategy was employed in a series of AZT and ddU analogues (e.g. 69) [46,47]. In this work, aryl amines were used to generate the monoamidates, and those based on pyridyl amines displayed notably low cytotoxicity and high antiviral activity. However, because no activity was observed in thymidine kinase deficient cell lines, further work will be necessary to clarify the specific mode of action.
5 Drug release
In addition to neutralizing negatively charged phosphorous moieties to allow diffusion across cellular membranes, it has become apparent that the appropriate selection of ester and/or amide prodrugs can be used to impart cell type and tissue type specificity which can impact the distribution of the free drug. Specifically, by choosing the processes that are entrusted to remove the protecting groups, whether chemical decomposition or enzymatic cleavage, one can control where and when the drug is released. Each drug/prodrug/target must be optimized individually as some masking groups can be more or less readily removed from different types of drugs [147]. However, we will here attempt to describe some general guidelines for planning drug release.
The majority of current prodrugs require enzymatic cleavage prior to biological function. Conceptually speaking, prodrugs designed to undergo chemical decomposition (e.g. simple esters, early cycloSal compounds, or thermolytic prodrugs) offer the least amount of control over tissue distribution. However, by altering the chemical stability to control drug release rates these compounds may offer advantages in terms of dosing, such as enhanced plasma stability and precisely controlled half-lives of elimination [148]. For example, some thermolytic prodrugs of DNA oligonucleotides have been examined [149]. Based on these studies, it was suggested that an optimal half-life of a thermolytic prodrug for animal work is between 100–200 hours at 37 °C, and that resistance to enzymatic cleavage allows better distribution of the drug to the cellular targets. In addition, depending on the masking group, thermolytic release has the potential to reduce accumulation of toxic intermediates and to overcome limitations in esterase activity on charged substrates when multiple groups need to be removed [150,151].
Ester-based compounds such as the POM analogs undergo relatively slow chemical decomposition but are generally susceptible to cleavage by non-specific esterases which can be found in most cells as well as the blood. For example, a representative bis-POM compound was demonstrated to have a five minute half-life in plasma [152]. The mechanism of cleavage involves an initial enzymatic step followed by spontaneous release of the linker. Interestingly, while the first POM group is rapidly removed leading to formation of the monoester, the mono-POM compound has a much longer half-life in plasma, and would likely offer advantages in terms of membrane permeability relative to the free diacid. Ultimately, the most promising use of POM-modified compounds may be to increase oral bioavailability, allowing accumulation of the free drug in the plasma at elevated levels, but not necessarily offering strong advantages in terms of tissue distribution relative to IV dosing.
When selecting which ester groups to use to increase oral absorption of a particular compound, it is important to keep in mind that the majority of neutral protecting groups will greatly increase oral absorption. Therefore, it may be more important to optimize the rate of release (i.e. esterase susceptibility) rather than attempting to maximize the rate of oral absorption. An excellent example of this rational was shown for various ester prodrugs of squalene synthase inhibitors [153]. Here, the authors clearly demonstrate a reduction in oral activity occurs when protecting groups are either removed too quickly or removed too slowly.
In contrast to the POM prodrugs, HDP esters require phospholipase C for removal of the protecting group and tend to have increased stability in the blood. This affords the actual drug with both increased oral availability and increased tissue distribution relative to IV dosing. The phospholipid masking groups tend to greatly increase the duration of exposure to the drugs, which can at times be detected even up to 10 days later [154]. Presumably drug release requires prodrug insertion in the cell membrane followed by slow cleavage intracellularly via phospholipase C metabolism. This combination of uptake, plasma stability, and long lasting cellular effects makes the mono phospholipid strategy especially effective for oral delivery of drugs such as brincidofovir, a highly promising antiviral agent.
Cleavage of the prodrugs of cyclic cidofovir and other related nucleotides requires several enzymatic steps to achieve the free drug. For example, the dipeptide prodrugs put forth by McKenna require removal of one of the amino acids by puromycin-sensitive aminopeptidase and spontaneous release of the remaining amino acid [155]. Modulating the enzymatic step that removes the first amino acid might provide an additional means to achieve tissue targeting depending on the expression of that enzyme. This hydrolysis likely must precede ring opening, which is also enzymatically-driven [156]. The specific mechanisms of ring opening are still emerging, with PDE7A recently identified as one enzyme important to this process [157].
The cyclic ester prodrugs have further enhanced tissue specificity of drug release, especially with the HepDirect system. Due to its specificity for metabolism by CYP3A4, accumulation of the free drug in the liver is favored, the predominant site of CYP3A4 expression. Likewise, while early generation cycloSal derivatives lacked selectivity, more recent modifications of these structures have afforded compounds that are stable in the presence of serum but not in cell extracts, thus offering some advantages over POM prodrugs [158].
The phosphoramidates also exhibit increased plasma stability relative to the acyloxyalkyl ester compounds. Whether diamidate (e.g. 70), or aryl amidate (71), these compounds appear to be released through an initial enzymatic hydrolysis of the amino acid carboxyl ester [159]. In the case of diamidates and aryloxy amino acid amidates this initial step is catalyzed by carboxypeptidase Y (cathepsin A) or carboxylesterase I [160], to afford the corresponding carboxylate (72 or 73). By increasing specificity for cathepsin A one can target the free drug to tissues such as peripheral blood mononuclear cells which have been found to highly express this enzyme [161,162]. The initial hydrolysis leads to a spontaneous elimination of one of the protecting groups, which may involve the cyclic intermediate 74 leaving a monoamide intermediate [163]. The amidate 75 subsequently is cleaved by cellular phosphoramidases such as Hint1 [160,164] to release the free drug (76). Mono phosphoramidates have been evaluated for enzymatic cleavage by human Hint1, and a set of guidelines for specific phosphoramidate cleavage by Hint enzymes has been established. For example, purine bases may be preferred but pyrimidine bases also can be used, phosphoramidate oxygen atoms are required, and an electrophilic group is required at the ribose 2′-position, while sterically-crowded amines reduce cleavage [165].
A set of phenol-containing phosphonoamidates with various amino acid esters has been tested against a panel of cellular serine and cysteine proteases. This study found large differences in the metabolism of the prodrugs, such that modifying the amino acid ester group can completely change the enzyme activity. It was noted that the alanine isopropyl amidate bearing a phenyl ester is metabolized by cathepsin A [166]. The authors suggest that based on differential tissue expression of these proteases, one might be able to design specific phosphonamidate prodrugs that will be preferentially metabolized in various tissues.
Some interesting differences in drug release also may occur based on the subcellular localization of the enzymes responsible for removal of the protecting groups. Much like prodrugs that target a specific cell type, prodrugs that target a particular subcellular locale would be best served by undergoing hydrolysis by a singular enzyme with known subcellular distribution. However, it should be noted that this approach is not without risks, as lack of redundant conversion pathways may increase the potential for drug interactions or sensitivity to genetic polymorphisms [167]. Nonetheless, there have been several attempts to develop single enzyme cleavage systems. For example, the phospholipase C driven release of HDP prodrugs might be expected to occur primarily in the cytoplasm or at the intracellular face of the plasma membrane [168]. Likewise, HepDirect prodrugs (which require CYP3A4) would be similarly released in the cytoplasm near the inner face of the plasma membrane [169]. On the other hand, release of phosphoramidates by Hint1 may occur in multiple compartments, as this enzyme has been reported to exhibit both cytoplasmic and nuclear localization [170]. However, phosphoramidates that are substrates for carboxypeptidase Y may be expected to be released in the lysosome, which would confer an advantage against targets that are lysosomal but would require membrane diffusion or active translocation of the free acid to reach cytoplasmic targets. Indeed, GS-9191, an amidate prodrug of 9-(2-phosphonylmethoxyethyl)-N6-cyclopropyl-2,6-diaminopurine, was demonstrated to be initially hydrolysed by cathepsin A in lysosomes followed by pH-dependent translocation to the cytoplasm [171].
Finally, in few select cases, it is not even necessary to remove the protecting groups in order to observe biological activity. For example, methylated phosphates were able to function as agonists for the adenosine A1 receptor, a GPCR, without removal of the methyl group, resulting in an enhanced in vivo bioavailability and pharmacodynamic effect [172]. Likewise, simple esters that block the ATP gated ion channel P2X were able to offer a cardioprotective effect [173]. Findings such as these suggest that while removal of the masking group is required for activity of antiviral agents and inhibitors of isoprenoid biosynthesis, both of which require coordination of the negatively charged phosphate in an enzyme active site, it is not necessary for certain phosphorous moieties including some receptor agonists. While binding to the receptor may be reduced by the presence of a small masking group, the pharmacokinetic advantages may outweigh a small loss in ligand binding affinity.
6 Current applications of prodrug technology
In recent years, there has been a dramatic increase in the synthesis of phosphate and phosphonate prodrugs. It could be argued that synthesis of prodrug forms is becoming the norm during generation of these drugs. In particular, two classes of prodrugs, the classic acyloxyalkyl phosphonates and the amidates [116,174], account for the majority of recent examples. Here, we attempt to highlight some important recent developments in three key areas (nucleotides, isoprenoids, and phosphorylated proteins) for which novel applications of prodrugs are revolutionizing their respective fields.
6.1 Nucleosides, nucleotides & nucleic acid metabolism
An intense area of research into phosphate and phosphonate prodrugs has centered on development of effective antiviral agents. The discovery of HIV stimulated tremendous efforts in this regard [59], and the continued importance of the disease encourages continued efforts in this area, including use of prodrugs such as tenofovir disoproxil as preventative agents [175]. Furthermore, increasing concern for viral infections such as HCV [100,135,176,177] and herpes virus [56] also motivates research. Because there are numerous reviews focused on prodrugs of nucleotides and nucleoside phosphonates [178–184], the following paragraphs focus primarily on other emerging applications in the nucleotide area.
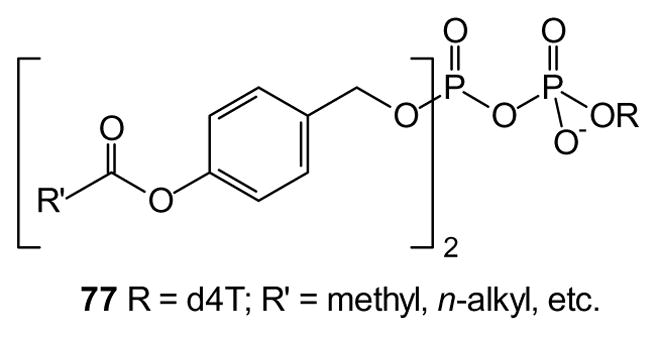
Much of the past research into nucleotide and nucleoside phosphonate prodrugs has been designed to afford drugs that include phosphorus and so do not require formation of a monophosphate through a kinase mediated reaction. This strategy, often referred to as a kinase bypass [15–18], can circumvent development of drug-resistant strains through mutations that delete one specific kinase. An intriguing alternative has been suggested through formation of prodrug forms based on nucleoside diphosphates (e.g. 77) [185,186]. Compounds with acyl residues greater than C6H13 were found to be highly active in a CEM/TK− assay, demonstrating that this is an effective strategy for intracellular delivery of nucleoside diphosphates. It remains to be seen whether this or a similar strategy may be extended to other types of diphosphates or even triphosphates.
At the same time, applications of the more traditional kinase bypass strategy to novel nucleoside analogues continue to appear. In this context, a recent paper described phenyl phosphoroamidate 78. After efforts to optimize the nucleoside analogue, both phosphorus stereoisomers were isolated and identified by crystallography. The more active SP isomer, compound 78, then became the first C-nucleoside to enter clinical development for treatment of HCV through inhibition of NS5B polymerase [187]. These findings illustrate that one consequence of the aryl phosphoramidates is the introduction of a new stereogenic center at phosphorus [188,189] which can dramatically impact biological activity. However, the majority of studies make no mention of this stereochemistry suggesting that assays were conducted on diastereomeric mixtures. Consistent with these results, another elegant study separated the phosphorus-centered diastereomers of compound 79, identified them by diffraction analysis, and assayed the individual isomers, reporting that the SP diastereomer shown displayed significantly greater activity [113,190]. The prodrug 79 produces high levels of the triphosphate in multiple species following oral dosing. Its toxicity is low with high potency towards HCV even in resistant cells and so it was recently approved for treatment of HCV (as Sofosbuvir) [191,192]. This success suggests that the issue of phosphorus stereochemistry will need to be addressed in other aryl phosphoramidates as well. It is interesting to note that in both cases the SP isomer was more potent, however neither the underlying cause of this phenomenon nor its prevalence are known at this time.
In addition, prodrug strategies are continuing to find application in other non-viral applications. For example, several prodrug forms of acyclic immucillin phosphonates have been prepared in an effort to identify anti-malarial agents that function through inhibition of purine biosynthesis in plasmodium. While the POM modified phosphonate 80 is able to penetrate erythrocytes, hydrolysis there limits drug entry into the parasite. However, lysophospholipid prodrugs (e.g. 81) are able to deliver the compound to the parasites, where they are cleaved by phospholipase A and C, releasing the free phosphonic acid [193]. This target also has been addressed with bisamidate prodrugs of bisphosphonates such as compound 82 as potential antimalarial agents [194] and antibiotics [195]. Taken together, these studies reiterate the importance of optimizing each prodrug-drug-target combination.
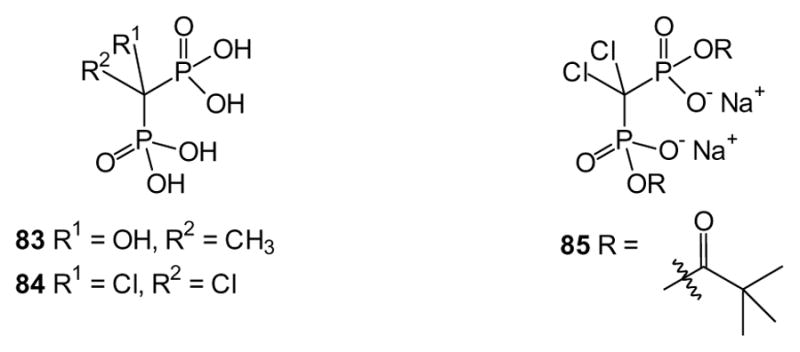
Some groups have evaluated prodrugs for their ability to prevent precipitation of phosphorus compounds. It has long been recognized that geminal bisphosphonates can be viewed as analogues of diphosphate which will not readily undergo hydrolysis. Derivatives with small substituents on the methylene carbon, compounds such as etidronate (83) and clodronate (84) are sometimes referred to as first generation bisphosphonates, because of their early clinical use [196]. In vivo these bisphosphonates are converted to ATP analogues that may directly trigger apoptosis [197,198], but as small, highly charged molecules they do not readily cross the cell membrane by means other than endocytosis. Various prodrug approaches have been examined to increase their ability to enter cells. For example, several bis-, tri-, and tetra-POM bisphosphonates including derivatives of clodronate [35] and etidronate [199] have been prepared, while a series of dianhydride derivatives of clodronate also has been made (e.g. 85) [200]. In the latter study, the acetyl, butyroyl, and benzoyl derivatives display half-lives in human serum of less than one minute, while the dipivaloyl bisphosphonate has a longer half-life of 3.3 hours. The authors demonstrate that while the solubility of the dianhydrides was less than clodronate, the compounds maintain their solubility in the presence of increasing concentrations of calcium, while clodronate does not. Therefore, compounds of this type may increase the oral bioavailability of clodronate and related bisphosphonates. More recently, others have examined a phosphonoamidate prodrug of clodronate [201]. However, while these approaches are likely to increase oral bioavailability of the compounds, their low predicted stability in the plasma may ultimately lead to cleavage of the prodrug and sequestration of the bisphosphonate to the bone rather than allow for distribution to other tissues.
6.2 Isoprenoids and inhibitors of isoprenoid metabolism
A second pathway based upon small molecule diphosphates, isoprenoid biosynthesis, may rival the importance of nucleotide metabolism in treatment of human disease [202]. Because isoprenoids have important roles in heart disease, bone disease, and cancer, various groups have investigated ways to elevate the concentration and target the delivery of isoprenoid pathway inhibitors using prodrug strategies.
Isoprenoid diphosphate synthases
At the heart of cellular isoprenoid biosynthesis pathways lie the isoprenoid diphosphate synthases, including farnesyl diphosphate synthase and geranylgeranyl diphosphate synthase [203]. A number of clinically successful agents used to treat osteoporosis and other bone diseases, collectively known as nitrogenous bisphosphonates and considered to be the second and third generations of methylene bisphosphonate derivatives, are inhibitors of farnesyl diphosphate synthase [196]. The nitrogenous bisphosphonates are highly effective inhibitors in part because their bisphosphonate substructure mimics the functionality of the natural di-phosphates. In addition to imparting potent enzyme inhibition, this highly charged bisphosphonate group serves to localize the nitrogenous bisphosphonates to the bone microenvironment, where they achieve long-lasting and local concentrations which are capable of inhibiting osteoclast function. Because bisphosphonate drugs exhibit anticancer effects in the bone, some groups have hypothesized that directing these drugs to other locales through synthesis of bisphosphonate prodrugs would lead to anti-proliferative effects against soft tissue cancers. Simple alkyl and aryl bisphosphonates have met with some success [204], but more readily cleaved prodrug forms may be more rewarding.
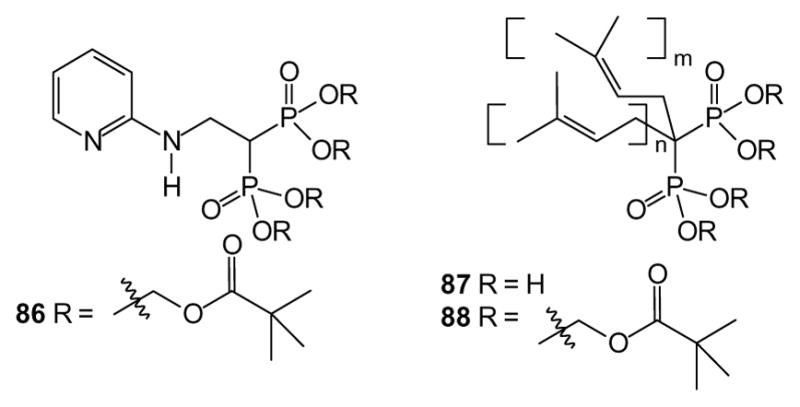
In 2006, Oldfield and colleagues published synthesis and evaluation of a tetra-POM bisphosphonate (86) that inhibits farnesyl diphosphate synthase. While not evaluated for oral availability, this compound was able to inhibit in vitro growth of cancer cells in a manner that was 20-fold more potent relative to the free acid [205]. This illustrates the anti-cancer potential of the target and also addresses the issue of cell permeability for this class of drugs. In 2008, we reported a series of isoprenoid bisphosphonates that function as geranylgeranyl diphosphate synthase inhibitors. Both the salt (87) and tetra-POM derivatives (88) were prepared, and growth inhibition and diminished protein geranylgeranylation were greater with the tetra POM compounds by up to 25-fold relative to their respective free acids [206]. The magnitude of the increase varied from compound to compound, with smaller compounds (with higher charge to mass ratios) benefiting more from POM protection. Taken together, these studies suggest that bisphosphonate inhibitors of both farnesyl and geranylgeranyl di-phosphate synthases exhibit difficulty entering cells, and neutralization of these compounds with a prodrug approach can increase their in vitro anti-cancer properties.
Prenyl transferases
Isoprenoid diphosphates such as farnesyl diphosphate and geranylgeranyl diphosphate are also post-translationally incorporated into proteins, increasing membrane affinity. Because some such prenylated proteins are known oncogenes, (e.g. Ras), there has been a substantial effort to develop inhibitors of the protein prenyl transferases farnesyl transferase and geranylgeranyl transferase I and II. Most efforts have focused on peptidomimetics, but a variety of add back experiments have shown that isoprenoid diphosphates enter cells and several isoprenoid diphosphate analogues also have been reported. While these analogues may suffer from the permeability problems attributed to other phosphates, there have been some reports of prodrug approaches with these compounds [207]. Recently, phosphoramidate prodrugs of the farnesyl [208] and geranyl-geranyl [209] monophosphate analogues 89 and 90 have been reported. Due to their similarity to the natural substrates, these compounds are among the most potent isoprenoid analogs reported for inhibition of the prenyltransferases. Their cellular activity was not strong, possibly due to further metabolism of the monophosphate. However, in combination with lovastatin these compounds exhibit some interesting effects on cancer cell viability, which suggests that further examination of the use of masking groups to deliver isoprenoid diphosphate analogues as transferase inhibitors may be warranted.
Inhibitors of the non-mevalonate pathway for isoprenoid biosynthesis
In contrast to humans, bacteria and parasites commonly utilize the non-mevalonate pathway of isoprenoid biosynthesis [210]. This allows one the opportunity to inhibit their growth selectively by therapeutic targeting of these enzymes. Analogous to statin drugs that inhibit the first enzyme of human isoprenoid synthesis, fosmidomycin (91) and its analog FR900098 (92) inhibit the first step of the non-mevalonate pathway, 1-deoxy-D-xylulose 5-phosphate (DOXP) reductoisomerase (Dxr). These drugs show some activity against malaria, but also exhibit low oral bioavailability. In 2001, Reichenberg synthesized a series of aryl ester modified FR900098 analogues (93) [211]. These compounds exhibited greater activity in a Plasmodium model when dosed orally relative to the free acids. Due to concerns about the potential for toxicity of the phenol metabolite, this group went on to produce a series of acyloxyalkyl (e.g. the POM compound 94) and then compounds reminiscent of POC derivatives (e.g. 95) as prodrugs of FR900098 [212,213]. At least one of these compounds shows improvement in activity over the free acid in the mouse model.
Later studies on analogues substituted at the C-1 or C-3 positions of fosmidomycin, or constrained by a ring system spanning C-1 and C-2, continued to employ bis-POM prodrugs to explore the SAR of FR900098 analogs against Plasmodium Dxr [214–217]. These compounds yielded several more potent inhibitors of Plasmodium growth in vitro, but use of the prodrug strategy in this system do not confer the magnitude of increase seen in other cellular systems. It is difficult to say if the limited efficacy is compound based or parasite specific. These results were echoed through studies of a series of fosmidomycin analogs [218], where again the bis-POM protecting strategy was only marginally effective. It is possible that fosmidomycin penetrates cells adequately of itself. The most recent studies reported a series of fosmidomycin analogs with enhanced potency against the purified enzyme and growth of the Plasmodium parasite in vitro [219]. While the activity was somewhat enhanced by bis-POM protection, the results parallel findings of limited activity of the tri-POM pro-drug squalene synthase inhibitor ER27856 in Leishmania. This again suggests either a decreased ability to penetrate the membrane of this organism or lack of ability to release the free drug [220].
Because fosmidomycin and FR900098 also are potent antibacterial agents, a prodrug approach has been examined to determine if it would increase efficacy in Gram-negative bacteria [221]. Several lipophilic esters were tested on a panel of Gram-positive and Gram-negative bacteria. The largest positive effects were seen in an M. tuberculosis strain (H37Rv), where several prodrugs were active at concentrations of 100 μg/mL versus >500 μg/mL for the free acid. Generally, the prodrugs did not enhance efficacy of FR900098 in Gram-negative pathogens, with only one out of six showing a modest increase in potency against E. coli k12. It is possible that these bacteria were not able to metabolize the prodrug or that the pro-drug is not effective at crossing the double membrane, because they are too lipophilic to transverse the hydrophilic transmembrane space. However, another group recently has reported activity of the bis-POM derivative 94 in mycobacterium [222]. All in all, future studies that focus on prodrugs of more metabolically stable compounds such as these would be well worthwhile.
6.3 Protein phosphorylation and interactions
The phosphorylation and dephosphorylation of amino acid residues in proteins is an essential aspect of many signal transduction pathways. The phosphorylated residues often promote protein-protein interactions via Src Homology 2 (SH2) domains and other protein domains that interact specifically with phosphorylated proteins. Therefore, compounds that interfere with protein to phosphorylated protein interactions are potential therapeutic agents.
Prodrugs of peptidomimetics that target the SH2 domain
Tyrosine phosphorylation is a primary component of signal transduction pathways such as those regulated by growth factor receptors. As the SH2 domain is the most prevalent of the phosphotyrosine recognition domains, it has emerged as a target for phosphotyrosine mimicking phosph(on)ate compounds and their respective prodrugs. One early study reported a series of SH2 domain blockers targeted at Src and Abl kinases [223]. The authors suggest that efficient SH2 blockers must contain a phosphate group, or a phosphonate substitute, to allow effect ligand binding. They successfully generated several dipeptide analogs including the phosphonic acid 96, the ethyl ester 97, and the di-POM compound 98 as phenylalanine phosphonates. Both esters exhibited much faster cellular uptake than the acid and the prodrug 98 was readily metabolized in cells to release the free acid.
A similar approach has been taken to prepare aryl phosphoramidates such as compound 99 [224], as prodrugs of SH2 domain analogs for Src/Lck inhibitors. The compounds exhibit low micromolar growth inhibition in Jurkat T cells, and undergo spontaneous hydrolysis with half-lives of approximately 30 minutes. The same masking groups have been applied in phosphonate 100 as a suspected SH2 domain blocker through inhibition of mitotic centromere-associated kinesin protein function in a panel of cell lines [225]. The compound inhibited cell growth but surprisingly the growth inhibition was not restricted to Src-dependent cells. The same prodrug format was used to investigate targeting protein tyrosine phosphatase 1B (PTP1B) as means of sensitizing cells to insulin signaling for treatment of diabetes and obesity [226]. In this study, cellular activity of the prodrug was seen at low nanomolar concentrations.
The goal of a similar study was to develop a phosphopeptide mimic that binds to the SH2 domain of Stat3, preventing its association with growth factor receptors and subsequent phosphorylation and translocation to the nucleus. In this case, a xenograft experiment was performed to assess activity in a MDA-MB-468 tumor model, and a ~30% decrease in tumor volume was observed after four weeks of treatment [227]. In contrast to the earlier findings [223], in this study the bis-POM compound 101, prepared as a phosphotyrosine surrogate, is significantly more effective than the mono-POM analog at producing cellular activity [228]. Subsequent studies therefore relied on the bis-POM approach to identify inhibitors of Stat3 [229,230] and Stat6 [231] binding interactions.
Finally, while most SH2 analogs have utilized POM or amidate masking groups, one lab recently has generated a cycloSal analog (compound 102) [232]. In this study, compounds were active in the micromolar range in a cell free assay system. It is not yet clear how rapidly such compounds are hydrolyzed and it may be unlikely that they would offer significant enhancement in cellular delivery of the phosphorylated peptide due to the chemical nature of the cycloSal hydrolysis. However, this report clearly demonstrates that many different prodrug strategies can be applied to obtain analogues of peptide phosphates.
Serine/threonine analogs
Only recently has the prodrug approach enabled cellular studies of phosphoserine and phosphothreonine analogs. After a phosphoserine analog with the ability to block interactions of 14-3-3 proteins was identified, a mixed phosphonoamidate ester prodrug of the corresponding difluoromethylene phosphonate was prepared (103). This prodrug exhibited approximately a 20-fold increase in activity over the free acid [233]. A recent report on phosphothreonine analogs targeted towards Plk1 also has relied upon a bis-POM strategy [234].
7 Conclusions
Incorporation of phosphates and phosphonates in potential drugs once may have been viewed as unrewarding, because of the inherent tension between a requirement for high negative charge density for bioactivity and the limited ability of such compounds to traverse the cell membrane. With the development of various prodrug forms it now has become routine. Understanding of both the chemistry and biological activity of different prodrugs has advanced tremendously since the earliest studies, and the pace of those advancements is increasing. From diesters of simple alcohols and symmetrical phosphorus species the art has advanced to necessarily asymmetric amidate esters. The phosphorus stereochemistry that once was avoided now is recognized as a potential design element and strategies for asymmetric synthesis have begun to appear. The increasing complexity of the prodrug forms has allowed evolution of drug release strategies and fosters drug targeting. These advances, together with the growing number of phosphorus-containing drugs in clinical use and clinical trials, makes clear that studies of prodrug forms will continue to be a vibrant research area.
Fig. 1.
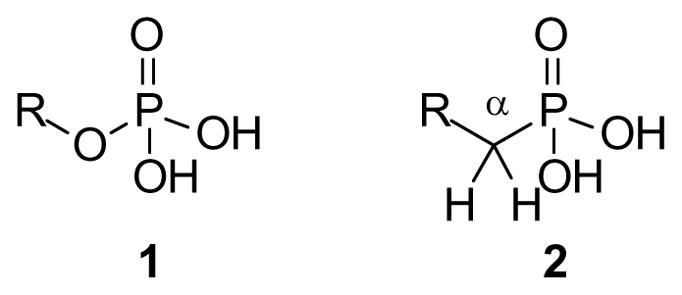
General structures of the phosphate and phosphonate groups
Fig. 2.

General structures of phosph(on)ate prodrugs
Fig. 3.

Use of phosphates to enhance water solubility of drugs
Fig. 4.

Examples of pivaloyloxymethyl (POM)-modified drugs
Fig. 5.
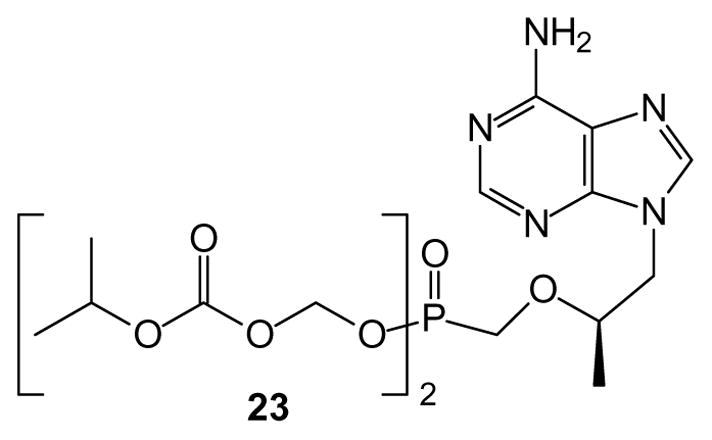
Tenofovir disoproxil, an isopropyloxycarbonyloxymethyl (POC)-modified drug
Fig. 6.

S-acylthioalkyl ester (SATE) modified nucleoside analogues.
Fig. 7.

CycloSal-PMEA (37) and some nonracemic cycloSal derivatives
Fig. 8.

The HepDirect strategy for phosph(on)ate prodrugs
Fig. 9.

Prodrugs derived from phosph(on)ate di(or mono) esters
Fig. 10.

Prodrugs including internal ester formation
Fig. 11.
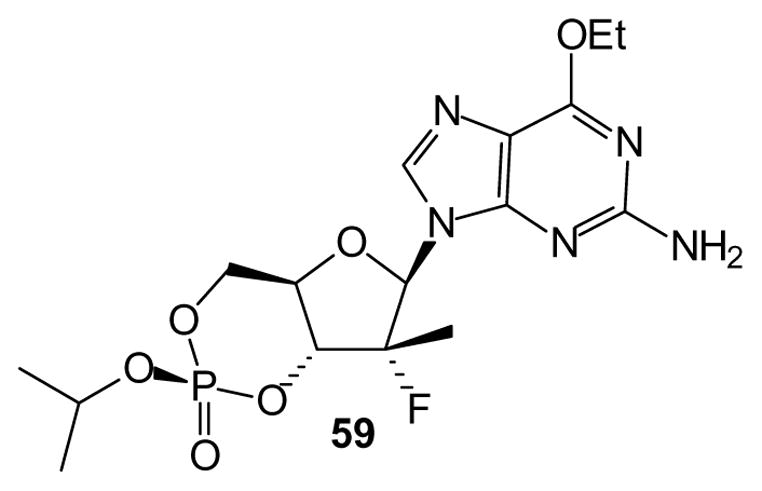
A phosphate prodrug including internal esterification
Fig. 12.

Phosphate and phosphonate prodrugs based on symmetrical di-amidates
Fig. 13.

Aryl phosphoramidate prodrugs
Fig. 14.

Alkyl phosphoramidate prodrugs
Fig. 15.
Mono phosphoramidate prodrugs
Fig. 16.

Drug release from diamidates and aryloxy amidates [120]
Fig. 17.
A diphosphate prodrug
Fig. 18.

Prodrugs assayed as the SP isomers
Fig. 19.

Prodrugs of some purine biosynthesis inhibitors
Fig. 20.
Examples of first generation bisphosphonates and a prodrug form
Fig. 21.
Prodrugs of farnesyl diphosphate synthase and geranylgeranyl di-phosphate synthase
Fig. 22.
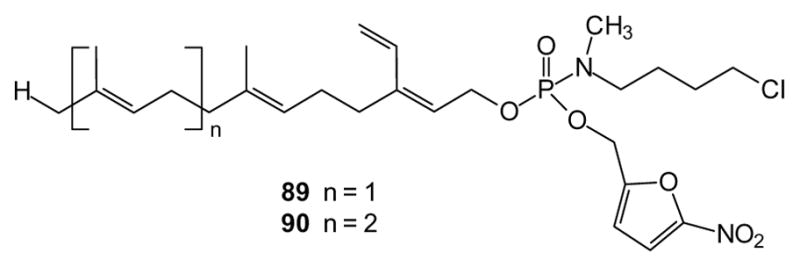
Monophosphate prodrugs that inhibit prenyl transferases.
Fig. 23.

Fosmidomycin and prodrug analogues
Fig. 24.

Bis-POM prodrug of an SH2 targeted peptidomimetic
Fig. 25.

Aryl phosphoramidate peptidomimetics
Fig. 26.
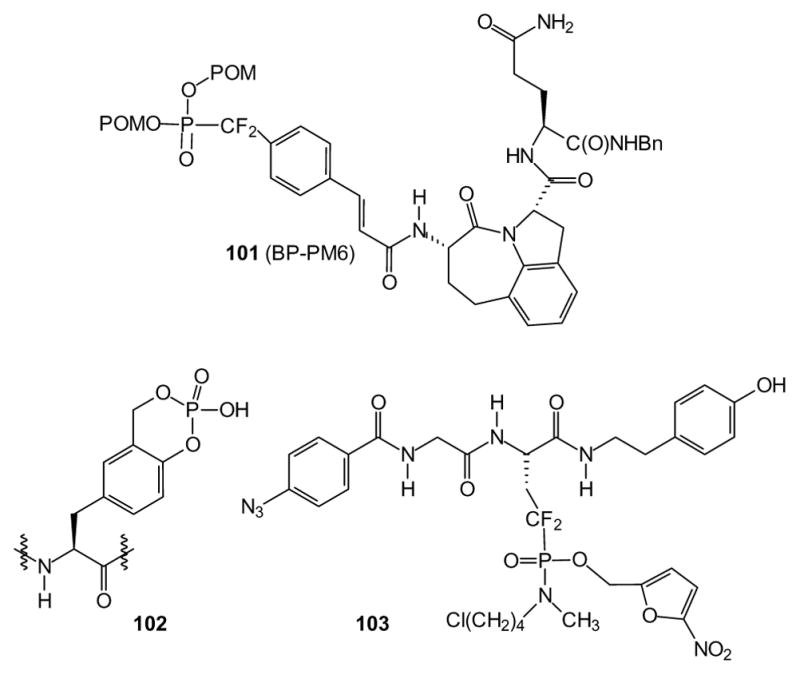
Some prodrug forms used in peptidomimetics
Scheme 2.

The products of HepDirect prodrug cleavage
Table 2.
Some prodrug variations based upon amidate linkages
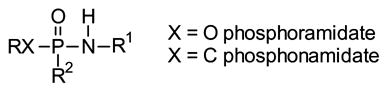
| ||
|---|---|---|
| R1 | R2 | |
| Symmetrical | ||
| Bisamidate | -CHCH3C(O)OCH(CH3)2 | -NH-R1 |
| Monoamidate- monoester | ||
| Amidate/phenyl ester | -CHCH3C(O)OCH(CH3)2 | -OC6H5 |
| Amidate/naphthyl ester | -CHCH3C(O)OCH(CH3)2 | -OC10H7 |
| Amidate/alkyl ester | -(CH2)4Cl (with NCH3) | -OCH2(C4H3NO3) |
| Monoamidates | ||
| Amidate | 2-pyridyl | -OH |
Acknowledgments
Financial support from the University of Connecticut, Department of Pharmaceutical Sciences (AJW) and from the Roy J. Carver Charitable Trust as a Research Program of Excellence and the NIH (DFW) is gratefully acknowledged.
List of Abbreviations
- AraC
arabinofuranosyl cytidine
- AZT
azidothymidine
- CMV
cytomegalovirus
- d4TMP
2′,3′-didehydro-3′-dideoxy-thymidine-5′-monophosphate
- DOXP
1-deoxy-D-xylulose 5-phosphate
- EBV
Epstein-Barr virus
- GCPR
G protein coupled receptor
- GemC
gemcitabine
- HBV
hepatitis B virus
- HCV
hepatitis C virus
- HDP
hexadecyloxypropyl
- HIV
human immunodeficiency virus
- HPMPA
9-(3-hydroxy-2-phosphonyl-methoxypropyl)adenine
- HSV
herpes-simplex virus
- NA
nucleoside analogue
- PMEA
9-[2-(phosphonomethoxy)ethyl]adenine
- POC
isopropyloxycarbonyloxymethyl
- POM
pivaloyloxymethyl
- RBV
Ribavirin
- SATE
S-acylthioalkyl ester
References
- 1.Westheimer FH. Why nature chose phosphates. Science. 1987;235:1173–8. doi: 10.1126/science.2434996. [DOI] [PubMed] [Google Scholar]
- 2.Engel R. Phosphonates as analogs of natural phosphates. Chem Rev. 1977;77:349–67. [Google Scholar]
- 3.Metcalf WW, van der Donk WA. Biosynthesis of phosphonic and phosphinic acid natural products. Annu Rev Biochem. 2009;78:65–94. doi: 10.1146/annurev.biochem.78.091707.100215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams R, Jencks WP, Westheimer FH. Table of pKa values. http://research.chem.psu.edu/brpgroup/pKa_compilation.pdf.
- 5.Wiemer DF. Synthesis of nonracemic phosphonates. Tetrahedron. 1997;53:16609–44. [Google Scholar]
- 6.Kornberg RD, McNamee MG, McConnell HM. Measurement of transmembrane potentials in phospholipid vesicles. Proc Natl Acad Sci USA. 1972;69:1508–13. doi: 10.1073/pnas.69.6.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghosh S, Chan JMW, Lea CR, Meints GA, Lewis JC, Tovian ZS, Flessner RM, Loftus TC, Bruchhaus I, Kendrick H, et al. Effects of bisphosphonates on the growth of entamoeba histolytica and plasmodium species in vitro and in vivo. J Med Chem. 2004;47:175–87. doi: 10.1021/jm030084x. [DOI] [PubMed] [Google Scholar]
- 8.Huttunen KM, Rautio J. Prodrugs - an efficient way to breach delivery and targeting barriers. Curr Top Med Chem. 2011;11:2265–87. doi: 10.2174/156802611797183230. [DOI] [PubMed] [Google Scholar]
- 9.Plunkett W, Huang P, Xu YZ, Heinemann V, Grunewald R, Gandhi V. Gemcitabine: Metabolism, mechanisms of action, and self-potentiation. Semin Oncol. 1995;22:3–10. [PubMed] [Google Scholar]
- 10.McGuigan C, Jones BCNM, Riley PA. Trans-esterification reactions yield novel masked phosphate derivatives of the anti-cancer agent araC. Bioorg Med Chem Lett. 1991;1:607–10. [Google Scholar]
- 11.Maiti M, Persoons L, Andrei G, Snoeck R, Balzarini J, Herdewijn P. Synthesis and anti-herpetic activity of phosphoramidate Pro-Tides. Chem Med Chem. 2013;8:985–93. doi: 10.1002/cmdc.201300035. [DOI] [PubMed] [Google Scholar]
- 12.Krise JP, Stella VJ. Prodrugs of phosphates, phosphonates, and phosphinates. Adv Drug Deliv Rev. 1996;19:287–310. [Google Scholar]
- 13.Cho A. Recent advances in oral prodrug discovery. Ann Rep Med Chem. 2006;41:395–407. [Google Scholar]
- 14.He GX, Krise JP, Oliyai R. In: Prodrugs of Phosphonates, Phosphinates, and Phosphates. Stella VJ, Borchardt RT, Hageman MJ, Oliyai R, Maag H, Tilley JW, editors. Springer; New York: 2007. pp. 923–64. [Google Scholar]
- 15.Meier C. Pro-nucleotides - recent advances in the design of efficient tools for the delivery of biologically active nucleoside monophosphates. Synlett. 1998:233–42. [Google Scholar]
- 16.Mackman RL, Cihlar T. Prodrug strategies in the design of nucleoside and nucleotide antiviral therapeutics. Ann Rep Med Chem. 2004;39:305–21. [Google Scholar]
- 17.Ariza ME. Current prodrug strategies for the delivery of nucleotides into cells. Drug Des Rev. 2005;2:373–87. [Google Scholar]
- 18.Ray AS, Hostetler KY. Application of kinase bypass strategies to nucleoside antivirals. Antiviral Res. 2011;92:277–91. doi: 10.1016/j.antiviral.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 19.Bobeck DR, Schinazi RF, Coats SJ. Advances in nucleoside monophosphate prodrugs as anti-HCV agents. Antivir Ther. 2010;15:935–50. doi: 10.3851/IMP1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Madela K, McGuigan C. Progress in the development of anti-hepatitis C virus nucleoside and nucleotide prodrugs. Fut Med Chem. 2012;4:625–50. doi: 10.4155/fmc.12.10. [DOI] [PubMed] [Google Scholar]
- 21.Sofia MJ, Chang W, Furman PA, Mosley RT, Ross BS. Nucleoside, nucleotide, and non-nucleoside inhibitors of hepatitis C virus NS5B RNA-dependent RNA-polymerase. J Med Chem. 2012;55:2481–531. doi: 10.1021/jm201384j. [DOI] [PubMed] [Google Scholar]
- 22.Hecker SJ, Erion MD. Prodrugs of phosphates and phosphonates. J Med Chem. 2008;51:2328–45. doi: 10.1021/jm701260b. [DOI] [PubMed] [Google Scholar]
- 23.Greene TW, Wuts PGM John Wiley and Sons, Inc, editor. Protective Groups in Organic Synthesis. 2. 1991. [Google Scholar]
- 24.Cho MJ, Kurtz RR, Lewis C, Machkovech SM, Houser DJ. Metronidazole phosphate--a water-soluble prodrug for parenteral solutions of metronidazole. J Pharm Sci. 1982;71:410–4. doi: 10.1002/jps.2600710409. [DOI] [PubMed] [Google Scholar]
- 25.Chung MC, Bosquesi PL, dos Santos JL. A prodrug approach to improve the physicochemical properties and decrease the genotoxicity of nitro compounds. Curr Pharm Des. 2011;17:3515–26. doi: 10.2174/138161211798194512. [DOI] [PubMed] [Google Scholar]
- 26.Palte MJ, Davis AKF, McGrath NA, Spiegel CA, Raines RT. Ribonucleoside 3′-phosphates as promoieties for an orally administered drug. Chem Med Chem. 2012;7:1361–4. doi: 10.1002/cmdc.201200243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ellis GA, McGrath NA, Palte MJ, Raines RT. Ribonuclease-activated cancer prodrug. ACS Med Chem Lett. 2012;3:268–72. doi: 10.1021/ml2002554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Boyle NM, Greene LM, Keely NO, Wang S, Cotter TS, Zisterer DM, Meegan MJ. Synthesis and biochemical activities of anti-proliferative amino acid and phosphate derivatives of microtubule-disrupting beta-lactam combretastatins. Eur J Med Chem. 2013;62:705–21. doi: 10.1016/j.ejmech.2013.01.016. [DOI] [PubMed] [Google Scholar]
- 29.Sweeny DJ, Li W, Clough J, Bhamidipati S, Singh R, Park G, Baluom M, Grossbard E, Lau DTW. Metabolism of fostamatinib, the oral methylene phosphate prodrug of the spleen tyrosine kinase inhibitor R406 in humans: Contribution of hepatic and gut bacterial processes to the overall biotransformation. Drug Metab Dispos. 2010;38:1166–76. doi: 10.1124/dmd.110.032151. [DOI] [PubMed] [Google Scholar]
- 30.Gu Y, Atwell GJ, Wilson WR. Metabolism and excretion of the novel bioreductive prodrug PR-104 in mice, rats, dogs, and humans. Drug Metab Dispos. 2010;38:498–508. doi: 10.1124/dmd.109.030973. [DOI] [PubMed] [Google Scholar]
- 31.Chou LC, Chen CT, Lee JC, Way TD, Huang CH, Huang SM, Teng CM, Yamori T, Wu TS, Sun CM, et al. Synthesis and preclinical evaluations of 2-(2-fluorophenyl)-6,7-methylenedioxyquinolin-4-one monosodium phosphate (CHM-1-P-na) as a potent antitumor agent. J Med Chem. 2010;53:1616–26. doi: 10.1021/jm901292j. [DOI] [PubMed] [Google Scholar]
- 32.Ferriz JM, Vinsova J. Prodrug design of phenolic drugs. Curr Pharm Des. 2010;16:2033–52. doi: 10.2174/138161210791293042. [DOI] [PubMed] [Google Scholar]
- 33.Dhareshwar SS, Stella VJ. A novel prodrug strategy for beta-dicarbonyl carbon acids: Syntheses and evaluation of the physicochemical characteristics of C-phosphoryloxymethyl (POM) and phosphoryloxymethyloxymethyl (POMOM) prodrug derivatives. J Pharm Sci. 2010;99:2711–23. doi: 10.1002/jps.22021. [DOI] [PubMed] [Google Scholar]
- 34.Gotthard G, Hiblot J, Gonzalez D, Elias M, Chabriere E. Structural and enzymatic characterization of the phosphotriesterase OPHC2 from pseudomonas pseudoalcaligenes. PLoS One. 2013;8:e77995. doi: 10.1371/journal.pone.0077995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niemi R, Vepsalainen J, Taipale H, Jarvinen T. Bisphosphonate prodrugs: Synthesis and in vitro evaluation of novel acyloxyalkyl esters of clodronic acid. J Med Chem. 1999;42:5053–8. doi: 10.1021/jm991109o. [DOI] [PubMed] [Google Scholar]
- 36.Niemi R, Turhanen P, Vepsalainen J, Taipale H, Jarvinen T. Bisphosphonate prodrugs: Synthesis and in vitro evaluation of alkyl and acyloxymethyl esters of etidronic acid as bioreversible prodrugs of etidronate. Eur J Pharm Sci. 2000;11:173–80. doi: 10.1016/s0928-0987(00)00099-3. [DOI] [PubMed] [Google Scholar]
- 37.Slatter JG, Feenstra KL, Hauer MJ, Kloosterman DA, Parton AH, Sanders PE, Scott G, Speed W. Metabolism of the bisphosphonate ester U-91502 in rats. Drug Metab Dispos. 1996;24:65–73. [PubMed] [Google Scholar]
- 38.Serafinowska HT, Ashton RJ, Bailey S, Harnden MR, Jackson SM, Sutton D. Synthesis and in vivo evaluation of prodrugs of 9-[2-(phosphonomethoxy)ethoxy]adenine. J Med Chem. 1995;38:1372–9. doi: 10.1021/jm00008a015. [DOI] [PubMed] [Google Scholar]
- 39.McGuigan C, Tollerfield SM, Riley PA. Synthesis and biological evaluation of some phosphate triester derivatives of the anti-viral drug AraA. Nucleic Acids Res. 1989;17:6065–75. doi: 10.1093/nar/17.15.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones BCNM, McGuigan C, Riley PA. Synthesis and biological evaluation of some phosphate triester derivatives of the anti-cancer drug AraC. Nucleic Acids Res. 1989;17:7195–201. doi: 10.1093/nar/17.18.7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lan S, Hsieh DC, Hillyer JW, Fancher RM, Rinehart KJ, Warrack BM, White RE. Metabolism of α-phosphonosulfonate squalene synthase inhibitors I. disposition of a farnesylethyl α-phosphonosulfonate and ester prodrugs in rats. Drug Metab Dispos. 1998;26:993–1000. [PubMed] [Google Scholar]
- 42.Dang Q, Liu Y, Rydzewski RM, Brown BS, Robinson E, van Poelje PD, Colby TJ, Erion MD. Bis(para-methoxy)benzyl] phosphonate prodrugs with improved stability and enhanced cell penetration. Bioorg Med Chem Lett. 2007;17:3412–6. doi: 10.1016/j.bmcl.2007.03.089. [DOI] [PubMed] [Google Scholar]
- 43.Dang Q, Brown BS, van Poelje PD, Colby TJ, Erion MD. Synthesis of phosphonate 3-phthalidyl esters as prodrugs for potential intracellular delivery of phosphonates. Bioorg Med Chem Lett. 1999;9:1505–10. doi: 10.1016/s0960-894x(99)00239-5. [DOI] [PubMed] [Google Scholar]
- 44.Topczewski JJ, Neighbors JD, Wiemer DF. Total synthesis of (+)-schweinfurthins B and E. J Org Chem. 2009;74:6965–72. doi: 10.1021/jo901161m. [DOI] [PubMed] [Google Scholar]
- 45.De Lombaert S, Erion MD, Tan J, Blanchard L, el-Chehabi L, Ghai RD, Sakane Y, Berry C, Trapani AJ. N-phosphonomethyl dipeptides and their phosphonate prodrugs, a new generation of neutral endopeptidase (NEP, EC 3.4.24.11) inhibitors. J Med Chem. 1994;37:498–511. doi: 10.1021/jm00030a009. [DOI] [PubMed] [Google Scholar]
- 46.Romanowska J, Sobkowski M, Szymanska-Michalak A, Kolodziej K, Dabrowska A, Lipniacki A, Piasek A, Pietrusiewicz ZM, Figlerowicz M, Guranowski A, et al. Aryl H-phosphonates 17: (N-aryl)phosphoramidates of pyrimidine nucleoside analogues and their synthesis, selected properties, and anti-HIV activity. J Med Chem. 2011;54:6482–91. doi: 10.1021/jm2001103. [DOI] [PubMed] [Google Scholar]
- 47.Romanowska J, Szymanska-Michalak A, Boryski J, Stawinski J, Kraszewski A, Loddo R, Sanna G, Collu G, Secci B, La Colla P. Aryl nucleoside H-phosphonates. part 16: Synthesis and anti-HIV-1 activity of di-aryl nucleoside phosphotriesters. Bioorg Med Chem. 2009;17:3489–98. doi: 10.1016/j.bmc.2009.02.033. [DOI] [PubMed] [Google Scholar]
- 48.De Clercq E, Holy A, Rosenberg I, Sakuma T, Balzarini J, Maudgal PC. A novel selective broad-spectrum anti-dna virus agent. Nature. 1986;323:464–7. doi: 10.1038/323464a0. [DOI] [PubMed] [Google Scholar]
- 49.Khandazhinskaya A, Yasko M, Shirokova E. The synthesis and antiviral properties of acyclic nucleoside analogues with a phosphonomethoxy fragment in the side chain. Curr Med Chem. 2006;13:2953–80. doi: 10.2174/092986706778521896. [DOI] [PubMed] [Google Scholar]
- 50.Farquhar D, Srivastva DN, Kattesch NJ, Saunders PP. Biologically reversible phosphate-protective groups. J Pharm Sci. 1983;72:324–5. doi: 10.1002/jps.2600720332. [DOI] [PubMed] [Google Scholar]
- 51.Srivastva DN, Farquhar D. Bioreversible phosphate protective groups - synthesis and stability of model acyloxymethyl phosphates. Bioorg Chem. 1984;12:118–29. [Google Scholar]
- 52.Iyer RP, Phillips LR, Biddle JA, Thakker DR, Egan W, Aoki S, Mitsuya H. Synthesis of acyloxyalkyl acylphosphonates as potential prodrugs of the antiviral, trisodium phosphonoformate (foscarnet sodium) Tetrahedron Lett. 1989;30:7141–4. [Google Scholar]
- 53.Saperstein R, Vicario PP, Strout HV, Brady E, Slater EE, Greenlee WJ, Ondeyka DL, Patchett AA, Hangauer DG. Design of a selective insulin receptor tyrosine kinase inhibitor and its effect on glucose uptake and metabolism in intact cells. Biochemistry. 1989;28:5694–701. doi: 10.1021/bi00439a053. [DOI] [PubMed] [Google Scholar]
- 54.Lee WA, Martin JC. Perspectives on the development of acyclic nucleotide analogs as antiviral drugs. Antiviral Res. 2006;71:254–9. doi: 10.1016/j.antiviral.2006.05.020. [DOI] [PubMed] [Google Scholar]
- 55.Atack JR, Prior AM, Fletcher SR, Quirk K, McKernan R, Ragan CI. Effects of L-690,488, a prodrug of the bisphosphonate inositol monophosphatase inhibitor L-690,330, on phosphatidylinositol cycle markers. J Pharmacol Exp Ther. 1994;270:70–6. [PubMed] [Google Scholar]
- 56.Topalis D, Pradere U, Roy V, Caillat C, Azzouzi A, Broggi J, Snoeck R, Andrei G, Lin J, Eriksson S, et al. Novel antiviral C5-substituted pyrimidine acyclic nucleoside phosphonates selected as human thymidylate kinase substrates. J Med Chem. 2011;54:222–32. doi: 10.1021/jm1011462. [DOI] [PubMed] [Google Scholar]
- 57.Naesens L, Bischofberger N, Augustijns P, Annaert P, Van den Mooter G, Arimilli MN, Kim CU, De Clercq E. Antiretroviral efficacy and pharmacokinetics of oral bis(isopropyloxycarbonyloxymethyl)-9-(2-phosphonylmethoxypropyl)adenine in mice. Antimicrob Agents Chemother. 1998;42:1568–73. doi: 10.1128/aac.42.7.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Arimilli MN, Kim CU, Dougherty J, Mulato A, Oliyai R, Shaw JP, Cundy KC, Bischofberger N. Synthesis, in vitro biological evaluation and oral bioavailability of 9-[2-(phosphonomethoxy)propyl]adenine (PMPA) prodrugs. Antivir Chem Chemother. 1997;8:557–64. [Google Scholar]
- 59.De Clercq E, Holy A. Acyclic nucleoside phosphonates: A key class of antiviral drugs. Nat Rev Drug Discovery. 2005;4:928–40. doi: 10.1038/nrd1877. [DOI] [PubMed] [Google Scholar]
- 60.Ripin DHB, Teager DS, Fortunak J, Basha SM, Bivins N, Boddy CN, Byrn S, Catlin KK, Houghton SR, Jagadeesh ST, et al. Process improvements for the manufacture of tenofovir disoproxil fumarate at commercial scale. Org Process Res Dev. 2010;14:1194–201. [Google Scholar]
- 61.Montagu A, Pradere U, Roy V, Nolan SP, Agrofoglio LA. Expeditious convergent procedure for the preparation of bis(POC) pro-drugs of new (E)-4-phosphono-but-2-en-1-yl nucleosides. Tetrahedron. 2011;67:5319–28. [Google Scholar]
- 62.Peyrottes S, Egron D, Lefebvre I, Gosselin G, Imbach JL, Perigaud C. SATE pronucleotide approaches: An overview. Mini-Rev Med Chem. 2004;4:395–408. doi: 10.2174/1389557043404007. [DOI] [PubMed] [Google Scholar]
- 63.Oh CH, Liu LJ, Hong JH. Design and synthesis of dually branched 5′-norcarbocyclic adenosine phosphonodiester analogue as a new anti-HIV prodrug. Nucleosides Nucleotides Nucleic Acids. 2010;29:721–33. doi: 10.1080/15257770.2010.509645. [DOI] [PubMed] [Google Scholar]
- 64.Liu LJ, Hong JH. Design and synthesis of novel sate derivatives of acyclic isocytosine and 9-deazaadenine C-nucleosides. Nucleosides Nucleotides Nucleic Acids. 2010;29:257–66. doi: 10.1080/15257771003745704. [DOI] [PubMed] [Google Scholar]
- 65.Fu X, Ou Y, Pei J, Liu Y, Li J, Zhou W, Lan Y, Wang A, Wang Y. Synthesis, anti-HBV activity and renal cell toxicity evaluation of mixed phosphonate prodrugs of adefovir. Eur J Med Chem. 2012;49:211–8. doi: 10.1016/j.ejmech.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 66.Meier C, Lorey M, De Clercq E, Balzarini J. Cyclic saligenyl phosphotriesters of 2′,3′-dideoxy-2′,3′-didehydrothymidine (d4T) - A new pro-nucleotide approach. Bioorg Med Chem Lett. 1997;7:99–104. [Google Scholar]
- 67.Meier C, Balzarini J. Application of the cycloSal-prodrug approach for improving the biological potential of phosphorylated biomolecules. Antiviral Res. 2006;71:282–92. doi: 10.1016/j.antiviral.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 68.Meier C, Knispel T, De Clercq E, Balzarini J. cycloSal-pronucleotides of 2′,3′-dideoxyadenosine and 2′,3′-dideoxy-2′,3′-didehydroadenosine: Synthesis and antiviral evaluation of a highly efficient nucleotide delivery system. J Med Chem. 1999;42:1604–14. doi: 10.1021/jm981096z. [DOI] [PubMed] [Google Scholar]
- 69.Meier C, Gorbig U, Muller C, Balzarini J. cycloSal-PMEA and cycloAmb-PMEA: Potentially new phosphonate prodrugs based on the cycloSal-pronucleotide approach. J Med Chem. 2005;48:8079–86. doi: 10.1021/jm050641a. [DOI] [PubMed] [Google Scholar]
- 70.Meier C, Ruppel MFH, Vukadinovic D, Balzarini J. “Lock-in”-cycloSal-pronucleotides - A new generation of chemical trojan horses? Mini-Rev Med Chem. 2004;4:383–94. doi: 10.2174/1389557043403972. [DOI] [PubMed] [Google Scholar]
- 71.Gisch N, Balzarini J, Meier C. 5-diacetoxymethyl-cycloSal-d4TMP - A prototype of enzymatically activated cycloSal-pronucleotides. Nucleosides, Nucleotides Nucleic Acids. 2007;26:861–4. doi: 10.1080/15257770701504025. [DOI] [PubMed] [Google Scholar]
- 72.Meier C, Lorey M, De Clercq E, Balzarini J. cycloSal-2′,3′-dideoxy-2′,3′-didehydrothymidine monophosphate (cycloSal-d4TMP): Synthesis and antiviral evaluation of a new d4TMP delivery system. J Med Chem. 1998;41:1417–27. doi: 10.1021/jm970664s. [DOI] [PubMed] [Google Scholar]
- 73.Morales EHR, Roman CA, Thomann JO, Meier C. Linear synthesis of chiral cycloSal-pronucleotides. Eur J Med Chem. 2011;2011:4397–408. [Google Scholar]
- 74.Rios Morales EH, Balzarini J, Meier C. Stereoselective synthesis and antiviral activity of methyl-substituted cycloSal-pronucleotides. J Med Chem. 2012;55:7245–52. doi: 10.1021/jm3008085. [DOI] [PubMed] [Google Scholar]
- 75.Wolf S, Warnecke S, Ehrit J, Freiberger F, Gerardy-Schahn R, Meier C. Chemical synthesis and enzymatic testing of CMP-sialic acid derivatives. Chembiochem. 2012;13:2605–15. doi: 10.1002/cbic.201200471. [DOI] [PubMed] [Google Scholar]
- 76.Huchting J, Ruthenbeck A, Meier C. Synthesis of cycloSal-(glycopyranosyl-6)-phosphates as activated sugar phosphates. Eur J Org Chem. 2013;2013:6907–16. [Google Scholar]
- 77.Erion MD, Bullough DA, Lin C, Hong Z. HepDirect prodrugs for targeting nucleotide-based antiviral drugs to the liver. Curr Opin Invest Drugs. 2006;7:109–17. [PubMed] [Google Scholar]
- 78.Erion MD, Reddy KR, Boyer SH, Matelich MC, Gomez-Galeno J, Lemus RH, Ugarkar BG, Colby TJ, Schanzer J, Van Poelje PD. Design, synthesis, and characterization of a series of cytochrome P(450) 3A-activated prodrugs (HepDirect prodrugs) useful for targeting phosph(on)ate-based drugs to the liver. J Am Chem Soc. 2004;126:5154–63. doi: 10.1021/ja031818y. [DOI] [PubMed] [Google Scholar]
- 79.Erion MD, van Poelje PD, Mackenna DA, Colby TJ, Montag AC, Fujitaki JM, Linemeyer DL, Bullough DA. Liver-targeted drug delivery using HepDirect prodrugs. J Pharmacol Exp Ther. 2005;312:554–60. doi: 10.1124/jpet.104.075903. [DOI] [PubMed] [Google Scholar]
- 80.Boyer SH, Sun Z, Jiang H, Esterbrook J, Gomez-Galeno JE, Craigo W, Reddy KR, Ugarkar BG, MacKenna DA, Erion MD. Synthesis and characterization of a novel liver-targeted prodrug of cytosine-1-beta-D-arabinofuranoside monophosphate for the treatment of hepatocellular carcinoma. J Med Chem. 2006;49:7711–20. doi: 10.1021/jm0607449. [DOI] [PubMed] [Google Scholar]
- 81.Hecker SJ, Reddy KR, van Poelje PD, Sun Z, Huang W, Varkhedkar V, Reddy MV, Fujitaki JM, Olsen DB, Koeplinger KA, et al. Liver-targeted prodrugs of 2′-C-methyladenosine for therapy of hepatitis C virus infection. J Med Chem. 2007;50:3891–6. doi: 10.1021/jm0701021. [DOI] [PubMed] [Google Scholar]
- 82.Bookser BC, Raffaele NB, Reddy KR, Fan K, Huang W, Erion MD. Synthesis of 3′-amino-3′-deoxyguanosine and 3′-amino-3′-deoxyxyloguanosine monophosphate HepDirect prodrugs from guanosine. Nucleosides, Nucleotides Nucleic Acids. 2009;28:969–86. doi: 10.1080/15257770903307151. [DOI] [PubMed] [Google Scholar]
- 83.Erion MD, Cable EE, Ito BR, Jiang H, Fujitaki JM, Finn PD, Zhang BH, Hou J, Boyer SH, van Poelje PD, et al. Targeting thyroid hormone receptor-beta agonists to the liver reduces cholesterol and triglycerides and improves the therapeutic index. Proc Natl Acad Sci USA. 2007;104:15490–5. doi: 10.1073/pnas.0702759104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Boyer SH, Jiang H, Jacintho JD, Reddy MV, Li H, Li W, Godwin JL, Schulz WG, Cable EE, Hou J, et al. Synthesis and biological evaluation of a series of liver-selective phosphonic acid thyroid hormone receptor agonists and their prodrugs. J Med Chem. 2008;51:7075–93. doi: 10.1021/jm800824d. [DOI] [PubMed] [Google Scholar]
- 85.Tsukada T, Tamaki K, Tanaka J, Takagi T, Yoshida T, Okuno A, Shiiki T, Takahashi M, Nishi T. A prodrug approach towards the development of tricyclic-based FBPase inhibitors. Bioorg Med Chem Lett. 2010;20:2938–41. doi: 10.1016/j.bmcl.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 86.Huttunen KM, Tani N, Juvonen RO, Raunio H, Rautio J. Design, synthesis, and evaluation of novel cyclic phosphates of 5-aminosalicylic acid as cytochrome p450-activated prodrugs. Mol Pharm. 2013;10:532–7. doi: 10.1021/mp300330v. [DOI] [PubMed] [Google Scholar]
- 87.Sun W, Peng W, Li G, Jiang T. Design, synthesis, and sustained-release property of 1,3-cyclic propanyl phosphate ester of 18 beta-glycyrrhetinic acid. Chem Biol Drug Des. 2011;77:206–11. doi: 10.1111/j.1747-0285.2010.01077.x. [DOI] [PubMed] [Google Scholar]
- 88.Remy DC, Sunthankar AV, Heidelberger C. Studies on fluorinated pyrimidines. XIV. the synthesis of derivatives of 5-fluoro-2′-deoxyuridine 5′-phosphate and related Compounds1. J Org Chem. 1962;27:2491–500. [Google Scholar]
- 89.Mukherjee KL, Heidelberger C. Studies of fluorinated pyrimidines. XV. inhibition of the incorporation of formate-C14 into DNA thymine of ehrlich ascites carcinoma cells by 5-fluoro-2′-deoxyuridine-5′-monophosphate and related compounds. Cancer Res. 1962;22:815–22. [PubMed] [Google Scholar]
- 90.De Napoli L, Di Fabio G, D’Onofrio J, Montesarchio D. An efficient solid phase synthesis of 5′-phosphodiester and phosphoramidate monoester nucleoside analogues. Chem Commun. 2005:2586–8. doi: 10.1039/b501043h. [DOI] [PubMed] [Google Scholar]
- 91.Hostetler KY. Alkoxyalkyl prodrugs of acyclic nucleoside phosphonates enhance oral antiviral activity and reduce toxicity: Current state of the art. Antiviral Res. 2009;82:A84–98. doi: 10.1016/j.antiviral.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hostetler KY. Synthesis and early development of hexadecyloxypropyl-cidofovir: An oral antipoxvirus nucleoside phosphonate. Viruses-Basel. 2010;2:2213–25. doi: 10.3390/v2102213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hostetler KY, Parker S, Sridhar CN, Martin MJ, Li JL, Stuhmiller LM, van Wijk GMT, van den Bosch H, Gardner MF, Aldern KA, et al. Acyclovir diphosphate dimyristoylglycerol - a phospholipid prodrug with activity against acyclovir-resistant herpes-simplex virus. Proc Natl Acad Sci USA. 1993;90:11835–9. doi: 10.1073/pnas.90.24.11835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hostetler KY, Beadle JR, Kini GD, Gardner MF, Wright KN, Wu TH, Korba BA. Enhanced oral absorption and antiviral activity of 1-O-octadecyl-sn-glycero-3-phospho-acyclovir and related compounds in hepatitis B virus infection, in vitro. Biochem Pharmacol. 1997;53:1815–22. doi: 10.1016/s0006-2952(97)82446-x. [DOI] [PubMed] [Google Scholar]
- 95.Ciesla S, Trahan J, Wan W, Beadle J, Aldern K, Painter G, Hostetler K. Esterification of cidofovir with alkoxyalkanols increases oral bioavailability and diminishes drug accumulation in kidney. Antiviral Res. 2003;59:163–71. doi: 10.1016/s0166-3542(03)00110-4. [DOI] [PubMed] [Google Scholar]
- 96.Hostetler KY, Aldern KA, Wan WB, Ciesla SL, Beadle JR. Alkoxyakl esters of (S)-9-[3-hydroxy-2-(phosphonomethoxy)propyl] adenine are potent inhibitors of the replication of wild-type and drug-resistant human immunodeficiency virus type 1 in vitro. Antimicrob Agents Chemother. 2006;50:2857–9. doi: 10.1128/AAC.01223-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dal Pozzo F, Andrei G, Lebeau I, Beadle JR, Hostetler KY, De Clercq E, Snoeck R. In vitro evaluation of the anti-orf virus activity of alkoxyalkyl esters of CDV, cCDV and (S)-HPMPA. Antiviral Res. 2007;75:52–7. doi: 10.1016/j.antiviral.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 98.Williams-Aziz SL, Hartline CB, Harden EA, Daily SL, Prichard MN, Kushner NL, Beadle JR, Wan WB, Hostetler KY, Kern ER. Comparative activities of lipid esters of cidofovir and cyclic cidofovir against replication of herpesviruses in vitro. Antimicrob Agents Chemother. 2005;49:3724–33. doi: 10.1128/AAC.49.9.3724-3733.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Marty FM, Winston DJ, Rowley SD, Vance E, Papanicolaou GA, Mullane KM, Brundage TM, Robertson AT, Godkin S, Mommeja-Marin H, et al. CMX001 to prevent cytomegalovirus disease in hematopoietic-cell transplantation. N Engl J Med. 2013;369:1227–36. doi: 10.1056/NEJMoa1303688. [DOI] [PubMed] [Google Scholar]
- 100.Valiaeva N, Wyles DL, Schooley RT, Hwu JB, Beadle JR, Prichard MN, Hostetler KY. Synthesis and antiviral evaluation of 9-(S)-[3-alkoxy-2-(phosphonomethoxy)propyl]nucleoside alkoxyalkyl esters: Inhibitors of hepatitis C virus and HIV-1 replication. Bioorg Med Chem. 2011;19:4616–25. doi: 10.1016/j.bmc.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dong SD, Lin C, Schroeder M. Synthesis and evaluation of a new phosphorylated ribavirin prodrug. Antiviral Res. 2013;99:18–26. doi: 10.1016/j.antiviral.2013.04.014. [DOI] [PubMed] [Google Scholar]
- 102.Krylov IS, Kashemirov BA, Hilfinger JM, McKenna CE. Evolution of an amino acid based prodrug approach: Stay tuned. Mol Pharm. 2013;10:445–58. doi: 10.1021/mp300663j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Oliyai R, Shaw JP, Sueoka-Lennen CM, Cundy KC, Arimilli MN, Jones RJ, Lee WA. Aryl ester prodrugs of cyclic HPMPC. I: PhysicochemicaI characterization and in vitro biological stability. Pharm Res. 1999;16:1687–93. doi: 10.1023/a:1018945713623. [DOI] [PubMed] [Google Scholar]
- 104.Oliyai R, Arimilli MN, Jones RJ, Lee WA. Pharmacokinetics of salicylate ester prodrugs of cyclic HPMPC in dogs. Nucleosides Nucleotides Nucleic Acids. 2001;20:1411–4. doi: 10.1081/NCN-100002566. [DOI] [PubMed] [Google Scholar]
- 105.Ruiz J, Beadle JR, Buller RM, Schreiwer J, Prichard MN, Keith KA, Lewis KC, Hostetler KY. Synthesis, metabolic stability and antiviral evaluation of various alkoxyalkyl esters of cidofovir and 9-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]adenine. Bioorg Med Chem. 2011;19:2950–8. doi: 10.1016/j.bmc.2011.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Keith KA, Wan WB, Ciesla SL, Beadle JR, Hostetler KY, Kern ER. Inhibitory activity of alkoxyalkyl and alkyl esters of cidofovir and cyclic cidofovir against orthopoxvirus replication in vitro. Anti-microb Agents Chemother. 2004;48:1869–71. doi: 10.1128/AAC.48.5.1869-1871.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lebeau I, Andrei G, Dal Pozzo F, Beadle JR, Hostetler KY, De Clercq E, van den Oord J, Snoeck R. Activities of alkoxvalkyl esters of cidofovir (CDV), cyclic CDV, and (S)-9-(3-hydroxy-2-phosphonylmethoxypropyl)adenine against orthopoxviruses in cell monolayers and in organotypic cultures. Antimicrob Agents Chemother. 2006;50:2525–9. doi: 10.1128/AAC.01489-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.McKenna CE, Kashemirov BA, Eriksson U, Amidon GL, Kish PE, Mitchell S, Kim JS, Hilfinger JM. Cidofovir peptide conjugates as prodrugs. J Organomet Chem. 2005;690:2673–8. [Google Scholar]
- 109.Eriksson U, Peterson LW, Kashemirov BA, Hilfinger JM, Drach JC, Borysko KZ, Breitenbach JM, Kim JS, Mitchell S, Kijek P, et al. Serine peptide phosphoester prodrugs of cyclic cidofovir: Synthesis, transport, and antiviral activity. Mol Pharm. 2008;5:598–609. doi: 10.1021/mp8000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Peterson LW, Sala-Rabanal M, Krylov IS, Serpi M, Kashemirov BA, McKenna CE. Serine side chain-linked peptidomimetic conjugates of cyclic HPMPC and HPMPA: Synthesis and interaction with hPEPT1. Mol Pharm. 2010;7:2349–61. doi: 10.1021/mp100186b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zakharova VM, Serpi M, Krylov IS, Peterson LW, Breitenbach JM, Borysko KZ, Drach JC, Collins M, Hilfinger JM, Kashemirov BA, et al. Tyrosine-based 1-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]cytosine and -adenine ((S)-HPMPC and (S)-HPMPA) prodrugs: Synthesis, stability, antiviral activity, and in vivo transport studies. J Med Chem. 2011;54:5680–93. doi: 10.1021/jm2001426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Krecmerova M, Holy A, Andrei G, Pomeisl K, Tichy T, Brehova P, Masojidkova M, Dracinsky M, Pohl R, Laflamme G, et al. Synthesis of ester prodrugs of 9-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]-2,6-diaminopurine (HPMPDAP) as anti-poxvirus agents. J Med Chem. 2010;53:6825–37. doi: 10.1021/jm901828c. [DOI] [PubMed] [Google Scholar]
- 113.Reddy PG, Bao D, Chang W, Chun B, Du J, Nagarathnam D, Rachakonda S, Ross BS, Zhang H, Bansal S, et al. 2′-deoxy-2′alpha-fluoro-2′-beta-C-methyl 3′,5′-cyclic phosphate nucleotide prodrug analogs as inhibitors of HCV NS5B polymerase: Discovery of PSI-352938. Bioorg Med Chem Lett. 2010;20:7376–80. doi: 10.1016/j.bmcl.2010.10.035. [DOI] [PubMed] [Google Scholar]
- 114.Du J, Bao D, Chun B, Jiang Y, Reddy PG, Zhang H, Ross BS, Bansal S, Bao H, Espiritu C, et al. Beta-D-2′-alpha-F-2′-beta-C-methyl-6-O-substituted 3′,5′-cyclic phosphate nucleotide prodrugs as inhibitors of hepatitis C virus replication: A structure-activity relationship study. Bioorg Med Chem Lett. 2012;22:5924–9. doi: 10.1016/j.bmcl.2012.07.066. [DOI] [PubMed] [Google Scholar]
- 115.Reddy PG, Chun B, Zhang H, Rachakonda S, Ross BS, Sofia MJ. Stereoselective synthesis of PSI-352938: A beta-D-2′-deoxy-2′-alpha-fluoro-2′-beta-C-methyl-3′,5′-cyclic phosphate nucleotide prodrug for the treatment of HCV. J Org Chem. 2011;76:3782–90. doi: 10.1021/jo200060f. [DOI] [PubMed] [Google Scholar]
- 116.Mehellou Y, Balzarini J, McGuigan C. Aryloxy phosphoramidate triesters: A technology for delivering monophosphorylated nucleosides and sugars into cells. Chemmedchem. 2009;4:1779–91. doi: 10.1002/cmdc.200900289. [DOI] [PubMed] [Google Scholar]
- 117.Devine KG, McGuigan C, O’Connor TJ, Nicholls SR, Kinchington D. Novel phosphate derivatives of zidovudine as anti-hiv compounds. AIDS. 1990;4:371–3. [PubMed] [Google Scholar]
- 118.Pertusati F, Serpi M, McGuigan C. Medicinal chemistry of nucleoside phosphonate prodrugs for antiviral therapy. Antivir Chem Chemother. 2012;22:181–203. doi: 10.3851/IMP2012. [DOI] [PubMed] [Google Scholar]
- 119.McGuigan C, Bourdin C, Derudas M, Hamon N, Hinsinger K, Kandil S, Madela K, Meneghesso S, Pertusati F, Serpi M, et al. Design, synthesis and biological evaluation of phosphorodiamidate pro-drugs of antiviral and anticancer nucleosides. Eur J Med Chem. 2013;70:326–40. doi: 10.1016/j.ejmech.2013.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.McGuigan C, Madela K, Aljarah M, Bourdin C, Arrica M, Barrett E, Jones S, Kolykhalov A, Bleiman B, Bryant KD, et al. Phosphorodiamidates as a promising new phosphate prodrug motif for antiviral drug discovery: Application to anti-HCV agents. J Med Chem. 2011;54:8632–45. doi: 10.1021/jm2011673. [DOI] [PubMed] [Google Scholar]
- 121.Jansa P, Baszczynski O, Dracinsky M, Votruba I, Zidek Z, Bahador G, Stepan G, Cihlar T, Mackman R, Holy A, et al. A novel and efficient one-pot synthesis of symmetrical diamide (bis-amidate) prodrugs of acyclic nucleoside phosphonates and evaluation of their biological activities. Eur J Med Chem. 2011;46:3748–54. doi: 10.1016/j.ejmech.2011.05.040. [DOI] [PubMed] [Google Scholar]
- 122.Erion MD, van Poelje PD, Dang Q, Kasibhatla SR, Potter SC, Reddy MR, Reddy KR, Jiang T, Lipscomb WN. MB06322 (CS-917): A potent and selective inhibitor of fructose 1,6-bisphosphatase for controlling gluconeogenesis in type 2 diabetes. Proc Natl Acad Sci USA. 2005;102:7970–5. doi: 10.1073/pnas.0502983102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dang Q, Liu Y, Cashion DK, Kasibhatla SR, Jiang T, Taplin F, Jacintho JD, Li H, Sun Z, Fan Y, et al. Discovery of a series of phosphonic acid-containing thiazoles and orally bioavailable diamide prodrugs that lower glucose in diabetic animals through inhibition of fructose-1,6-bisphosphatase. J Med Chem. 2011;54:153–65. doi: 10.1021/jm101035x. [DOI] [PubMed] [Google Scholar]
- 124.van Poelje PD, Potter SC, Erion MD. Fructose-1, 6-bisphosphatase inhibitors for reducing excessive endogenous glucose production in type 2 diabetes. Handb Exp Pharmacol. 2011;203:279–301. doi: 10.1007/978-3-642-17214-4_12. [DOI] [PubMed] [Google Scholar]
- 125.Pertusati F, Hinsinger K, Flynn ÁS, Powell N, Tristram A, Balzarini J, McGuigan C. PMPA and PMEA prodrugs for the treatment of HIV infections and human papillomavirus (HPV) associated neoplasia and cancer. Eur J Med Chem. 2014;78:259–68. doi: 10.1016/j.ejmech.2014.03.051. [DOI] [PubMed] [Google Scholar]
- 126.Saboulard D, Naesens L, Cahard D, Salgado A, Pathirana R, Velazquez S, McGuigan C, De Clercq E, Balzarini J. Characterization of the activation pathway of phosphoramidate triester prodrugs of stavudine and zidovudine. Mol Pharmacol. 1999;56:693–704. [PubMed] [Google Scholar]
- 127.Lee WA, He GX, Eisenberg E, Cihlar T, Swaminathan S, Mulato A, Cundy KC. Selective intracellular activation of a novel prodrug of the human immunodeficiency virus reverse transcriptase inhibitor tenofovir leads to preferential distribution and accumulation in lymphatic tissue. Antimicrob Agents Chemother. 2005;49:1898–906. doi: 10.1128/AAC.49.5.1898-1906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Eisenberg EJ, He GX, Lee WA. Metabolism of GS-7340, a novel phenyl monophosphoramidate intracellular prodrug of PMPA, in blood. Nucleosides, Nucleotides Nucleic Acids. 2001;20:1091–8. doi: 10.1081/NCN-100002496. [DOI] [PubMed] [Google Scholar]
- 129.Ballatore C, McGuigan C, De Clercq E, Balzarini J. Synthesis and evaluation of novel amidate prodrugs of PMEA and PMPA. Bioorg Med Chem Lett. 2001;11:1053–6. doi: 10.1016/s0960-894x(01)00128-7. [DOI] [PubMed] [Google Scholar]
- 130.McGuigan C, Hassan-Abdallah A, Srinivasan S, Wang Y, Siddiqui A, Daluge SM, Gudmundsson KS, Zhou H, McLean EW, Peckham JP, et al. Application of phosphoramidate ProTide technology significantly improves antiviral potency of carbocyclic adenosine derivatives. J Med Chem. 2006;49:7215–26. doi: 10.1021/jm060776w. [DOI] [PubMed] [Google Scholar]
- 131.Leisvuori A, Aiba Y, Lonnberg T, Poijarvi-Virta P, Blatt L, Beigelman L, Lonnberg H. Chemical and enzymatic stability of amino acid derived phosphoramidates of antiviral nucleoside 5′-monophosphates bearing a biodegradable protecting group. Org Biomol Chem. 2010;8:2131–41. doi: 10.1039/b924321f. [DOI] [PubMed] [Google Scholar]
- 132.McGuigan C, Murziani P, Slusarczyk M, Gonczy B, Vande Voorde J, Liekens S, Balzarini J. Phosphoramidate ProTides of the anti-cancer agent FUDR successfully deliver the preformed bioactive monophosphate in cells and confer advantage over the parent nucleoside. J Med Chem. 2011;54:7247–58. doi: 10.1021/jm200815w. [DOI] [PubMed] [Google Scholar]
- 133.Mehellou Y, Valente R, Mottram H, Walsby E, Mills KI, Balzarini J, McGuigan C. Phosphoramidates of 2′-beta-D-arabinouridine (AraU) as phosphate prodrugs; design, synthesis, in vitro activity and metabolism. Bioorg Med Chem. 2010;18:2439–46. doi: 10.1016/j.bmc.2010.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.McGuigan C, Derudas M, Gonczy B, Hinsinger K, Kandil S, Pertusati F, Serpi M, Snoeck R, Andrei G, Balzarini J, et al. ProTides of N-(3-(5-(2′-deoxyuridine))prop-2-ynyl)octanamide as potential anti-tubercular and anti-viral agents. Bioorg Med Chem. 2014;22:2816–24. doi: 10.1016/j.bmc.2014.02.056. [DOI] [PubMed] [Google Scholar]
- 135.Perrone P, Luoni GM, Kelleher MR, Daverio F, Angell A, Mulready S, Congiatu C, Rajyaguru S, Martin JA, Leveque V, et al. Application of the phosphoramidate ProTide approach to 4′-azidouridine confers sub-micromolar potency versus hepatitis C virus on an inactive nucleoside. J Med Chem. 2007;50:1840–9. doi: 10.1021/jm0613370. [DOI] [PubMed] [Google Scholar]
- 136.Vanpouille C, Lisco A, Derudas M, Saba E, Grivel JC, Brichacek B, Scrimieri F, Schinazi R, Schols D, McGuigan C, et al. A new class of dual-targeted antivirals: Monophosphorylated acyclovir pro-drug derivatives suppress both human immunodeficiency virus type 1 and herpes simplex virus type 2. J Infect Dis. 2010;201:635–43. doi: 10.1086/650343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ruda GF, Wong PE, Alibu VP, Norval S, Read KD, Barrett MP, Gilbert IH. Aryl phosphoramidates of 5-phospho erythronohydroxamic acid, A new class of potent trypanocidal compounds. J Med Chem. 2010;53:6071–8. doi: 10.1021/jm1004754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Vernachio JH, Bleiman B, Bryant KD, Chamberlain S, Hunley D, Hutchins J, Ames B, Gorovits E, Ganguly B, Hall A, et al. INX-08189, a phosphoramidate prodrug of 6-O-methyl-2′-C-methyl guanosine, is a potent inhibitor of hepatitis C virus replication with excellent pharmacokinetic and pharmacodynamic properties. Antimicrob Agents Chemother. 2011;55:1843–51. doi: 10.1128/AAC.01335-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.McGuigan C, Perrone P, Madela K, Neyts J. The phosphoramidate ProTide approach greatly enhances the activity of beta-2′C-methylguanosine against hepatitis C virus. Bioorg Med Chem Lett. 2009;19:4316–20. doi: 10.1016/j.bmcl.2009.05.122. [DOI] [PubMed] [Google Scholar]
- 140.McGuigan C, Madela K, Aljarah M, Gilles A, Battina SK, Ramamurty CVS, Srinivas Rao C, Vernachio J, Hutchins J, Hall A, et al. Dual pro-drugs of 2′-C-methyl guanosine monophosphate as potent and selective inhibitors of hepatitis C virus. Bioorg Med Chem Lett. 2011;21:6007–12. doi: 10.1016/j.bmcl.2011.06.013. [DOI] [PubMed] [Google Scholar]
- 141.Liederer BM, Borchardt RT. Enzymes involved in the bioconversion of ester-based prodrugs. J Pharm Sci. 2006;95:1177–95. doi: 10.1002/jps.20542. [DOI] [PubMed] [Google Scholar]
- 142.Lim SM, Westover KD, Ficarro SB, Harrison RA, Choi HG, Pacold ME, Carrasco M, Hunter J, Kim ND, Xie T, et al. Therapeutic targeting of oncogenic K-ras by a covalent catalytic site inhibitor. Angew Chem, Int Ed. 2014;53:199–204. doi: 10.1002/anie.201307387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Tobias SC, Borch RF. Synthesis and biological studies of novel nucleoside phosphoramidate prodrugs. J Med Chem. 2001;44:4475–80. doi: 10.1021/jm010337r. [DOI] [PubMed] [Google Scholar]
- 144.Tobias SC, Borch RF. Synthesis and biological evaluation of a cytarabine phosphoramidate prodrug. Mol Pharm. 2004;1:112–6. doi: 10.1021/mp034019v. [DOI] [PubMed] [Google Scholar]
- 145.Lu P, Liu J, Wang Y, Chen X, Yang Y, Ji R. Design, synthesis and evaluation of novel oxazaphosphorine prodrugs of 9-(2-phosphonomethoxyethyl)adenine (PMEA, adefovir) as potent HBV inhibitors. Bioorg Med Chem Lett. 2009;19:6918–21. doi: 10.1016/j.bmcl.2009.10.072. [DOI] [PubMed] [Google Scholar]
- 146.Gardelli C, Attenni B, Donghi M, Meppen M, Pacini B, Harper S, Di Marco A, Fiore F, Giuliano C, Pucci V, et al. Phosphoramidate prodrugs of 2′-C-methylcytidine for therapy of hepatitis C virus infection. J Med Chem. 2009;52:5394–407. doi: 10.1021/jm900447q. [DOI] [PubMed] [Google Scholar]
- 147.Valette G, Pompon A, Girardet JL, Cappellacci L, Franchetti P, Grifantini M, La Colla P, Loi AG, Perigaud C, Gosselin G, et al. Decomposition pathways and in vitro HIV inhibitory effects of isoddA pronucleotides: Toward a rational approach for intracellular delivery of nucleoside 5′-monophosphates. J Med Chem. 1996;39:1981–90. doi: 10.1021/jm9507338. [DOI] [PubMed] [Google Scholar]
- 148.Grajkowski A, Ausin C, Kauffman JS, Snyder J, Hess S, Lloyd JR, Beaucage SL. Solid-phase synthesis of thermolytic DNA oligonucleotides functionalized with a single 4-hydroxy-1-butyl or 4-phosphato-/thiophosphato-1-butyl thiophosphate protecting group. J Org Chem. 2007;72:805–15. doi: 10.1021/jo062087y. [DOI] [PubMed] [Google Scholar]
- 149.Ausin C, Grajkowski A, Cieslak J, Gapeev A, Beaucage SL. Time-dependent thermocontrol of the hydrophilic and lipophilic properties of DNA oligonucleotide prodrugs. Curr Protoc Nucleic Acid Chem. 2010;Chapter 4(Unit 4.42) doi: 10.1002/0471142700.nc0442s43. [DOI] [PubMed] [Google Scholar]
- 150.Kiuru E, Ahmed Z, Lonnberg H, Beigelman L, Ora M. 2,2-disubstituted 4-acylthio-3-oxobutyl groups as esterase- and thermolabile protecting groups of phosphodiesters. J Org Chem. 2013;78:950–9. doi: 10.1021/jo302421u. [DOI] [PubMed] [Google Scholar]
- 151.Ora M, Mantyvaara A, Lonnberg H. 3-acetyloxy-2-cyano-2-(alkylaminocarbamoyl)propyl groups as biodegradable protecting groups of nucleoside 5′-monophosphates. Molecules. 2011;16:552–66. doi: 10.3390/molecules16010552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Farquhar D, Khan S, Srivastva DN, Saunders PP. Synthesis and antitumor evaluation of bis[(pivaloyloxy)methyl] 2′-deoxy-5-fluorouridine 5′-monophosphate (FdUMP): A strategy to introduce nucleotides into cells. J Med Chem. 1994;37:3902–9. doi: 10.1021/jm00049a009. [DOI] [PubMed] [Google Scholar]
- 153.Dickson JK, Biller SA, Magnin DR, Petrillo EW, Hillyer JW, Hsieh DC, Lan SJ, Rinehart JK, Gregg RE, Harrity TW, et al. Orally active squalene synthase inhibitors: Bis((acyloxy)alkyl) prodrugs of the alpha-phosphonosulfonic acid moiety. J Med Chem. 1996;39:661–4. doi: 10.1021/jm950735s. [DOI] [PubMed] [Google Scholar]
- 154.Aldern KA, Ciesla SL, Winegarden KL, Hostetler KY. Increased antiviral activity of 1-O-hexadecyloxypropyl-[ 2-(14)C] cidofovir in MRC-5 human lung fibroblasts is explained by unique cellular uptake and metabolism. Mol Pharmacol. 2003;63:678–81. doi: 10.1124/mol.63.3.678. [DOI] [PubMed] [Google Scholar]
- 155.Tehler U, Nelson CH, Peterson LW, Provoda CJ, Hilfinger JM, Lee KD, McKenna CE, Amidona GL. Puromycin-sensitive aminopeptidase: An antiviral prodrug activating enzyme. Antiviral Res. 2010;85:482–9. doi: 10.1016/j.antiviral.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mendel DB, Cihlar T, Moon K, Chen MS. Conversion of 1-[((S)-2-hydroxy-2-oxo-1,4,2-dioxaphosphorinan-5-yl)methyl]cytosine to cidofovir by an intracellular cyclic CMP phosphodiesterase. Anti-microb Agents Chemother. 1997;41:641–6. doi: 10.1128/aac.41.3.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Schneider E, Kuhn M, Reinecke D, Wolter S, Burhenne H, Kaever V, Seifert R. Fishing for elusive cCMP-degrading phosphodiesterases. BMC Pharmacol Toxicol. 2013;14:1–2. [Google Scholar]
- 158.Gisch N, Balzarini J, Meier C. Enzymatically activated cycloSal-d4T-monophosphates: The third generation of cycloSal-pronucleotides. J Med Chem. 2007;50:1658–67. doi: 10.1021/jm0613267. [DOI] [PubMed] [Google Scholar]
- 159.Birkus G, Kutty N, He GX, Mulato A, Lee W, McDermott M, Cihlar T. Activation of 9-[(R)-2-[[(S)-[[(S)-1-(isopropoxycarbonyl)ethyl]amino] phenoxyphosphinyl]-methoxy]propyl]adenine (GS-7340) and other tenofovir phosphonoamidate prodrugs by human proteases. Mol Pharmacol. 2008;74:92–100. doi: 10.1124/mol.108.045526. [DOI] [PubMed] [Google Scholar]
- 160.Furman PA, Murakami E, Niu C, Lam AM, Espiritu C, Bansal S, Bao H, Tolstykh T, Steuer HM, Keilman M, et al. Activity and the metabolic activation pathway of the potent and selective hepatitis C virus pronucleotide inhibitor PSI-353661. Antiviral Res. 2011;91:120–32. doi: 10.1016/j.antiviral.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ray AS, Vela JE, Boojamra CG, Zhang L, Hui H, Callebaut C, Stray K, Lin KY, Gao Y, Mackman RL, et al. Intracellular metabolism of the nucleotide prodrug GS-9131, a potent anti-human immunodeficiency virus agent. Antimicrob Agents Chemother. 2008;52:648–54. doi: 10.1128/AAC.01209-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Mackman RL, Ray AS, Hui HC, Zhang L, Birkus G, Boojamra CG, Desai MC, Douglas JL, Gao Y, Grant D, et al. Discovery of GS-9131: Design, synthesis and optimization of amidate prodrugs of the novel nucleoside phosphonate HIV reverse transcriptase (RT) inhibitor GS-9148. Bioorg Med Chem. 2010;18:3606–17. doi: 10.1016/j.bmc.2010.03.041. [DOI] [PubMed] [Google Scholar]
- 163.Lonnberg T, Ora M, Lonnberg H. Hydrolytic reactions of nucleoside phosphoramidates: Kinetics and mechanisms. Mini-Rev Org Chem. 2010;7:33–43. [Google Scholar]
- 164.Murakami E, Tolstykh T, Bao H, Niu C, Micolochick Steuer HM, Bao D, Chang W, Espiritu C, Bansal S, Lam AM, et al. Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977. J Biol Chem. 2010;285:34337–47. doi: 10.1074/jbc.M110.161802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Chou TF, Baraniak J, Kaczmarek R, Zhou X, Cheng J, Ghosh B, Wagner CR. Phosphoramidate pronucleotides: A comparison of the phosphoramidase substrate specificity of human and escherichia coli histidine triad nucleotide binding proteins. Mol Pharm. 2007;4:208–17. doi: 10.1021/mp060070y. [DOI] [PubMed] [Google Scholar]
- 166.Birkus G, Wang R, Liu X, Kutty N, MacArthur H, Cihlar T, Gibbs C, Swaminathan S, Lee W, McDermott M. Cathepsin A is the major hydrolase catalyzing the intracellular hydrolysis of the antiretroviral nucleotide phosphonoamidate prodrugs GS-7340 and GS-9131. Antimicrob Agents Chemother. 2007;51:543–50. doi: 10.1128/AAC.00968-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Eichenbaum G, Skibbe J, Parkinson A, Johnson MD, Baumgardner D, Ogilvie B, Usuki E, Tonelli F, Holsapple J, Schmitt-Hoffmann A. Use of enzyme inhibitors to evaluate the conversion pathways of ester and amide prodrugs: A case study example with the prodrug ceftobiprole medocaril. J Pharm Sci. 2012;101:1242–52. doi: 10.1002/jps.22816. [DOI] [PubMed] [Google Scholar]
- 168.Rhee SG. Reflections on the days of phospholipase C. Adv Biol Regul. 2013;53:223–31. doi: 10.1016/j.jbior.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 169.Baylon JL, Lenov IL, Sligar SG, Tajkhorshid E. Characterizing the membrane-bound state of cytochrome P450 3A4: Structure, depth of insertion, and orientation. J Am Chem Soc. 2013;135:8542–51. doi: 10.1021/ja4003525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Weiske J, Huber O. The histidine triad protein Hint1 interacts with pontin and reptin and inhibits TCF-beta-catenin-mediated transcription. J Cell Sci. 2005;118:3117–29. doi: 10.1242/jcs.02437. [DOI] [PubMed] [Google Scholar]
- 171.Birkus G, Kutty N, Frey CR, Shribata R, Chou T, Wagner C, McDermott M, Cihlar T. Role of cathepsin A and lysosomes in the intracellular activation of novel antipapillomavirus agent GS-9191. Antimicrob Agents Chemother. 2011;55:2166–73. doi: 10.1128/AAC.01603-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Korboukh I, Hull-Ryde EA, Rittiner JE, Randhawa AS, Coleman J, Fitzpatrick BJ, Setola V, Janzen WP, Frye SV, Zylka MJ, et al. Orally active adenosine A(1) receptor agonists with antinociceptive effects in mice. J Med Chem. 2012;55:6467–77. doi: 10.1021/jm3004834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kumar TS, Yang T, Mishra S, Cronin C, Chakraborty S, Shen JB, Liang BT, Jacobson KA. 5′-phosphate and 5′-phosphonate ester derivatives of (N)-methanocarba adenosine with in vivo cardioprotective activity. J Med Chem. 2013;56:902–14. doi: 10.1021/jm301372c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Mehellou Y. Phosphoramidate prodrugs deliver with potency against hepatitis C virus. Chemmedchem. 2010;5:1841–2. doi: 10.1002/cmdc.201000310. [DOI] [PubMed] [Google Scholar]
- 175.Donnell D, Baeten JM, Bumpus NN, Brantley J, Bangsberg DR, Haberer JE, Mujugira A, Mugo N, Ndase P, Hendrix C, et al. HIV protective efficacy and correlates of tenofovir blood concentrations in a clinical trial of PrEP for HIV prevention. J Acquir Immune Defic Syndr. 2014 doi: 10.1097/QAI.0000000000000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Chang W, Bao D, Chun BK, Naduthambi D, Nagarathnam D, Rachakonda S, Reddy PG, Ross BS, Zhang H, Bansal S, et al. Discovery of PSI-353661, a novel purine nucleotide prodrug for the treatment of HCV infection. ACS Med Chem Lett. 2011;2:130–5. doi: 10.1021/ml100209f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Du J, Chun B, Mosley RT, Bansal S, Bao H, Espiritu C, Lam AM, Murakami E, Niu C, Micolochick Steuer HM, et al. Use of 2′-spirocyclic ethers in HCV nucleoside design. J Med Chem. 2014;57:1826–35. doi: 10.1021/jm401224y. [DOI] [PubMed] [Google Scholar]
- 178.Jordheim LP, Durantel D, Zoulim F, Dumontet C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat Rev Drug Discovery. 2013;12:447–64. doi: 10.1038/nrd4010. [DOI] [PubMed] [Google Scholar]
- 179.De Clercq E. The clinical potential of the acyclic (and cyclic) nucleoside phosphonates. the magic of the phosphonate bond. Biochem Pharmacol. 2011;82:99–109. doi: 10.1016/j.bcp.2011.03.027. [DOI] [PubMed] [Google Scholar]
- 180.De Clercq E. Highlights in antiviral drug research: Antivirals at the horizon. Med Res Rev. 2013;33:1215–48. doi: 10.1002/med.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Poijarvi-Virta P, Lonnberg H. Prodrug approaches of nucleotides and oligonucleotides. Curr Med Chem. 2006;13:3441–65. doi: 10.2174/092986706779010270. [DOI] [PubMed] [Google Scholar]
- 182.Hurwitz SJ, Schinazi RF. Prodrug strategies for improved efficacy of nucleoside antiviral inhibitors. Curr Opin HIV AIDS. 2013;8:556–64. doi: 10.1097/COH.0000000000000007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Miralles-Lluma R, Figueras A, Busque F, Alvarez-Larena A, Balzarini J, Figueredo M, Font J, Alibes R, Marechal J. Synthesis, antiviral evaluation, and computational studies of cyclobutane and cyclobutene L- nucleoside analogues. Eur J Med Chem. 2013;2013:7761–75. [Google Scholar]
- 184.Oliveira FM, Barbosa LCA, Ismail FMD. The diverse pharmacology and medicinal chemistry of phosphoramidates - a review. RSC Adv. 2014;4:18998–9012. [Google Scholar]
- 185.Schulz T, Balzarini J, Meier C. The DiPPro approach: Synthesis, hydrolysis, and antiviral activity of lipophilic d4T diphosphate prodrugs. Chem Med Chem. 2014;9:762–75. doi: 10.1002/cmdc.201300500. [DOI] [PubMed] [Google Scholar]
- 186.Jessen HJ, Schulz T, Balzarini J, Meier C. Bioreversible protection of nucleoside diphosphates. Angew Chem Int Ed. 2008;47:8719–22. doi: 10.1002/anie.200803100. [DOI] [PubMed] [Google Scholar]
- 187.Cho A, Zhang L, Xu J, Lee R, Butler T, Metobo S, Aktoudianakis V, Lew W, Ye H, Clarke M, et al. Discovery of the first C-nucleoside HCV polymerase inhibitor (GS-6620) with demonstrated antiviral response in HCV infected patients. J Med Chem. 2014;57:1812–25. doi: 10.1021/jm400201a. [DOI] [PubMed] [Google Scholar]
- 188.Arbelo Roman C, Wasserthal P, Balzarini J, Meier C. Diastereoselective synthesis of (aryloxy)phosphoramidate prodrugs. Eur J Org Chem. 2011;2011:4899–909. [Google Scholar]
- 189.Arbelo Roman C, Balzarini J, Meier C. Diastereoselective synthesis of aryloxy phosphoramidate prodrugs of 3′-deoxy-2′,3′-didehydrothymidine monophosphate. J Med Chem. 2010;53:7675–81. doi: 10.1021/jm100817f. [DOI] [PubMed] [Google Scholar]
- 190.Sofia MJ, Bao D, Chang W, Du J, Nagarathnam D, Rachakonda S, Reddy PG, Ross BS, Wang P, Zhang HR, et al. Discovery of a beta-d-2′-deoxy-2′-alpha-fluoro-2′-beta-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J Med Chem. 2010;53:7202–18. doi: 10.1021/jm100863x. [DOI] [PubMed] [Google Scholar]
- 191.Gane EJ, Stedman CA, Hyland RH, Ding X, Svarovskaia E, Symonds WT, Hindes RG, Berrey MM. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med. 2013;368:34–44. doi: 10.1056/NEJMoa1208953. [DOI] [PubMed] [Google Scholar]
- 192.Sulkowski MS, Gardiner DF, Rodriguez-Torres M, Reddy KR, Hassanein T, Jacobson I, Lawitz E, Lok AS, Hinestrosa F, Thuluvath PJ, et al. Daclatasvir plus sofosbuvir for previously treated or untreated chronic HCV infection. N Engl J Med. 2014;370:211–21. doi: 10.1056/NEJMoa1306218. [DOI] [PubMed] [Google Scholar]
- 193.Hazleton KZ, Ho M, Cassera MB, Clinch K, Crump DR, Rosario I, Merino EF, Almo SC, Tyler PC, Schramm VL. Acyclic immucillin phosphonates: Second-generation inhibitors of plasmodium falciparum hypoxanthine- guanine-xanthine phosphoribosyltransferase. Chem Biol. 2012;19:721–30. doi: 10.1016/j.chembiol.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Keough DT, Spacek P, Hockova D, Tichy T, Vrbkova S, Slavetinska L, Janeba Z, Naesens L, Edstein MD, Chavchich M, et al. Acyclic nucleoside phosphonates containing a second phosphonate group are potent inhibitors of 6-oxopurine phosphoribosyltransferases and have antimalarial activity. J Med Chem. 2013;56:2513–26. doi: 10.1021/jm301893b. [DOI] [PubMed] [Google Scholar]
- 195.Keough DT, Hockova D, Rejman D, Spacek P, Vrbkova S, Krecmerova M, Eng WS, Jans H, West NP, Naesens LMJ, et al. Inhibition of the escherichia coli 6-oxopurine phosphoribosyltransferases by nucleoside phosphonates: Potential for new antibacterial agents. J Med Chem. 2013;56:6967–84. doi: 10.1021/jm400779n. [DOI] [PubMed] [Google Scholar]
- 196.Ebetino FH, Hogan AL, Sun S, Tsoumpra MK, Duan X, Triffitt JT, Kwaasi AA, Dunford JE, Barnett BL, Oppermann U, et al. The relationship between the chemistry and biological activity of the bisphosphonates. Bone. 2011;49:20–33. doi: 10.1016/j.bone.2011.03.774. [DOI] [PubMed] [Google Scholar]
- 197.Rogers MJ, Ji XH, Russell RGG, Blackburn GM, Williamson MP, Bayless AV, Ebetino FH, Watts DJ. Incorporation of bisphosphonates into adenine-nucleotides by amebas of the cellular slime-mold dictyostelium-discoideum. Biochem J. 1994;303:303–11. doi: 10.1042/bj3030303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Frith JC, Monkkonen J, Blackburn GM, Russell RGG, Rogers MJ. Clodronate and liposome-encapsulated clodronate are metabolized to a toxic ATP analog, adenosine 5′-(beta,gamma-dichloromethylene) triphosphate, by mammalian cells in vitro. J Bone Miner Res. 1997;12:1358–67. doi: 10.1359/jbmr.1997.12.9.1358. [DOI] [PubMed] [Google Scholar]
- 199.Niemi R, Turhanen P, Vepsalainen J, Taipale H, Jarvinen T. Bisphosphonate prodrugs: Synthesis and in vitro evaluation of alkyl and acyloxymethyl esters of etidronic acid as bioreversible prodrugs of etidronate. Eur J Pharm Sci. 2000;11:173–80. doi: 10.1016/s0928-0987(00)00099-3. [DOI] [PubMed] [Google Scholar]
- 200.Ahlmark M, Vepsalainen J, Taipale H, Niemi R, Jarvinen T. Bisphosphonate prodrugs: Synthesis and in vitro evaluation of novel clodronic acid dianhydrides as bioreversible prodrugs of clodronate. J Med Chem. 1999;42:1473–6. doi: 10.1021/jm9810809. [DOI] [PubMed] [Google Scholar]
- 201.Webster MR, Zhao M, Rudek MA, Hann CL, Freel Meyers CL. Bisphosphonamidate clodronate prodrug exhibits potent anti-cancer activity in non-small-cell lung cancer cells. J Med Chem. 2011;54:6647–56. doi: 10.1021/jm200521a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Wiemer AJ, Hohl RJ, Wiemer DF. The intermediate enzymes of isoprenoid metabolism as anticancer targets. Anticancer Agents Med Chem. 2009;9:526–42. doi: 10.2174/187152009788451860. [DOI] [PubMed] [Google Scholar]
- 203.Wiemer AJ, Wiemer DF, Hohl RJ. Geranylgeranyl diphosphate synthase: An emerging therapeutic target. Clin Pharmacol Ther. 2011;90:804–12. doi: 10.1038/clpt.2011.215. [DOI] [PubMed] [Google Scholar]
- 204.Monteil M, Migianu-Griffoni E, Sainte-Catherine O, Di Benedetto M, Lecouvey M. Bisphosphonate prodrugs: Synthesis and biological evaluation in HuH7 hepatocarcinoma cells. Eur J Med Chem. 2014;77:56–64. doi: 10.1016/j.ejmech.2014.02.054. [DOI] [PubMed] [Google Scholar]
- 205.Zhang Y, Leon A, Song Y, Studer D, Haase C, Koscielski LA, Oldfield E. Activity of nitrogen-containing and non-nitrogen-containing bisphosphonates on tumor cell lines. J Med Chem. 2006;49:5804–14. doi: 10.1021/jm060280e. [DOI] [PubMed] [Google Scholar]
- 206.Wiemer AJ, Yu JS, Shull LW, Barney RJ, Wasko BM, Lamb KM, Hohl RJ, Wiemer DF. Pivaloyloxymethyl-modified isoprenoid bisphosphonates display enhanced inhibition of cellular geranyl-geranylation. Bioorg Med Chem. 2008;16:3652–60. doi: 10.1016/j.bmc.2008.02.016. [DOI] [PubMed] [Google Scholar]
- 207.Troutman JM, Chehade KA, Kiegiel K, Andres DA, Spielmann HP. Synthesis of acyloxymethyl ester prodrugs of the transferable protein farnesyl transferase substrate farnesyl methylenediphosphonate. Bioorg Med Chem Lett. 2004;14:4979–82. doi: 10.1016/j.bmcl.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 208.Clark MK, Scott SA, Wojtkowiak J, Chirco R, Mathieu P, Reiners JJ, Jr, Mattingly RR, Borch RF, Gibbs RA. Synthesis, biochemical, and cellular evaluation of farnesyl monophosphate prodrugs as farnesyltransferase inhibitors. J Med Chem. 2007;50:3274–82. doi: 10.1021/jm0701829. [DOI] [PubMed] [Google Scholar]
- 209.Sane KM, Mynderse M, LaLonde DT, Dean IS, Wojtkowiak JW, Fouad F, Borch RF, Reiners JJ, Jr, Gibbs RA, Mattingly RR. A novel geranylgeranyl transferase inhibitor in combination with lovastatin inhibits proliferation and induces autophagy in STS-26T MPNST cells. J Pharmacol Exp Ther. 2010;333:23–33. doi: 10.1124/jpet.109.160192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Wiemer AJ, Hsiao CH, Wiemer DF. Isoprenoid metabolism as a therapeutic target in gram-negative pathogens. Curr Top Med Chem. 2010;10:1858–71. doi: 10.2174/156802610793176602. [DOI] [PubMed] [Google Scholar]
- 211.Reichenberg A, Wiesner J, Weidemeyer C, Dreiseidler E, Sanderbrand S, Altincicek B, Beck E, Schlitzer M, Jomaa H. Diaryl ester prodrugs of FR900098 with improved in vivo antimalarial activity. Bioorg Med Chem Lett. 2001;11:833–5. doi: 10.1016/s0960-894x(01)00075-0. [DOI] [PubMed] [Google Scholar]
- 212.Ortmann R, Wiesner J, Reichenberg A, Henschker D, Beck E, Jomaa H, Schlitzer M. Acyloxyalkyl ester prodrugs of FR900098 with improved in vivo anti-malarial activity. Bioorg Med Chem. 2003;13:2163–6. doi: 10.1016/s0960-894x(03)00354-8. [DOI] [PubMed] [Google Scholar]
- 213.Ortmann R, Wiesner J, Reichenberg A, Henschker D, Beck E, Jomaa H, Schlitzer M. Alkoxycarbonyloxyethyl ester prodrugs of FR900098 with improved in vivo antimalarial activity. Arch Pharm (Weinheim) 2005;338:305–14. doi: 10.1002/ardp.200500976. [DOI] [PubMed] [Google Scholar]
- 214.Kurz T, Schlueter K, Kaula U, Bergmann B, Walter RD, Geffken D. Synthesis and antimalarial activity of chain substituted pivaloyloxymethyl ester analogues of fosmidomycin and FR900098. Bioorg Med Chem. 2006;14:5121–35. doi: 10.1016/j.bmc.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 215.Kurz T, Behrendt C, Kaula U, Bergmann B, Walter RD. A-phenylethyl substituted bis(pivaloyloxymethyl) ester analogs of fosmidomycin and FR900098. Aust J Chem. 2007;60:154–8. doi: 10.1002/ardp.200700107. [DOI] [PubMed] [Google Scholar]
- 216.Schluter K, Walter RD, Bergmann B, Kurz T. Arylmethyl substituted derivatives of fosmidomycin: Synthesis and antimalarial activity. Eur J Med Chem. 2006;41:1385–97. doi: 10.1016/j.ejmech.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 217.Kurz T, Schluter K, Pein M, Behrendt C, Bergmann B, Walter RD. Conformationally restrained aromatic analogues of fosmidomycin and FR900098. Arch Pharm (Weinheim) 2007;340:339–44. doi: 10.1002/ardp.200700013. [DOI] [PubMed] [Google Scholar]
- 218.Haemers T, Wiesner J, Giessmann D, Verbrugghen T, Hillaert U, Ortmann R, Jomaa H, Link A, Schlitzer M, Van C, Serge Synthesis of beta- and gamma-oxa isosteres of fosmidomycin and FR900098 as antimalarial candidates. Bioorg Med Chem. 2008;16:3361–71. doi: 10.1016/j.bmc.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 219.Brucher K, Illarionov B, Held J, Tschan S, Kunfermann A, Pein MK, Bacher A, Graewert T, Maes L, Mordmueller B, et al. A-substituted β-oxa isosteres of fosmidomycin: Synthesis and biological evaluation. J Med Chem. 2012;55:6566–75. doi: 10.1021/jm300652f. [DOI] [PubMed] [Google Scholar]
- 220.Granthon AC, Braga MV, Rodrigues JCF, Cammerer S, Lorente SO, Gilbert IH, Urbina JA, de S, Wanderley Alterations on the growth and ultrastructure of leishmania chagasi induced by squalene synthase inhibitors. Vet Parasitol. 2007;146:25–34. doi: 10.1016/j.vetpar.2006.12.022. [DOI] [PubMed] [Google Scholar]
- 221.Uh E, Jackson ER, San Jose G, Maddox M, Lee RE, Lee RE, Boshoff HI, Dowd CS. Antibacterial and antitubercular activity of fosmidomycin, FR900098, and their lipophilic analogs. Bioorg Med Chem Lett. 2011;21:6973–6. doi: 10.1016/j.bmcl.2011.09.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Ponaire S, Zingle C, Tritsch D, Grosdemange-Billiard C, Rohmer M. Growth inhibition of mycobacterium smegmatis by prodrugs of deoxyxylulose phosphate reducto-isomerase inhibitors, promising anti-mycobacterial agents. Eur J Med Chem. 2012;51:277–85. doi: 10.1016/j.ejmech.2012.02.031. [DOI] [PubMed] [Google Scholar]
- 223.Stankovic CJ, Surendran N, Lunney EA, Plummer MS, Para KS, Shahripour A, Fergus JH, Marks JS, Herrera R, Hubbell SE, et al. The role of 4-phosphonodifluoromethyl- and 4-phosphono-phenylalanine in the selectivity and cellular uptake of SH2 domain ligands. Bioorg Med Chem Lett. 1997;7:1909–14. [Google Scholar]
- 224.Garrido-Hernandez H, Moon KD, Geahlen RL, Borch RF. Design and synthesis of phosphotyrosine peptidomimetic prodrugs. J Med Chem. 2006;49:3368–76. doi: 10.1021/jm060142p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Huang R, Oh H, Arrendale A, Martin VA, Galan J, Workman EJ, Stout JR, Walczak CE, Tao WA, Borch RF, et al. Intracellular targets for a phosphotyrosine peptidomimetic include the mitotic kinesin, MCAK. Biochem Pharmacol. 2013;86:597–611. doi: 10.1016/j.bcp.2013.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Boutselis IG, Yu X, Zhang ZY, Borch RF. Synthesis and cell-based activity of a potent and selective protein tyrosine phosphatase 1B inhibitor prodrug. J Med Chem. 2007;50:856–64. doi: 10.1021/jm061146x. [DOI] [PubMed] [Google Scholar]
- 227.Auzenne EJ, Klostergaard J, Mandal PK, Liao WS, Lu Z, Gao F, Bast RC, Jr, Robertson FM, McMurray JS. A phosphopeptide mimetic prodrug targeting the SH2 domain of Stat3 inhibits tumor growth and angiogenesis. J Exp Ther Oncol. 2012;10:155–62. [PMC free article] [PubMed] [Google Scholar]
- 228.Mandal PK, Liao WSL, McMurray JS. Synthesis of phosphatase-stable, cell-permeable peptidomimetic prodrugs that target the SH2 domain of Stat3. Org Lett. 2009;11:3394–7. doi: 10.1021/ol9012662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Mandal PK, Gao F, Lu Z, Ren Z, Ramesh R, Birtwistle JS, Kaluarachchi KK, Chen X, Bast RC, Liao WSL, et al. Potent and selective phosphopeptide mimetic prodrugs targeted to the src homology 2 (SH2) domain of signal transducer and activator of transcription 3. J Med Chem. 2011;54:3549–63. doi: 10.1021/jm2000882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Mandal PK, Ren Z, Chen X, Kaluarachchi K, Liao WSL, McMurray JS. Structure-activity studies of phosphopeptidomimetic pro-drugs targeting the src homology 2 (SH2) domain of signal transducer and activator of transcription 3 (Stat3) Int J Pept Res Ther. 2013;19:3–12. doi: 10.1007/s10989-012-9313-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Morlacchi P, Mandal PK, McMurray JS. Synthesis and in vitro evaluation of a peptidomimetic inhibitor targeting the src homology 2 (SH2) domain of STAT6. ACS Med Chem Lett. 2014;5:69–72. doi: 10.1021/ml4003919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Chu CY, Chang CP, Chou YT, Handoko, Hu YL, Lo LC, Lin JJ. Development and evaluation of novel phosphotyrosine mimetic inhibitors targeting the src homology 2 domain of signaling lymphocytic activation molecule (SLAM) associated protein. J Med Chem. 2013;56:2841–9. doi: 10.1021/jm301610q. [DOI] [PubMed] [Google Scholar]
- 233.Arrendale A, Kim K, Choi JY, Li W, Geahlen RL, Borch RF. Synthesis of a phosphoserine mimetic prodrug with potent 14-3-3 protein inhibitory activity. Chem Biol. 2012;19:764–71. doi: 10.1016/j.chembiol.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Qian WJ, Burke TR, Jr, Terrence R. Design and synthesis of a reagent for solid-phase incorporation of the phosphothreonine mimetic (2S,3R)-2-amino-3-methyl-4-phosphonobutyric acid (pmab) into peptides in a bio-reversible phosphonyl-bis-pivaloyloxymethyl (POM) prodrug form. Amino Acids. 2013;45:1143–8. doi: 10.1007/s00726-013-1567-0. [DOI] [PMC free article] [PubMed] [Google Scholar]