

Abstract
Glucocorticoid and glucocorticoid receptor (GC/GR) interactions alter numerous aspects of neuronal function. These consequences (e.g., anti-inflammatory vs. pro-inflammatory) can vary depending on the duration of GC exposure or central nervous system (CNS) injury model. In this review we discuss how GC/GR interactions impact neuronal recovery after a central or peripheral nerve injury and discuss how GC exposure duration can produce divergent CNS neuronal growth responses. Finally we consider how new findings on gender specific immune cell responses after a nerve injury could intersect with GC/GR interactions to impact pain processing.
Keywords: glucocorticoids, plasticity, stress, neuropathic pain
Introduction
Nerve injury, stress, and inflammation can activate the hypothalamus-pituitary-adrenal (HPA) axis, which causes the release of glucocorticoids (GCs) into the bloodstream that bind to the widely expressed glucocorticoid or mineralocorticoid nuclear receptors (GRs, MRs). Unbound GR is in the cytoplasm but when GC binds GR, it translocates into the nucleus where it initiates gene transcription (Revollo and Cidlowski, 2009). At basal levels, GCs are more likely to bind to MRs. However, at times of elevated GC concentration (e.g., stress) GCs will bind GRs.
GC/GR interactions can affect chronic conditions such as neuropathic pain. Neuropathic pain develops after nerve injury and results from abnormal pain processing. Even though nerve injury is necessary to produce neuropathic pain, it is not causal and there are many factors that contribute to the neuropathic pain phenotype (Costigan et al., 2009). Neuropathic pain can be characterized by the presence of allodynia, a pain response to a previously non-noxious stimulus, and hyperalgesia, an exaggerated response to an already painful stimulus.
Neuropathic pain is associated with structural changes to the somatosensory system (Figure 1; Costigan et al., 2009). These changes can be at the level of the synapse or the cell. At the synapse central sensitization, a type of use-dependent synaptic plasticity, arises from changes in excitatory amino acids, ion channels, ionotropic receptor density, and activation of presynaptic and postsynaptic kinases (Figure 1C; Costigan et al., 2009). The onset of central sensitization is caused by synaptic strengthening to the point that input activity that was previously below threshold will activate nociceptors (Costigan et al., 2009), inducing an ectopic pain response. In addition to synaptic plasticity, there is evidence of anatomical plasticity (e.g., sprouting) after the onset of neuropathic pain. Sprouting helps injured neurons reconnect with their targets, but also produces maladaptive changes. While controversial, general sensory neuron sprouting and increased input into nociceptive pathways is thought to contribute to increased pain (Figure 1B; Costigan et al., 2009). In contrast, nerve injury also promotes neurodegeneration and death in primary sensory neurons, a permanent alteration in the sensory processing pathway (Figure 1B, E; Costigan et al., 2009).
Figure 1.

Plasticity in the somatosensory system after injury and/or glucocorticoid (GC) exposure.
The majority of studies are in rodents, however similar changes to the human somatosensory system and brain can be deduced. Sensory neurons and nociceptors enter the central nervous system (CNS) through the dorsal root ganglia and synapse in the dorsal horn of the spinal cord. Each pathway routes through the medulla to the thalamus and ends in the primary somatosensory cortex. (A) GCs affect the hippocampus and amygdala differently. Hippocampal CA3 pyramidal dendrites atrophy while basolateral amygdala pyramidal and stellate dendrites increase in complexity. (B) The dorsal horn of the spinal cord is a region of divergent effects after nerve injury. Nociceptive sensory neurons sprout and increase pain, or undergo neuronal death. (C) Synaptic plasticity contributes to central sensitization from changes in excitatory amino acid release, ion channel receptor expression, ionotropic receptor density, and activation of pre- and postsynaptic kinases. (D) Neuropathic pain associated with trigeminal neuropathy is correlated with reduced gray matter in the ipsilateral spinal trigeminal nucleus. (E) After nerve injury, primary sensory neurons degenerate. (F) Tight junction protein expression is reduced with GC exposure and ischemia and seizure. (G) Middle cerebral artery occlusion and GC exposure increase inflammation and infarct size. (H) Microglial reactivity increases in the dorsal horn of the spinal cord after spared nerve injury and GC exposure. (I) Chronic constriction injury produces a time-dependent upregulation of N-methyl-D-aspartate (NMDA) receptor subunits within the spinal cord dorsal horn.
Neuropathic pain induces changes in both the peripheral and central nervous systems (CNS), often with cell-specific changes (Figure 1). For example, elevated GCs induce plasticity in the CA3 region of the hippocampus, the basolateral amygdala (BLA), and the spinal trigeminal nucleus, all changes that can increase central sensitization (Figure 1A, B, D). This review will explore these interactions and speculate about GC induced plasticity in the central and peripheral nervous systems.
Glucocorticoids and Their Relation to Pain
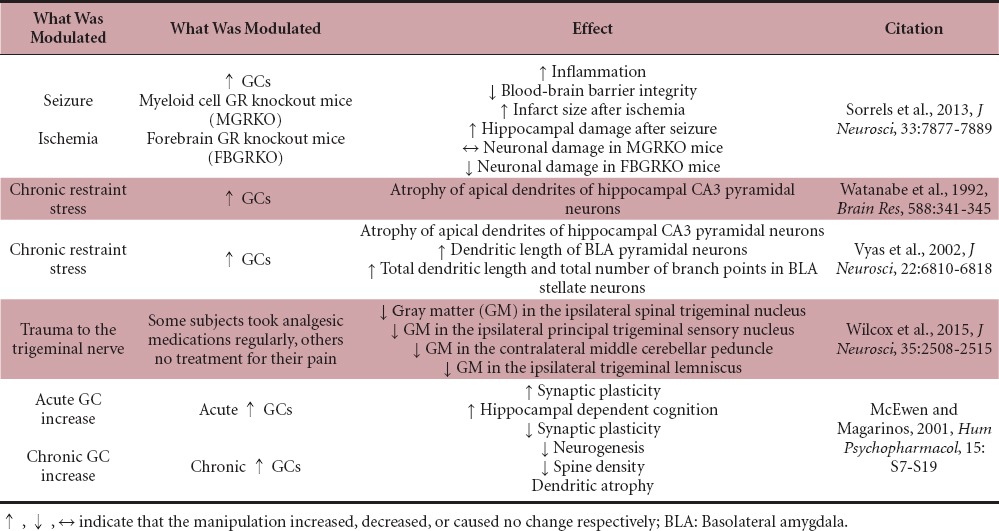
GCs are anti-inflammatory, and as such, synthetic GCs are used to treat nerve injury (Bracken et al., 1992), but growing evidence suggests that GCs can also be pro-inflammatory (Table 1; Sorrells et al., 2013). For example, in mouse models of seizure and ischemia, when GCs were delivered prior to kainic acid treatment or middle cerebral artery occlusion microglial activation was increased (e.g., increased Iba-1 and CD68 staining as well as nuclear p65 levels; Figure 1A, G; Sorrells et al., 2013). Increased GC/GR signaling in endothelial cells reduced levels of several principal proteins in blood-brain barrier (BBB) tight junctions which compromised BBB integrity (Table 1 and Figure 1F; Sorrells et al., 2013). Neuronal death induced from GC signaling in myeloid cells outweighed GC/GR neuroprotective effects. This was shown in myeloid GR knockout mice and neuronal forebrain GR knockout mice (Sorrells et al., 2013). GC treatment prior to MCAO increased infarct size and hippocampal damage following KA excitotoxic injury regardless of forebrain neuronal GR expression whereas loss of GR in myeloid cells did not change infarct size or hippocampal damage (Sorrells et al., 2013).
Table 1.
Effect of glucocorticoid (GC)/glucocorticoid receptor (GR) on the central nervous system
GCs are also being studied in the context of peripheral nerve injury. It has been shown that neuropathic pain caused by chronic constriction injury (CCI) is inhibited by the GR antagonist RU38486 or antisense mediated GR knockdown (Table 2; Shuxing Wang et al., 2004). Similarly neuropathic pain caused by CCI is inhibited by RU486 or the more specific GR antagonist dexamethasone 21-mesylate (Table 2; Takasaki et al., 2005). While the exact molecular mechanism is unknown, a loss of endogenous GCs after adrenalectomy weakened, and synthetic GCs reestablished the neuropathic pain response (e.g., dexamethasone; Table 2; Wang et al., 2004). Interestingly, increasing endogenous GCs increased neuropathic pain. Mice receiving acute restraint stress (60 minutes), which produces an approximately 12–24 hours increase in GCs prior to spared nerve injury (SNI) showed increased mechanical allodynia that was prevented by the GR antagonist RU486 (Alexander et al., 2009). Administration of corticosterone to non-stressed mice produced the same effect as acute restraint stress (Table 2). Stress also increased dorsal horn microglial reactivity (Figure 1H; Alexander et al., 2009). These studies suggest that GC/GR interactions play a significant role in the generation of neuropathic pain, but exact cell type and place of GC/GR interaction that is mediating this response needs further investigation.
Table 2.
Effect of glucocorticoid (GC)/glucocorticoid receptor (GR) signaling on peripheral nerve injury and pain

N-methyl-D-aspartate (NMDA) receptors have a key role in GR-dependent neuropathic pain. CCI produced a time-dependent upregulation of NMDA receptor subunits within the spinal cord dorsal horn (Figure 1I; Wang et al., 2005). This upregulation was significantly reduced when given the GR antagonist RU486 or an antisense oligonucleotide against GR (Wang et al., 2005). In Alexander et al. (2009), they also explored the idea that NMDA receptors play a role in stress-exacerbated nerve injury-induced neuropathic pain. Administration of the NMDA receptor antagonist memantine prior to acute restraint stress and SNI in mice diminished injury-induced allodynia, similarly to the GR antagonist (Table 2; Alexander et al., 2009). These studies show that drugs that block either GC or NMDA signaling also block GC-induced increases in allodynia.
Plasticity Induced by Stress or Neuropathic Pain has Divergent Effects
Increased GCs induce structural plasticity in the CNS, which is dependent on the duration of the stress. Chronic stress which results in prolonged and sustained GC elevation (six hours of restraint stress daily for three weeks) induced atrophy of apical dendrites of hippocampal CA3 pyramidal neurons (Table 1 and Figure 1A; Watanabe et al., 1992). However, a later study found contrasting effects of stress on dendritic plasticity in neurons in the hippocampus and neurons in the amygdala (Vyas et al., 2002). Vyas et al. (2002) showed that chronic immobilization stress (two hours per day for 10 days) induced dendritic atrophy in CA3 pyramidal cells in the hippocampus, similar to what was observed previously by Watanabe et al. (1992), but simultaneously increased dendritic length of BLA pyramidal neurons. Total dendritic length and total number of branch points also increased in BLA stellate neurons after chronic stress (Table 1 and Figure 1A; Vyas et al., 2002). In the same study, chronic unpredictable stress (10 days of randomly chosen stressors) was also examined (Vyas et al., 2002). Chronic unpredictable stress induced dendritic atrophy in CA3 pyramidal cells in the hippocampus, however not to the same degree as chronic immobilization stress (Vyas et al., 2002). Interestingly, chronic unpredictable stress caused the opposite effect in BLA neurons than what was observed from chronic immobilization stress (Vyas et al., 2002). Unpredictable stress caused a decrease in dendritic length in bipolar/bitufted BLA neurons (Vyas et al., 2002). Even though anatomical plasticity was evaluated extensively, the physiological consequences were not investigated. However, a study that examined psychosocial stress in tree shrews found changes in neuronal excitability of hippocampal CA3 pyramidal neurons (Kole et al., 2004). Taken together, these studies suggest that the effects of GCs/stress on plasticity are not always repressive and are cell-type dependent. As discussed here, elevated GCs cause degeneration in some neurons, yet promote outgrowth in others.
Chronic restraint stress caused decreased dendritic length and decreased dendritic spine density of apical, but not basilar dendrites in the medial prefrontal cortex, specifically layer II/III pyramidal neurons (Cook and Wellman, 2004; Radley et al., 2005). Early life stress (e.g., maternal separation) also induced changes in the medial prefrontal cortex (Chocyk et al., 2013). Basal dendritic tree atrophy as well as reduced spine density on the second-order branches of both apical and basal dendrites in layer II/III pyramidal neurons occurred after maternal separation (Chocyk et al., 2013). This early life stress also caused an impairment of long-term potentiation (Chocyk et al., 2013). Another study found that an NMDA receptor antagonist blocked dendritic atrophy and induced hypertrophy of apical dendrites in medial prefrontal cortex (Martin and Wellman, 2011).
The length of GC exposure also differentially affected synaptic plasticity. Acutely raised GC levels increased synaptic plasticity and facilitated hippocampal dependent cognition (McEwen and Magarinos, 2001), while long-term increased GCs impaired synaptic plasticity and cognition, decreased neurogenesis and spine density, and caused dendritic atrophy (Table 1; McEwen and Magarinos, 2001). These data suggest that acute stress can promote positive changes, such as growth and improved learning, while prolonged stress can promote negative ones, such as cell death.
A recent study showed for the first time that there are changes at the primary synapse level in humans following the onset of neuropathic pain (Wilcox et al., 2015). Using magnetic resonance imaging and diffusion tensor imaging it was revealed that subjects with painful trigeminal neuropathy have decreased gray matter in the ipsilateral spinal trigeminal nucleus (Figure 1D), the ipsilateral principal trigeminal sensory nucleus, the contralateral middle cerebellar peduncle, and the ipsilateral trigeminal lemniscus, which correlated with anatomical changes in the region of the primary synapse in these areas (Table 1; Wilcox et al., 2015). This study again reiterates that neuropathic pain is associated with structural plasticity in the brain, and showed that these changes did not correlate to intensity of pain, or to use of analgesic medications (Wilcox et al., 2015). Trigeminal neuropathy is extremely painful, which implies that anatomical changes associated with neuropathy could induce injurious alterations in synaptic wiring leading to an increase in pain. It is not clear if the plasticity is what increases pain perception, or if the plasticity is a response to the pain. It is important that Wilcox et al. (2015) addresses plasticity at the level of the primary synapse because sensory neurons are the first order nociceptors that arborize into the spinal cord and make synapses with the periphery. It remains unknown if synapse number or neuronal connections and survival are directly responsible for changing pain perception.
Sex Differences in Pain Hypersensitivity
GC/GR interactions have divergent effects based on cell type and CNS region, and new findings suggest large differences exist between males and females in the cell types that contribute to allodynia. For example, it has been recently demonstrated that microglia in male mice contribute to the development of neuropathic pain, but do not in females (Sorge et al., 2015). This study utilized SNI to induce neuropathic pain and depleted glial cells with minocycline, flourocitrate, or propentofylline. A reversal of the mechanical allodynia phenotype was found only in males, with no change in females (Table 2; Sorge et al., 2015). These three glial inhibitors can have unintended side effects, so the experimenters next depleted spinal cord microglia with intrathecal saporin toxin. They again observed reversed mechanical allodynia in male mice, and not females (Table 2; Sorge et al., 2015). The P2X4 receptor is required in microglia for the development of SNI induced neuropathic pain. Differential dorsal horn gene expression was observed between the sexes, and an upregulation of the P2X4 receptor was observed in males, but not females. The expression of other genes associated with microglial activity was equally upregulated in both sexes (Sorge et al., 2015). This suggests that the injury induced microgliosis occurs equally in males and females but that the allodynia-generating P2X4 signaling pathway is specific to the male microglia response. This study also explored NMDA receptor mediated neuropathic pain and found that allodynia was reversed equally in males and females following intrathecal NMDA receptor blockade (Table 2). Together these data show that there are multiple interacting pathways contributing to the development of neuropathic pain, and that these pathways may be gender specific.
These gender specific findings are extremely important because the majority of pain studies have been conducted in males. The foundation of what we know about neuropathic pain is based on how it is mediated in males. Future studies that explore these sex differences are needed to determine if many “answered” questions about neuropathic pain are truly answered when considering both sexes. Sex differences in pain hypersensitivity cause further concern for the effects of stress because we have seen that stress can induce changes in cell morphology and increase pain (Watanabe et al., 1992; Vyas et al., 2002; Alexander et al., 2009). It is necessary then to consider how and if there are sex differences in GC/GR interactions that could affect pain perception.
Neuronal Plasticity – Causal or Reactionary?
The balance of the findings discussed in this review suggest there is an “optimal level’ of GCs that can promote positive changes, but once that threshold is exceeded, there are detrimental changes. This could mean that acute stress can produce this optimal level of GCs, whereas chronic stress surpasses the GC threshold to produce different outcomes depending on the cell type and possibly onthe length of GC/GR interactions and downstream transcriptional activity. It will be a challenge to find the balance when the effects of GC/GR interactions are divergent depending on cell type and perhaps sex specific. It is currently unknown how both acute and chronic stress affects sensory neuron plasticity. It is known that there are structural changes in brain regions following the onset of neuropathic pain or elevated GCs, which are dependent on the timing and length of GC exposure, but several questions remain to be answered: Is the plasticity causal to neuropathic pain? Where does the plasticity occur? How do GC/GR interactions in specific cell types affect neuronal plasticity and growth? And finally, are there GC/GR induced sex specific differences in pain sensitivity? Future research will address these questions to provide a deeper understanding of how GC/GR interactions affect neuronal plasticity.
Footnotes
Conflicts of interest: None declared.
References
- Alexander JK, DeVries AC, Kigerl KA, Dahlman JM, Popovich PG. Stress exacerbates neuropathic pain via glucocorticoid and NMDA receptor activation. Brain Behav Immun. 2009;23:851–860. doi: 10.1016/j.bbi.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken MB, Shepard MJ, Holford TR, Leo-Summers L, Aldrich EF, Fazl M, Fehlings MG, Herr DL, Hitchon PW, Marshall LF, Nockels RP, Pascale V, Perot PL, Jr, Piepmeier J, Sonntag VKH, Wagner F, Wilberger JE, Winn HR, Young W. Methylprednisolone or naloxone treatment after acute spinal cord injury: 1-year follow-up data. J Neurosurg. 1992;76:23–31. doi: 10.3171/jns.1992.76.1.0023. [DOI] [PubMed] [Google Scholar]
- Chocyk A, Bobula B, Dudys D, Przyborowska A, Majcher-Maślanka I, Hess G, Wedzony K. Early-life stress affects the structural and functional plasticity of the medial prefrontal cortex in adolescent rats? Eur J Neurosci. 2013;38:2089–2107. doi: 10.1111/ejn.12208. [DOI] [PubMed] [Google Scholar]
- Cook SC, Wellman CL. Chronic stress alters dendritic morphology in rat medial prefrontal cortex. J Neurobiol. 2004;60:236–248. doi: 10.1002/neu.20025. [DOI] [PubMed] [Google Scholar]
- Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Neural Plast. 2010:1–32. doi: 10.1146/annurev.neuro.051508.135531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole MH, Czeh B, Fuchs E. Homeostatic maintenance in excitability of tree shrew hippocampal CA3 pyramidal neurons after chronic stress. Hippocampus. 2004;14:742–751. doi: 10.1002/hipo.10212. [DOI] [PubMed] [Google Scholar]
- Martin KP, Wellman CL. NMDA receptor blockade alters stress-induced dendritic remodeling in medial prefrontal cortex. Cereb Cortex. 2011;21:2366–2373. doi: 10.1093/cercor/bhr021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS, Magarinos AM. Stress and hippocampal plasticity: Implications for the pathophysiology of affective disorders. Hum Psychopharmacol. 2001:16. doi: 10.1002/hup.266. [DOI] [PubMed] [Google Scholar]
- Radley JJ, Rocher AB, Miller M, Janssen WGM, Liston C, Hof PR, McEwen BS, Morrison JH. Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cereb Cortex. 2005;16:313–320. doi: 10.1093/cercor/bhi104. [DOI] [PubMed] [Google Scholar]
- Revollo JR, Cidlowski JA. Mechanisms generating diversity in glucocorticoid receptor signaling. Ann N Y Acad Sci. 2009;1179:167–178. doi: 10.1111/j.1749-6632.2009.04986.x. [DOI] [PubMed] [Google Scholar]
- Sorge RE, Mapplebeck JC, Rosen S, Beggs S, Taves S, Alexander JK, Martin LJ, Austin JS, Sotocinal SG, Chen D, Yang M, Shi XQ, Huang H, Pillon NJ, Bilan PJ, Tu Y, Klip A, Ji RR, Zhang J, Salter MW, et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci. 2015:5. doi: 10.1038/nn.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrells SF, Caso JR, Munhoz CD, Sapolsky RM. The stressed CNS: when glucocorticoids aggravate inflammation. Neuron. 2009;64:33–39. doi: 10.1016/j.neuron.2009.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrells SF, Caso JR, Munhoz CD, Hu CK, Tran K V, Miguel ZD, Chien BY, Sapolsky RM. Glucocorticoid signaling in myeloid cells worsens acute CNS injury and inflammation. J Neurosci. 2013;33:7877–7889. doi: 10.1523/JNEUROSCI.4705-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasaki I, Kurihara T, Saegusa H, Zong S, Tanabe T. Effects of glucocorticoid receptor antagonists on allodynia and hyperalgesia in mouse model of neuropathic pain. Eur J Pharmacol. 2005;524:80–83. doi: 10.1016/j.ejphar.2005.09.045. [DOI] [PubMed] [Google Scholar]
- Vyas A, Mitra R, Shankaranarayana Rao BS, Chattarji S. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci. 2002;22:6810–6818. doi: 10.1523/JNEUROSCI.22-15-06810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Lim G, Zeng Q, Sung B, Yang L, Mao J. Central glucocorticoid receptors modulate the expression and function of spinal NMDA receptors after peripheral nerve injury. J Neurosci. 2005;25:488–495. doi: 10.1523/JNEUROSCI.4127-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Lim G, Zeng Q, Sung B, Ai Y, Guo G, Yang L, Mao J. Expression of central glucocorticoid receptors after peripheral nerve injury contributes to neuropathic pain behaviors in rats. J Neurosci. 2004;24:8595–8605. doi: 10.1523/JNEUROSCI.3058-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Gould E, McEwen BS. Stress induces atrophy of apical dendrites of hippocampal CA3 pyramidal neurons. Brain Res. 1992;588:341–345. doi: 10.1016/0006-8993(92)91597-8. [DOI] [PubMed] [Google Scholar]
- Wilcox SL, Gustin SM, Macey PM, Peck CC, Murray GM, Henderson LA. Anatomical changes at the level of the primary synapse in neuropathic pain: evidence from the spinal trigeminal nucleus. J Neurosci. 2015;35:2508–2515. doi: 10.1523/JNEUROSCI.3756-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]