

Abstract
The generation of mice lacking SCYL1 or SCYL2 and the identification of Scyl1 as the causative gene in the motor neuron disease mouse model muscle deficient (Scyl1mdf/mdf) demonstrated the importance of the SCY1-like family of protein pseudokinases in neuronal function and survival. Several essential cellular processes such as intracellular trafficking and nuclear tRNA export are thought to be regulated by SCYL proteins. However, whether deregulation of these processes contributes to the neurodegenerative processes associated with the loss of SCYL proteins is still unclear. Here, I briefly review the evidence supporting that SCYL proteins play a role in these processes and discuss their possible involvement in the neuronal functions of SCYL proteins. I also propose ways to determine the importance of these pathways for the functions of SCYL proteins in vivo.
Keywords: SCY1-like, SCYL1, SCYL2, SCYL3, motor neuron, hippocampal neuron, pseudokinase, neuro-degeneration
Introduction
The SCY1-like family of protein pseudokinases comprises 3 members: SCYL1, also known as p105 or protein with an N-terminal kinase-like protein domain (NTKL); SCYL2, also known as a coated vesicle-associated kinase of 104 kDa (CVAK104); and SCYL3, also known as the protein-associating with the C-terminal domain of Ezrin (PACE-1) (Figure 1). SCYL proteins are evolutionarily conserved and ubiquitously expressed proteins that share common structural features, such as an N-terminal kinase domain; centrally localized Huntingtin, elongation factor 3, protein phosphatase 2A and yeast kinase TOR1 (HEAT) repeats; and a C-terminal segment containing one or more coiled-coil domains. Unique to SCYL3 is a myristoylation consensus sequence at its N-terminus. Unlike conventional kinases, which possess phosphotransferase activity, the kinase domains of SCYL proteins are predicted to be inactive because of alterations in key residues essential for catalytic activity and might serve other functions.
Figure 1.
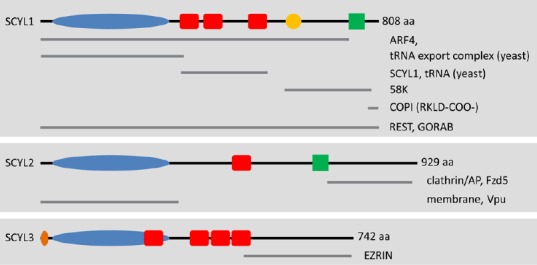
Schematic representation of SCYL family members and their potential interacting partners.
The SCYL family of protein pseudokinases comprises SCYL1, SCYL2, and SCYL3. Several SCYL1 isoforms have been reported in humans, most of which display variable C-terminal regions. These isoforms are not presented here. The kinase domains (blue ovals) of SCYL proteins are predicted to be inactive because of alterations of residues that are essential for catalytic activity. The red boxes represent the predicted HEAT repeats. The crystal structure of Cex1p (the yeast ortholog of SCYL1) reveals the presence of 6 pairs of antiparallel α-helices. The orange circle represents a proline-rich region of unknown function in SCYL1. The green boxes represent predicted coiled-coil regions. The orange oval represents the myristoylation consensus sequence found in SCYL3. Gray lines underneath the SCYL proteins highlight the regions of SCYL proteins that mediate interactions with their protein partners (labeled on the right of each gray line). For example, SCYL1 interacts with COPI via dibasic residues found in the C-terminal region of SCYL1 (RKLD-COO-). Other regions of SCYL1 are proposed to mediate the interactions with various other macromolecules, such as the small GTPases Arf4 and Ran (the tRNA export complex); tRNAs, Golgi-associated proteins GORAB and 58K; and itself, SCYL1. Homo-oligomerization of SCYL1 and tRNA binding are mediated by the central HEAT repeats. The N-terminal pseudokinase domain of SCYL2 can interact with the HIV protein Vpu and is capable of interacting with membrane structures within cultured cells. The C-terminal region of SCYL2 can also interact with clathrin/AP complexes (AP1 and AP2) as well as Fzd5. The C terminus of SCYL3 mediates the interaction between SCYL3 and the membrane-associated protein EZRIN. The functional relevance of these interactions and their implications in SCYL proteins functions in vivo (in particular SCYL1 and SCYL2) is unclear. NextProt (http://www.nextprot.org/db/) was used to identify domains and regions of the human SCYL proteins.
aa: Amino acids; ARF4: ADP-ribosylation factor 4; COPI: coat protein complex coatomer I; REST: RE-1 silencing transcription factor.
The targeted inactivation of Scyl1 in mice causes an early-onset progressive motor neuron disorder with features characteristic of amyotrophic lateral sclerosis (ALS) (Pelletier et al., 2012). Skeletal muscles of Scyl1-deficient mice show neurogenic atrophy, fiber type switching, and disuse atrophy. The changes in muscle fibers are associated with the progressive loss of large caliber axons in the periphery and lower motor neurons in the ventral horn of the spinal cord. We also demonstrated that SCYL1 acts in a neural cell-autonomous manner to maintain motor neuron functions and survival. Remarkably, loss of SCYL1 function leads to the mislocalization of TDP-43 from its normal, predominantly nuclear, location to cytosolic aggregates and accumulation of ubiquilin-2 within cytoplasmic inclusions in lower motor neurons, both of which are hallmarks of ALS and many neurodegenerative conditions (Chen-Plotkin et al., 2010).
Similarly, the targeted disruption of Scyl2 in mice causes severe neurologic disorders, leading to premature death in the majority of newborn mice and degeneration of CA3 pyramidal neurons in surviving mice. The loss of CA3 neurons is neuron-autonomous, occurs during functional maturation of the hippocampus and is the result of excitotoxicity, an apoptotic cell death caused by excessive excitatory signaling (Gingras et al., 2015). Mechanistically, we found that excitatory receptors KA1 and NR1, which are key mediators of excitotoxicity (Hara and Snyder, 2007), are overrepresented at synapses of Scyl2-deficient mice and thus may contribute to the neurodegenerative process associated with the loss of SCYL2 (Gingras et al., 2015).
While studies have begun to elucidate the biologic functions of SCYL1 and SCYL2, the physiological role of SCYL3 is still unknown. Mice deficient for SCYL3 have been generated in my laboratory and are now being studied.
Molecular and Cellular Functions of SCYL Proteins
Although our studies suggest a role for SCYL1 in regulating TDP-43 and ubiquilin-2 homeostasis and a role for SCYL2 in regulating synaptic expression of excitatory neurotransmitter receptors (Pelletier et al., 2012; Gingras et al., 2015), the molecular events leading to these biochemical changes and the molecular bases underlying the neurodegenerative processes associated with the loss of SCYL proteins have remained elusive.
By virtue of their subcellular localization and molecular interactions, SCYL proteins are proposed to regulate intracellular protein trafficking. Initially identified in a yeast two-hybrid screen for proteins interacting with protein kinase B, SCYL1 was later shown to localize to the ER-Golgi intermediate compartment (ERGIC), interact with the membrane coat protein complex coatomer I (COPI) via its C-terminal tail (Figure 1), facilitate retrograde transport of the KDEL (Lys-Asp-Glu-Leu) endoplasmic reticulum protein retention receptor (Burman et al., 2008), and regulate Golgi morphology (Burman et al., 2010; Hamlin et al., 2014). RNAi-mediated depletion of SCYL1 was shown to increase the size of the Golgi and tubulation of the ERGIC (Burman et al., 2010; Hamlin et al., 2014). In addition to interacting with COPI, SCYL1 also interacts with several Golgi-associated proteins: the formiminotransferase cyclodeaminase 58K (FTCD), a bifunctional Golgi-associated enzyme that couples histidine metabolism with folate metabolism and candidate protein integrating the Golgi apparatus with the intermediate filament cytoskeleton; the SCYL1 binding protein 1 (SCYL1-BP1; also known as NTKL-BP1 or GORAB), a candidate golgin mutated in gerodermia osteodysplastica, a rare autosomal recessive disorder of the connective tissue; and Arf4, a class II Arf GTPase involved in COPI-mediated membrane trafficking (Hamlin et al., 2014). The binding of these proteins through distinct SCYL1 domains or motifs (Figure 1) and the demonstration that both RNAi-mediated depletion of SCYL1 and overexpression of SCYL1 in HeLa cells promote tubulation of the ERGIC suggest that SCYL1 may function as a scaffolding protein, linking Golgi-associated proteins to COPI coat complexes (Hamlin et al., 2014).
SCYL2 was first identified through subcellular proteomics as a component of clathrin-coated vesicles and proposed to regulate clathrin-mediated endocytosis by phosphorylating the β2-subunit of the plasma membrane adaptor AP2 (Conner and Schmid, 2005). Although subsequent studies confirmed the interaction of SCYL2 with components of the clathrin machinery, they failed to detect any kinase activity associated with immunoprecipitated or bacterially expressed SCYL2 (Duwel and Ungewickell, 2006; Borner et al., 2007). Instead, they found that the N-terminal pseudokinase domain of SCYL2 mediates interaction with intracellular membranes whereas its C-terminal segment interacts with components of clathrin-AP1 complex. These studies also suggested a role for SCYL2 in regulating clathrin functions at the trans-Golgi network and endosomal compartments rather than the plasma membrane (Duwel and Ungewickell, 2006; Borner et al., 2007). Consistent with a role for SCYL2 in regulating intracellular trafficking, independent studies demonstrated that SCYL2 can promotelysosomal degradation of Frizzled 5 (Terabayashi et al., 2009) and limit the release of HIV-1 particles (Miyakawa et al., 2012).
Unlike SCYL1 and SCYL2, much less is known about the biochemical and cell biological properties of SCYL3. SCYL3 was identified in a yeast two-hybrid screen for proteins associating with Ezrin, a member of the ezrin/radixin/moesin (ERM) family of proteins, which links the cytoplasmic tails of transmembrane proteins to the actin cytoskeleton. Based on its subcellular localization, SCYL3 is proposed to regulate membrane protein or lipid trafficking at the Golgi and plasma membrane (Sullivan et al., 2003).
Given that intracellular trafficking plays a critical role in the normal development, function, and survival of the nervous system and that deregulation of these pathways is associated with various neurologic disorders such as Alzheimer's disease, Down syndrome, Parkinson's disease, and motor neuron diseases (e.g., ALS and spinal muscular atrophy), it is reasonable to assume that deregulation of these pathways contributes to the neurodegenerative processes seen in Scyl1- and Scyl2-deficient mice. Unexpectedly however, we found that intracellular trafficking and membrane protein turnover proceeded normally in Scyl1-and Scyl2-deficient fibroblasts. Similarly the overall organization of intracellular membranes appears normal in Scyl1- and Scyl2-deficient fibroblasts (Pelletier et al., 2012; Gingras et al., 2015). These findings contrast substantially with previous findings in cultured cells (Duwel and Ungewickell, 2006; Borner et al., 2007; Burman et al., 2008, 2010; Hamlin et al., 2014) and suggest that SCYL proteins play at most an accessory role in regulating intracellular trafficking, if any. In line with this, we found that unlike the targeted deletion of core components of the clathrin or COPI complexes, which causes embryonic lethality, targeted deletion of Scyl1 or Scyl2 has milder consequences (Pelletier et al., 2012; Gingras et al., 2015). Thus, the contribution of SCYL proteins in regulating intracellular trafficking and membrane protein turnover remains unclear, and whether the deregulation of these processes contributes to the neurodegenerative processes associated with the loss of SCYL functions is unknown.
Another fundamental process thought to be regulated by SCYL1 is the nucleocytoplasmic shuttling of tRNAs. Specifically, SCYL1 interacts with the nuclear pore complex proteins Nup98 and Nup107; the nuclear tRNA export receptors XPOT and XPO5;as well as the GTP-bound form of Ran, which is a member of the Ras superfamily of small GTPases involved in the nuclear cytoplasmic shuttling of RNA and the reassembly of the nuclear envelope after chromosome separation (Chafe and Mangroo, 2010). Although knockdown studies failed to show an essential nonredundant function for SCYL1 in regulating nuclear tRNA export in cultured cells (Chafe and Mangroo, 2010), the possible relation between SCYL and RNA metabolism is quite interesting and should not be dismissed. RNA metabolism defects have long been associated with a wide range of human neurologic disorders, and motor neurons of Scyl1-deficient mice exhibit TDP-43 pathology (Pelletier et al., 2012). Interestingly, recent findings also suggest a role for COPI in inter-compartmental trafficking of RNA in neurons (Todd et al., 2013), raising the possibility that SCYL1 regulates the trafficking of ribonucleoprotein particles rather than (or in addition to) regulating membrane protein trafficking.
Recent findings also implicate SCYL1 in regulating the turnover of the RE-1 silencing transcription factor (REST)(Karlin et al., 2014). REST is a transcriptional repressor that is actively degraded by the proteasome during the transition from pluripotent to neural/progenitor cells. Its degradation, mediated by the ubiquitin-proteasome system, allows expression of genes that are critical to nervous system development. Early studies showed that aberrant expression of REST in the spinal cord of chicken embryos causes axonal pathfinding errors. Thus, the regulation of REST turnover by SCYL1 may significantly affect motor neuron pathfinding and survival. Studies to confirm these findings are under way.
To date, up to 7 isoforms of human SCYL1 have been identified, 2 of which have been characterized. These isoforms have divergent C-terminal segments resulting from the alternative splicing of exons at the 3′ end of the SCYL1 gene and are proposed to regulate various functions other than membrane protein and RNA trafficking. TEIF (telomerase transcriptional elements-interacting factor, SCYL1-isoform 7) was identified in a yeast one-hybrid screen for transcriptional regulators of the human telomerase reverse transcriptase. TEIF was shown to regulate the transcription of the human telomerase reverse transcriptase via its highly divergent C-terminal segment which exhibits homology to the transcription factor Jun-B (Tang et al., 2004). Another variant (SCYL1-isoform3) localizes to the centrosomes and regulates centrosome number and function (Kato et al., 2002). Whether these splice variants are found in mice and whether they contribute to the function of SCYL1 in motor neurons remains unknown.
Perspective
Although mutations in SCYL genes have not been associated with human diseases, it is likely that the downstream pathways affected by the loss of SCYL proteins in mice are also affected in human neurodegenerative diseases. Thus, identifying the pathways that are critical for the neuronal functions of SCYL proteins will not only further our understanding of the functions of SCYL proteins and disease pathophysiology but also provide insight into targets for drug development. One way to identify these pathways is by performing in vivo structure-function studies. The systematic mutagenesis of SCYL proteins to identify mutants that preferentially disrupt the interaction with components of one pathway and introduction of these mutations in mice will allow the identification of pathways that are critically involved in the neuronal functions of SCYL proteins. Protein truncation and site-directed mutagenesis studies have already provided insights into the molecular interactions between SCYL proteins and the various pathways possibly regulated by the pseudokinases (Figure 1) and recent advances in genetic engineering technologies (Pelletier et al., 2015) now provide fast and efficient ways to perform structure-function relationship studies in the context of native chromosomal environment.
Acknowledgments:
The author thanks Dr. Vani Shanker for editing the paper.
Footnotes
Funding: The author received support from the American Lebanese Syrian Associated Charities.
Conflicts of interest: The author declares no competing financial interests.
References
- Borner GH, Rana AA, Forster R, Harbour M, Smith JC, Robinson MS. CVAK104 is a novel regulator of clathrin-mediated SNARE sorting. Traffic. 2007;8:893–903. doi: 10.1111/j.1600-0854.2007.00576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burman JL, Hamlin JN, McPherson PS. Scyl1 regulates Golgi morphology. PLoS One. 2010;5:e9537. doi: 10.1371/journal.pone.0009537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burman JL, Bourbonniere L, Philie J, Stroh T, Dejgaard SY, Presley JF, McPherson PS. Scyl1, mutated in a recessive form of spinocerebellar neurodegeneration, regulates COPI-mediated retrograde traffic. J Biol Chem. 2008;283:22774–22786. doi: 10.1074/jbc.M801869200. [DOI] [PubMed] [Google Scholar]
- Chafe SC, Mangroo D. Scyl1 facilitates nuclear tRNA export in mammalian cells by acting at the nuclear pore complex. Mol Biol Cell. 2010;21:2483–2499. doi: 10.1091/mbc.E10-03-0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Plotkin AS, Lee VM, Trojanowski JQ. TAR DNA-binding protein 43 in neurodegenerative disease. Nat Rev Neurol. 2010;6:211–220. doi: 10.1038/nrneurol.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner SD, Schmid SL. CVAK104 is a novel poly-L-lysine-stimulated kinase that targets the beta2-subunit of AP2. J Biol Chem. 2005;280:21539–21544. doi: 10.1074/jbc.M502462200. [DOI] [PubMed] [Google Scholar]
- Duwel M, Ungewickell EJ. Clathrin-dependent association of CVAK104 with endosomes and the trans-Golgi network. Mol Biol Cell. 2006;17:4513–4525. doi: 10.1091/mbc.E06-05-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras S, Earls LR, Howell S, Smeyne RJ, Zakharenko SS, Pelletier S. SCYL2 protects CA3 pyramidal neurons from excitotoxicity during functional maturation of the mouse hippocampus. J Neurosci. 2015;35:10510–10522. doi: 10.1523/JNEUROSCI.2056-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamlin JN, Schroeder LK, Fotouhi M, Dokainish H, Ioannou MS, Girard M, Summerfeldt N, Melancon P, McPherson PS. Scyl1 scaffolds class II Arfs to specific subcomplexes of coatomer through the gamma-COP appendage domain. J Cell Sci. 2014;127:1454–1463. doi: 10.1242/jcs.136481. [DOI] [PubMed] [Google Scholar]
- Hara MR, Snyder SH. Cell signaling and neuronal death. Ann Rev Pharmacol Toxicol. 2007;47:117–141. doi: 10.1146/annurev.pharmtox.47.120505.105311. [DOI] [PubMed] [Google Scholar]
- Karlin KL, Mondal G, Hartman JK, Tyagi S, Kurley SJ, Bland CS, Hsu TY, Renwick A, Fang JE, Migliaccio I, Callaway C, Nair A, Dominguez-Vidana R, Nguyen DX, Osborne CK, Schiff R, Yu-Lee LY, Jung SY, Edwards DP, Hilsenbeck SG, et al. The oncogenic STP axis promotes triple-negative breast cancer via degradation of the REST tumor suppressor. Cell Rep. 2014;9:1318–1332. doi: 10.1016/j.celrep.2014.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Yano K, Morotomi-Yano K, Saito H, Miki Y. Identification and characterization of the human protein kinase-like gene NTKL: mitosis-specific centrosomal localization of an alternatively spliced isoform. Genomics. 2002;79:760–767. doi: 10.1006/geno.2002.6774. [DOI] [PubMed] [Google Scholar]
- Miyakawa K, Sawasaki T, Matsunaga S, Tokarev A, Quinn G, Kimura H, Nomaguchi M, Adachi A, Yamamoto N, Guatelli J, Ryo A. Interferon-induced SCYL2 limits release of HIV-1 by triggering PP2A-mediated dephosphorylation of the viral protein Vpu. Sci Signal. 2012;5:ra73. doi: 10.1126/scisignal.2003212. [DOI] [PubMed] [Google Scholar]
- Pelletier S, Gingras S, Green DR. Mouse genome engineering via CRISPR-Cas9 for study of immune function. Immunity. 2015;42:18–27. doi: 10.1016/j.immuni.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier S, Gingras S, Howell S, Vogel P, Ihle JN. An early onset progressive motor neuron disorder in Scyl1-deficient mice is associated with mislocalization of TDP-43. J Neurosci. 2012;32:16560–16573. doi: 10.1523/JNEUROSCI.1787-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan A, Uff CR, Isacke CM, Thorne RF. PACE-1, a novel protein that interacts with the C-terminal domain of ezrin. Exp Cell Res. 2003;284:224–238. doi: 10.1016/s0014-4827(02)00054-x. [DOI] [PubMed] [Google Scholar]
- Tang Z, Zhao Y, Mei F, Yang S, Li X, Lv J, Hou L, Zhang B. Molecular cloning and characterization of a human gene involved in transcriptional regulation of hTERT. Biochem Biophys Res Commun. 2004;324:1324–1332. doi: 10.1016/j.bbrc.2004.09.201. [DOI] [PubMed] [Google Scholar]
- Terabayashi T, Funato Y, Fukuda M, Miki H. A coated vesicle-associated kinase of 104 kDa (CVAK104) induces lysosomal degradation of frizzled 5 (Fzd5) J Biol Chem. 2009;284:26716–26724. doi: 10.1074/jbc.M109.039313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd AG, Lin H, Ebert AD, Liu Y, Androphy EJ. COPI transport complexes bind to specific RNAs in neuronal cells. Hum Mol Genet. 2013;22:729–736. doi: 10.1093/hmg/dds480. [DOI] [PMC free article] [PubMed] [Google Scholar]