

The development of ischemic brain damage is dramatically affected by the immune system, whose activation occurs immediately after the insult and may last for several days, involving a complex interplay between soluble and cellular mediators (Amantea et al., 2015). Accordingly, recent expression profiling studies have revealed that the majority of the genes modulated in the blood of stroke patients participate in the regulation of innate immune responses (Brooks et al., 2014). Moreover, in the clinical setting, serum levels of markers of acute inflammation correlate with the severity of ischemic brain damage and neurological deficit.
Both local and systemic inflammatory responses, involving soluble messengers and specialized cells activated in the brain or recruited from the periphery, exert a dualistic role on the development of ischemic cerebral injury. Brain resident microglia and blood-borne immune cells crucially contribute to the acute and chronic processes implicated in tissue injury, as well as to the regenerative and reparative mechanisms that limit the damage and provide tissue healing and recovery. In this context, an attractive approach to improve successful clinical translation of stroke therapeutics would consist in achieving a rational modulation of the immune system, by blocking its detrimental inflammatory responses while promoting its beneficial components. This perspective commentary will focus on the most recent findings regarding relevant targets and drugs for immunomodulation in stroke and their potential application in patients.
The reduction of cerebral blood flow caused by the ischemic insult prompts rapid neuronal death in the ischemic core regions and triggers the release of adenosine triphosphate (ATP) and danger associated molecular patterns (DAMPs) that stimulate purinergic and specific pattern recognition receptors (e.g., toll-like receptors), respectively, causing activation of astrocytes and microglia. Depending on the specific phenotype triggered by the environmental stimuli, microglia may be prompted to release inflammatory molecules, such as interleukin (IL)-1 and tumor necrosis factor (TNF), or to acquire an amoeboid morphology endowed with phagocytic activity that clears the damage and promotes repair. Moreover, activated microglia, damaged neurons and virtually all the other components of the neurovascular unit release toxic mediators, including cytokines, proteases and free radicals that prompt blood-brain barrier rupture and brain infiltration of circulating leukocytes. Thus, signals generated from the brain are implicated in the peripheral activation and in the cerebral recruitment of neutrophils, monocytes and lymphocytes that actively participate in the detrimental inflammatory processes that contribute to ischemic tissue damage, as well as in the beneficial and immunoregulatory mechanisms that provide tissue repair. In this context, cerebral ischemia is no longer considered a pathology that affects solely the brain, but represents a complex disease in which the neuro-immune crosstalk plays a crucial role. This underscores that, beyond neuronal mechanisms of toxicity, understanding the spatio-temporal evolution of the recruitment of distinct immune cells and their polarization towards specific subtypes is crucial to define their role in ischemic brain damage and to identify novel targets for an effective immunomodulation.
Notably, the majority of the genes acutely regulated in the blood of stroke patients are expressed in neutrophils and, to a lesser extent, in macrophages (Brooks et al., 2014). Neutrophils are the first blood-borne cells to be recruited in the ischemic brain and, although some studies have argued that they are unable to penetrate the brain parenchyma, their pivotal role in the pathogenesis of ischemic brain damage is clearly established. In patients, higher peripheral leukocyte and neutrophil counts, but not lymphocyte counts, are associated with larger infarct volumes, and brain accumulation of neutrophils correlates with poor neurological outcome and brain damage severity both in humans and in rodents. Indeed, neutrophils exert detrimental effects by microvessel obstruction/thrombosis and by releasing a plethora of cytotoxic molecules, including reactive oxygen species (ROS), reactive nitrogen species (particularly peroxynitrite) and proteases (such as matrix metalloproteinase-9, MMP-9) endowed with injurious effects, as demonstrated by the evidence that neutrophils depletion, as well as blockade of their trafficking, is neuroprotective in focal stroke. Nevertheless, neutrophils also possess the ability to polarize towards beneficial N2 phenotypes (Figure 1). Accordingly, in a recent study, Cuartero et al. (2013) have originally demonstrated that activation of the nuclear peroxisome proliferator-activated receptor (PPAR)-γ prompts neutrophil reprogramming toward the N2 phenotype in the setting of stroke (Cuartero et al., 2013). Thus, the PPAR-γ agonist rosiglitazone exerts neuroprotection and promotes resolution of inflammation by triggering N2-polarization and increased neutrophil clearance after experimental stroke induced by permanent middle cerebral artery occlusion (MCAo) in rodents (Cuartero et al., 2013). Although the exact mechanism by which N2 neutrophils rescue the ischemic brain is not clear yet, these findings suggest that the use of N2-polarizing drugs may be beneficial for stroke therapy.
Figure 1.
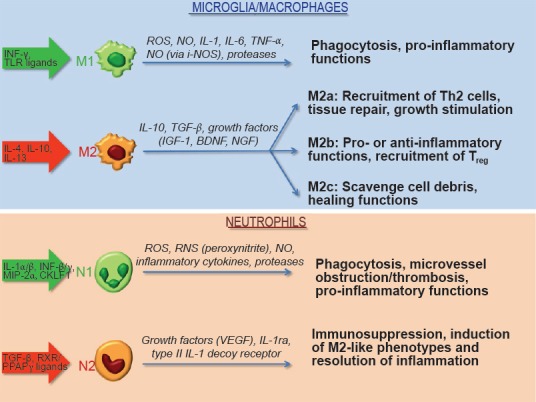
Polarization of innate immune cells and their function in stroke.
Various stimuli, including cytokines and certain receptor ligands, promote polarization of microglia/macrophages and neutrophils towards specific phenotypes. Based on their ability to release pro-inflammatory and detrimental mediators (M1 or N1 phenotypes) or immunomodulatory and pro-survival factors (M2 or N2 phenotypes), innate immune cells participate in the development of ischemic tissue damage or provide tissue healing and recovery especially during the late phases of the disease. BDNF: Brain-derived neurotrophic factor; CKLF-1: chemokine-like factor 1; IGF: insulin-like growth factor; IL: interleukin; IL-1ra: interleukin-1 receptor antagonist; INF: interferon; i-NOS: inducible nitric oxide synthase; MIP: macrophage inflammatory protein; NGF: nerve growth factor; NO: nitric oxide; PPAR: peroxisome proliferator-activated receptor; RNS: reactive nitrogen species; ROS: reactive oxygen species; RXR: retinoid X receptors; TGF: transforming growth factor; TLR: toll-like receptors; TNF: tumor necrosis factor; VEGF: vascular endothelial growth factor.
At variance with the studies focussing on neutrophils, polarization of microglia/macrophages has been more extensively investigated (Figure 1). Their switch towards a multitude of phenotypes is strongly dependent on specific microenvironmental signals related to the spatio-temporal progression of ischemic brain damage (Fumagalli et al., 2015). Early after an ischemic insult, local microglia and newly recruited macrophages assume the M2 “beneficial” phenotype, to then develop into a pro-inflammatory M1 phenotype upon priming by ischemic neurons. Microglial cells then shift from a ramified to an ameboid macrophage-like shape, associated to phagocytosis and to the release of inflammatory mediators (Amantea et al., 2010). At late differentiation state, microglia assumes a phenotype indistinguishable from blood-borne macrophages, bearing similar antigenic and morphological features (Fumagalli et al., 2015). Although phagocytosis clears cell debris and contributes to the resolution of inflammation, microglia have also been shown to engulf salvageable neurons thus increasing brain damage.
Likewise microglia, macrophages infiltrating the ischemic brain may exert a dualistic role on the development of tissue damage depending on their polarization status (Figure 1). The M1 phenotype initiates and sustains inflammation by releasing neurotoxic factors (i.e., TNF-α, IL-1β, monocyte chemoattractant protein (MCP)-1, macrophage inflammatory protein (MIP)-1α, and IL-6) and ROS that underlie macrophage/microglia-mediated neurotoxicity after stroke; whereas, M2-polarized cells are involved in beneficial responses by clearing debris and by promoting angiogenesis, tissue remodeling and repair.
The dualistic role exerted by the cellular mediators of the innate immune reaction may explain why most anti-inflammatory approaches, conceived disregarding the potential beneficial function of the target, have failed to reach the clinical setting (Amantea et al., 2015). In this context, an attractive opportunity to develop novel effective stroke therapeutics consists in reducing the detrimental responses, while promoting the endogenous neuroprotective reactions of the innate immune system. Recently, this concept has been validated in the preclinical setting by studies demonstrating the neuroprotective effects of shifting the polarization of immune cells towards non-inflammatory phenotypes in stroke animal models.
Endogenous production of the M2-polarizing cytokine IL-4, triggered by MCAo in mice, promotes Th2 polarization and, thus, beneficial effects on stroke outcome (Xiong et al., 2011). Moreover, Gliem et al. (2012) have reported that a subpopulation of bone marrow-derived monocytes/macrophages, recruited via CCR2 and acting through transforming growth factor (TGF)-β1, preserves the integrity of the neurovascular unit in murine stroke models. A decreased expression of M1 markers, together with a partial preservation of the ischemia-induced expression of M2 markers has been suggested to underlie the amelioration of stroke outcome observed in myeloid-specific mineralcorticoid receptor knockout mice (Frieler et al., 2011). Similarly, deficiency of the fractalkine receptor CX3CR1 triggers a protective inflammatory milieu, characterized by the elevation of markers of M2 polarization (Fumagalli et al., 2015). Despite this little evidence, the molecular processes that regulate macrophage polarization in stroke have not been completely clarified, and further studies are needed to understand whether the acquisition of a specific phenotype stems from recruitment of circulating precursors or in situ cell re-instruction.
Moreover, to date, only few experimental studies have evaluated the therapeutic benefits of drugs acting by increasing the M2/M1 ratio in stroke. Among these, minocycline has been reported to promote neurovascular remodeling during stroke recovery by facilitating alternative activation of microglia/macrophages towards a non-inflammatory protective phenotype (Yang et al., 2015). By following the concept of drug repurposing, we have recently demonstrated that acute treatment with the macrolide antibiotic azithromycin attenuates blood-brain barrier damage and cerebral ischemic damage in rodents subjected to MCAo, with a significant amelioration of neurological deficits up to 7 days after the insult (Amantea et al., 2016). Up-regulation of M2 markers has also been shown to underlie neuroprotection by Exendin-4, a glucagon-like receptor 1 agonist clinically used against type 2 diabetes in young healthy and in aged diabetic/obese mice subjected to middle cerebral artery occlusion (Darsalia et al., 2014). Interestingly, another drug widely used for the treatment of type 2 diabetes, metformin, has shown promising results in stroke animal models based on its immunomodulatory properties. Metformin is well-recognized as an activator of adenosine 5’-monophosphate-activated protein kinase (AMPK). In mice subjected to MCAo, chronic metformin treatment promotes functional recovery and tissue repair via AMPK-dependent skewing of microglia/macrophages toward an M2 phenotype (Jin et al., 2014).
Thus, the preclinical findings highlighting the neuroprotective potential of M2- or N2-polarizing agents in stroke are increasing, although further studies are needed to better investigate the molecular targets that mediate immune cell shift towards beneficial phenotypes. In order to add significance to such findings, the relevance of M1-to-M2 or N1-to-N2 polarization for stroke outcome should also be validated in the clinical setting. In fact, although ischemic stroke is a leading cause of mortality and long-term disability worldwide, the only treatments available to date consist in blood flow restoration by lysis or removal of the thrombus. This, together with the fact that these procedures can be applied to less that 10% of patients due to their narrow therapeutic window, highlights the urgent need of more effective and safe stroke therapeutics. In this context, it is interesting to note that the majority of the studies discussed herein, concerning effective immunomodulatory compounds in stroke models, are based on repurposing existing drugs characterised by a well established safety profile in human. This drug discovery approach allows to dramatically reduce the risk of clinical failure due to undesired side effects or unsuccessful validation of the target, and is therefore a promising strategy to implement the clinical translation of immunomodulatory therapies for stroke.
The author thanks all the collaborators who contributed to the research papers upon which the present commentary is based. Due to space limitations, the author regrets the omission of many important studies and their corresponding references.
References
- Amantea D, Bagetta G, Tassorelli C, Mercuri NB, Corasaniti MT. Identification of distinct cellular pools of interleukin-1beta during the evolution of the neuroinflammatory response induced by transient middle cerebral artery occlusion in the brain of rat. Brain Res. 2010;1313:259–269. doi: 10.1016/j.brainres.2009.12.017. [DOI] [PubMed] [Google Scholar]
- Amantea D, Micieli G, Tassorelli C, Cuartero MI, Ballesteros I, Certo M, Moro MA, Lizasoain I, Bagetta G. Rational modulation of the innate immune system for neuroprotection in ischemic stroke. Front Neurosci. 2015;9:147. doi: 10.3389/fnins.2015.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amantea D, Certo M, Petrelli F, Tassorelli C, Micieli G, Corasaniti MT, Puccetti P, Fallarino F, Bagetta G. Azithromycin protects mice against ischemic stroke injury by promoting macrophage transition towards M2 phenotype. Exp Neurol. 2016;1:116–125. doi: 10.1016/j.expneurol.2015.10.012. [DOI] [PubMed] [Google Scholar]
- Brooks SD, Van Gilder R, Frisbee JC, Barr TL. Genomics for the advancement of clinical translation in stroke. In: Micieli G, Amantea D, editors. Rational Basis for Clinical Translation in Stroke Therapy. Boca Raton, FL: CRC Press; 2014. pp. 123–136. [Google Scholar]
- Cuartero MI, Ballesteros I, Moraga A, Nombela F, Vivancos J, Hamilton JA, Corbí ÁL, Lizasoain I, Moro MA. N2 neutrophils, novel players in brain inflammation after stroke: modulation by the PPARγ agonist, rosiglitazone. Stroke. 2014;44:3498–3508. doi: 10.1161/STROKEAHA.113.002470. [DOI] [PubMed] [Google Scholar]
- Darsalia V, Hua S, Larsson M, Mallard C, Nathanson D, Nyström T, Sjöholm Å, Johansson ME, Patrone C. Exendin-4 reduces ischemic brain injury in normal and aged type 2 diabetic mice and promotes microglial M2 polarization. PLoS One. 2014;9:e103114. doi: 10.1371/journal.pone.0103114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieler RA, Meng H, Duan SZ, Berger S, Schütz G, He Y, Xi G, Wang MM, Mortensen RM. Myeloid-specific deletion of the mineralocorticoid receptor reduces infarct volume and alters inflammation during cerebral ischemia. Stroke. 2011;42:179–185. doi: 10.1161/STROKEAHA.110.598441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli S, Perego C, Pischiutta F, Zanier ER, De Simoni MG. The ischemic environment drives microglia and macrophage function. Front Neurol. 2015;6:81. doi: 10.3389/fneur.2015.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gliem M, Mausberg AK, Lee JI, Simiantonakis I, van Rooijen N, Hartung HP, Jander S. Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann Neurol. 2012;71:743–752. doi: 10.1002/ana.23529. [DOI] [PubMed] [Google Scholar]
- Jin Q, Cheng J, Liu Y, Wu J, Wang X, Wei S, Zhou X, Qin Z, Jia J, Zhen X. Improvement of functional recovery by chronic metformin treatment is associated with enhanced alternative activation of microglia/macrophages and increased angiogenesis and neurogenesis following experimental stroke. Brain Behav Immun. 2014;40:131–142. doi: 10.1016/j.bbi.2014.03.003. [DOI] [PubMed] [Google Scholar]
- Xiong X, Barreto GE, Xu L, Ouyang YB, Xie X, Giffard RG. Increased brain injury and worsened neurological outcome in interleukin-4 knockout mice after transient focal cerebral ischemia. Stroke. 2011;42:2026–2032. doi: 10.1161/STROKEAHA.110.593772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Salayandia VM, Thompson JF, Yang LY, Estrada EY, Yang Y. Attenuation of acute stroke injury in rat brain by minocycline promotes blood-brain barrier remodeling and alternative microglia/macrophage activation during recovery. J Neuroinflammation. 2015;12:26. doi: 10.1186/s12974-015-0245-4. [DOI] [PMC free article] [PubMed] [Google Scholar]