

Abstract
Objective
Gaucher disease (GD) is a lysosomal storage disease characterized by a deficiency of glucocerebrosidase. Although enzyme‐replacement and substrate‐reduction therapies are available, their efficacies in treating the neurological manifestations of GD are negligible. Pharmacological chaperone therapy is hypothesized to offer a new strategy for treating the neurological manifestations of this disease. Specifically, ambroxol, a commonly used expectorant, has been proposed as a candidate pharmacological chaperone. The purpose of this study was to evaluate the safety, tolerability, and neurological efficacy of ambroxol in patients with neuronopathic GD.
Methods
This open‐label pilot study included five patients who received high‐dose oral ambroxol in combination with enzyme replacement therapy. Safety was assessed by adverse event query, physical examination, electrocardiography, laboratory studies, and drug concentration. Biochemical efficacy was assessed through evidence of glucocerebrosidase activity in the lymphocytes and glucosylsphingosine levels in the cerebrospinal fluid. Neurological efficacy was evaluated using the Unified Myoclonus Rating Scale, Gross Motor Function Measure, Functional Independence Measure, seizure frequency, pupillary light reflex, horizontal saccadic latency, and electrophysiologic studies.
Results
High‐dose oral ambroxol had good safety and tolerability, significantly increased lymphocyte glucocerebrosidase activity, permeated the blood–brain barrier, and decreased glucosylsphingosine levels in the cerebrospinal fluid. Myoclonus, seizures, and pupillary light reflex dysfunction markedly improved in all patients. Relief from myoclonus led to impressive recovery of gross motor function in two patients, allowing them to walk again.
Interpretation
Pharmacological chaperone therapy with high‐dose oral ambroxol shows promise in treating neuronopathic GD, necessitating further clinical trials.
Introduction
Gaucher disease (GD) is an autosomal recessive lysosomal storage disease with glucocerebrosidase (GCase, EC3.2.1.45) deficiency caused by its gene (GBA1) mutations, which leads to prominent accumulation of glucosylceramide (GlcCer) and its deacylated form, glucosylsphingosine (GlcSph). GlcSph accumulates in the brain and is assumed to be responsible for the neurological manifestations of GD.1, 2, 3 GD has traditionally been classified into three subtypes based on the absence or presence of primary central nervous system (CNS) involvement.4 GD type 1 (GD1) is classified as a nonneuronopathic form and comprises approximately 95% of GD cases in Western countries, such as the United States, Europe, Israel, and other places where Ashkenazi Jewish population is present.5, 6 GD type 2 (GD2) and type 3 (GD3) are the acute and chronic neuronopathic forms, respectively, and are collectively known as neuronopathic GD (nGD). GD2 is characterized by severe and progressive CNS involvement, and patients usually die before reaching 2 years of age. Compared with GD2, GD3 is considerably more heterogeneous. The onset of neurological manifestations occurs later, and the disease usually progresses more slowly. In Asian countries, nGD comprises approximately 60% of GD cases. The difference in nGD prevalence between Asia and Western countries is believed to result from a dissimilar GBA1 mutation distribution.7, 8
To date, enzyme replacement therapy (ERT) and substrate reduction therapy (SRT) have been approved for the treatment of GD. These therapies ameliorate visceral, hematologic, and skeletal abnormalities. Nevertheless, their efficacy for neurological manifestations is mostly negligible.9, 10 Pharmacological chaperone therapy (PCT) is a new approach for GD. In GD, the mutant enzyme protein fails to fold correctly (i.e., it misfolds), which results in the acceleration of endoplasmic reticulum (ER)‐associated degradation, even if the functional potential is maintained. Pharmacological chaperones (PCs) selectively bind to the misfolded GCase in the ER, facilitating the correct folding of the protein and inducing functional recovery. Small‐molecule PCs can cross the blood–brain barrier; therefore, PCT has emerged as a promising therapy for nGD. Pioneering studies on PCT were first reported in Fabry disease,11, 12 and the effectiveness in CNS involvement was confirmed in GM1‐gangliosidosis.13 .
Several groups, including ours, have reported that some compounds are useful as PCs in vivo.14, 15, 16, 17, 18 Ambroxol, a commercially available expectorant, was identified as a PC candidate for GD19 and was shown to enhance endogenous GCase activity in the murine CNS.20 Therefore, we aimed to analyze the potential efficacy of ambroxol in patients with nGD. Here, we report that PCT with high‐dose oral ambroxol is well tolerated and effective for the treatment of neurological manifestations, particularly for the disabling myoclonus and pupillary light reflex (PLR) dysfunction, in five patients with nGD.
Methods
Study design and patients
This was a multicenter, open‐label pilot study conducted to investigate the safety, tolerability, and efficacy of high‐dose ambroxol in patients with nGD. Patient inclusion criteria were as follows: (1) having a diagnosis that was confirmed by measuring GCase activity and GBA1 mutation analysis; (2) having a known GBA1 mutation on which the chaperone effects of ambroxol could be detected (i.e., F213I, N188S, G193W, R120W, or G202R);20, 21 or (3) exhibiting significant chaperone effects confirmed by an in vitro test with patient‐derived cultured skin fibroblasts. Exclusion criteria were as follows: (1) use of any other investigational treatment for GD within 3 months prior to the initiation of the study; and (2) patients with serious hepatic, renal, or cardiovascular disorders. Treatment plans were approved by institutional review boards of the Tottori University School of Medicine, Shiga Medical Center for Children, and Kurume University School of Medicine. All patients or their legal representatives provided written informed consent.
Treatment
Ambroxol (Mucosolvan; Teijin Pharma, Tokyo, Japan) was commercially purchased as 15 mg ambroxol hydrochloride tablets. Oral ambroxol was administered at the target dose (25 mg/kg/day or a maximum dose of 1300 mg/day) divided into three equal doses. Ambroxol suspension was administered through feeding tubes when swallowing was impaired. Details regarding the dosing regimen for each patient are shown in Figure 1 and Data S1.
Figure 1.
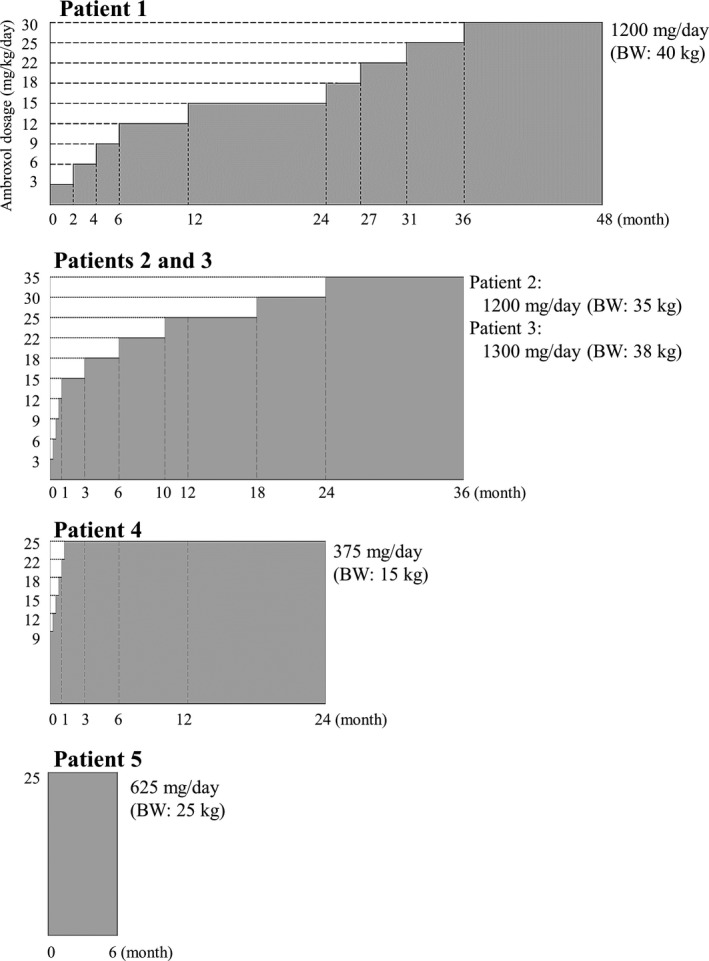
Overview of the dosing regimen for each patient. Ambroxol administration was initiated with a starting dose of 3 mg/kg/day divided into three equal doses in the first three patients (patients 1, 2, and 3), because safety information in humans about long‐term high‐dose ambroxol administration was limited. The dose was subsequently increased in increments of 3 mg/kg to reach the target doses (25 mg/kg/day or a maximum dose of 1300 mg/day) over several months to years. For the remaining two patients, ambroxol was initiated at 9 mg/kg/day in patient 4 or 25 mg/kg/day in patient 5. ERT and concomitant medications were continued during the study.
Assessments
Because PCs, including ambroxol, are not always effective on all mutations, we first investigated the chaperone activity of ambroxol in patient‐derived cultured skin fibroblasts using a method modified from earlier research,21 as described in Data S1. After the in vitro screening, patients were assessed at baseline and scheduled follow‐up. Assessments included safety, biochemical efficacy, and neurological efficacy.
Safety assessments, which were the primary outcome measures, included data on adverse event queries, physical examination, electrocardiography, laboratory studies, and ambroxol concentrations. The ambroxol trough serum concentration was measured just before the next dose, and the peak serum concentration was measured 2.5 ± 0.5 h after the medications were given. Lumbar puncture was performed immediately after peak blood samples were collected to assess the rate of penetration of ambroxol into the cerebrospinal fluid (CSF). Serum and CSF ambroxol concentrations were quantified using liquid chromatography‐tandem mass spectrometry (LC/MS/MS), as previously described.22, 23 Biochemical efficacy was assessed through mean percentage changes from baseline of GCase activity in lymphocytes. Additionally, CSF GlcSph levels were determined by the LC/MS/MS method (methods see Data S1). Because of the phenotypic heterogeneity of nGD, the assessment of neurological efficacy was dependent on a combination of tests as follows: the Unified Myoclonus Rating Scale (UMRS);24 the Gross Motor Function Measure (GMFM);25 the Functional Independence Measure (FIM);26 seizure frequency (per 28 days); neuro‐ophthalmological testing (i.e., PLR and horizontal saccadic latency); and electrophysiological studies (somatosensory‐evoked potential [SEP] and visual‐evoked potential [VEP]). UMRS is a statistically validated, quantitative clinical rating instrument designed to evaluate the response of patients undergoing antimyoclonic therapies. Changes in the myoclonus scores at rest were evaluated in all patients. Changes in the myoclonus scores with action, functional tests, and stimulus sensitivity were evaluated in patients who could follow the instructions. PLR was assessed in all patients using an infrared pupillometer, as reported in an earlier study.27 In this study, we selected a 1‐s red stimulus of 270 cd/m2 and evaluated the mean values of the initial constriction rate and latency. In testable patients, horizontal saccadic latency was measured, using a DC‐electrooculogram (EOG) technique (methods see Data S1).
Statistical analysis
Paired comparisons of all parameters for safety and efficacy assessments were calculated using standard descriptive statistical measures (i.e., mean and 95% confidence interval [CI]) and the Wilcoxon signed‐rank test. The Mann–Whitney U‐test was used for the following comparisons: effects of ambroxol on in vitro chaperone tests, and baseline horizontal saccadic latency in control subjects versus patients with nGD. Statistical analysis was performed using GraphPad Prism version 6.0 for Windows (GraphPad Software, San Diego, CA, USA).
Results
Patient characteristics
Five patients with nGD with positive in vitro chaperone tests (Fig. 2) were recruited from multicenter physician referrals. Clinical characteristics at baseline are shown in Table 1. Patient 1 was bedridden with persistent facial myoclonus. Generalized myoclonus was easily induced by touch or postural change (i.e., stimulus‐sensitive), which made it difficult for caregivers to change diapers and turn over the patient to prevent bedsores. Myoclonus evolved into a characteristic pattern of status epilepticus, characterized by prolonged increasing muscle tone with intense rhythmic myoclonus of limbs. These seizures frequently occurred and were labeled as “myoclonic‐generalized status epilepticus (MGSE)”28 in this study. Patients 2 and 3 had severe myoclonus of the limbs. Myoclonus was easily increased by voluntary movement, and patients were no longer able to perform activities of daily living by themselves. Additionally, patient 2 had myoclonic seizures of the trunk every day, which forced her to be confined to a wheelchair. Patient 3 had generalized tonic‐clonic seizures (GTCs) almost every day. Patient 4 was bedridden with generalized relentless myoclonus. Patient 5 was diagnosed with GD3 at the age of 3 years and received allogeneic bone marrow transplantation (BMT) at age 4. BMT was successful and GCase activity in lymphocytes returned to the normal range; however, neurological manifestations appeared 10 years after BMT, and she had eventually become bedridden at age 18. Progression of myoclonus and axial dystonia was observed, and antiepileptic treatment was ineffective.
Figure 2.
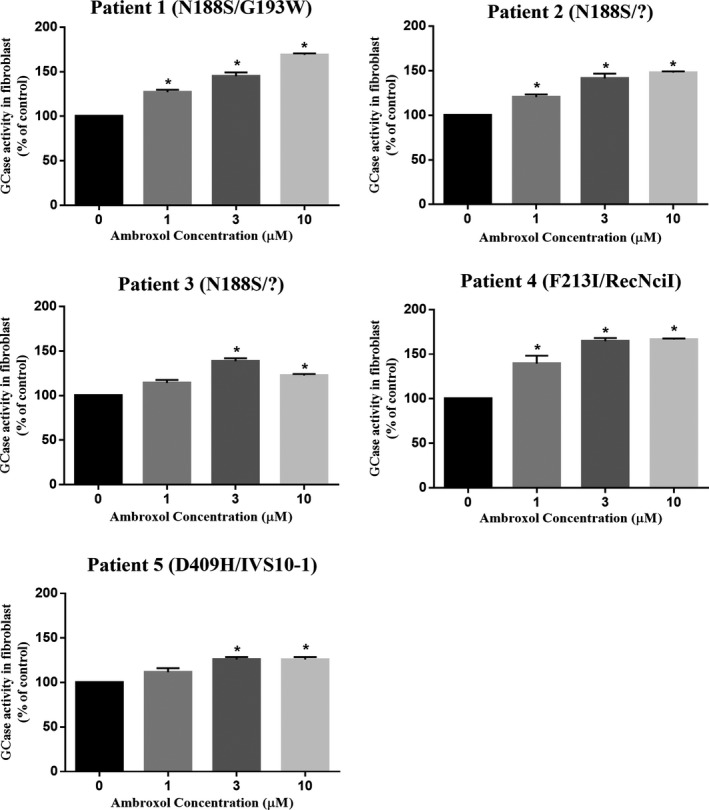
Chaperone effect of ambroxol on mutant GCase activities in GD fibroblasts. Primary skin fibroblasts derived from each patient were incubated with the indicated concentrations of ambroxol for 4 days. In all panels, the data were expressed as the relative increase in GCase activity in the presence of ambroxol compared with that in untreated cells. The results represent the mean ± SEM of three independent experiments. Statistically significant differences between the treated and untreated fibroblasts were elicited, using the Mann–Whitney U‐test. *P < 0.05.
Table 1.
Overview of clinical characteristics of patients at initiation of treatment
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | |
|---|---|---|---|---|---|
| Age (sex) | 28 years (female) | 20 years (female) | 15 years (female) | 3 years (female) | 25 years (female) |
| Genotype | N188S/G193W | N188S/? | N188S/? | F213I/RecNciI | D409H/IVS10‐1 |
| Phenotype | GD3 | GD3 | GD3 | GD2 | GD3 |
| Age at diagnosis | 20 years | 19 years | 14 years | 11 months | 3 years |
| First neurological symptom (age) | PME (12 years) | PME (7 years) | PME (8 years) | HSIF, head thrusting (3 months) | Apneic spells (6 months) |
| Presenting symptoms |
Bedridden (since 20 years) Tube feeding (since 20 years) Tracheostomy (since 27 years) |
Unable to sit without support Use of wheelchair (since 17 years) Total assistance |
Barely standing with support for a short time only Use of wheelchair (since 14 years) Total assistance |
Bedridden Tube feeding (since 13 months) Tracheostomy and ventilation (since 20 months) |
Bedridden (since 18 years) Tube feeding (since 25 years) Tracheostomy (since 25 years) Generalized dystonia |
| Communication | Unable to utter words | Communicates well | Communicates well | Unable to utter words | Communicates well |
| Wechsler Scale | NA | VIQ: 59, PIQ: NA (due to myoclonus) | VIQ: 79, PIQ: 62 (poor study due to myoclonus) | NA | VIQ: 55, PIQ: NA (due to myoclonus and dystonia) |
| Myoclonus | Myoclonus of the face(at rest) | Myoclonus of the limbs (at rest/with action) | Myoclonus of the trunk and limbs (at rest/with action) | Generalized myoclonus (at rest) | Myoclonus of the limbs and face (at rest) |
| Seizures (frequency) | MGSE (daily, uncountable) | Myoclonic seizures with falling (daily, uncountable)GTCs (4.8 days/28 days) | GTCs (26 days/28 days) | Tonic (daily, uncountable)GCSE (3 times/year) | GTCs (12 days/28 days) |
| Oculomotor abnormalities | Gaze palsy in all directions | HSIF | HSIF | Gaze palsy in all directions | HSIF and VGP |
| SEP at baseline | Disappeared cortical waves (N18, N20, N30) | Giant SEP | Giant SEP | Prolonged latency (N18, N20) | Normal |
| VEP at baseline | Giant VEP | normal | normal | normal | Giant VEP |
| Prior treatments |
ERT (for 8 years) AED (VPA, PB, NZP, TPM, CBZ, ZNS, VGB, CZP) Piracetam Baclofen |
ERT (for 4 months) AED (VPA, CBZ, ZNS, TPM, LEV) Piracetam |
ERT (for 4 months) AED (VPA, CZP, TPM, LEV) Piracetam |
ERT (for 2 years) AED (VPA, PB, TPM,CBZ,LEV, potassium bromide) Piracetam, Tizanidine Dantrolene |
BMT (at 4 years) AED (CBZ, CZP, DZP) Dantrolene |
AED, antiepileptic drug; BMT, bone marrow transplantation; CBZ, carbamazepine; CZP, clonazepam; DZP, diazepam; ERT, enzyme replacement therapy; GTCs, generalized tonic‐clonic seizures; GCSE, generalized convulsive status epilepticus; HSIF, horizontal saccadic initiation failure; LEV, levetiracetam; MGSE, myoclonic‐generalized status epilepticus; NA, not available; NZP, nitrazepam; PB, phenobarbital; PIQ, performance IQ; PME, progressive myoclonus epilepsy; SEP, somatosensory evoked potential; TPM, topiramate; VEP, visual evoked potential; VGB, vigabatrin; VGP, vertical gaze palsy; VIQ, verbal IQ; VPA, sodium valproate; ZNS, zonisamide.
Safety and biochemical efficacy
Patient 1 developed self‐limited skin rashes in months 3 and 4. Two patients exhibited hypouricemia as previously described;29 however, high‐dose ambroxol had no influence on cardiac QTc intervals or other laboratory data (Table 2). The mean serum trough concentration of ambroxol was 1.4 μmol/L, mean serum peak concentration was 2.2 μmol/L, mean CSF peak concentration was 0.4 μmol/L, and mean penetration rate into the CSF was 15.6% (Table 3). The CSF trough concentration was calculated from each patient's penetration rate; the mean CSF trough concentration was 0.24 μmol/L (range, 0.06–0.62 μmol/L). GCase activity in the lymphocytes increased by 171.1% (95% CI, 61.9–280.3; P = 0.03) and reached levels observed in either carriers or control subjects. CSF GlcSph levels were below the lower limit of quantification (10.0 pg/mL) in all control subjects. In contrast, CSF GlcSph levels in patients with nGD were elevated at baseline and fell by 25.7% (95% CI, 8.0–43.4, P = 0.03) after therapy.
Table 2.
Systemic disease parameters and safety variables
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | |
|---|---|---|---|---|---|
| Hemoglobin, g/dL (RV: 11.0–15.0) | |||||
| Baseline | 13.8 | 13.1 | 13.4 | 12.0 | 12.8 |
| Posttreatment | 13.2 | 12.5 | 13.9 | 13.2 | 13.6 |
| Platelets, ×109/L (RV: 12.5–34.3) | |||||
| Baseline | 18.5 | 11.5 | 11.2 | 19.0 | 12.2 |
| Posttreatment | 17.0 | 7.1 | 9.4 | 18.4 | 18.8 |
| Angiotensin‐converting enzyme (ACE), U/L (RV: 8.3–21.4) | |||||
| Baseline | 8.2 | 9.8 | 13.1 | 46.4 | 8.7 |
| Posttreatment | 7.8 | 7.0 | 8.8 | 19.2 | 8.5 |
| Uric acid, mg/dL (RV: 2.3–7.0) | |||||
| Baseline | 2.6 | 5.1 | 5.2 | 4.7 | 2.0 |
| Posttreatment | 0.7 | 2.2 | 2.4 | 3.9 | 3.7 |
| QTc interval (Fridericia), ms | |||||
| Baseline | 370 | 402 | 404 | 421 | 413 |
| Posttreatment (peak) | 401 | 387 | 394 | 402 | 435 |
RV, reference values. The effects of treatment were analyzed at the following times: patient 1, month 48; patients 2 and 3, month 36; patient 4, month 24; and patient 5, month 6.
Table 3.
Pharmacokinetics and biochemical efficacy of ambroxol at 25 mg/kg/day
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Concentration of ambroxol, μmol/L | Trough | Peak | Trough | Peak | Trough | Peak | Trough | Peak | Trough | Peak |
| Serum | 0.99 | 1.75 | 2.00 | 2.96 | 3.17 | 4.24 | 0.41a | 0.70a | 0.51 | 1.32 |
| CSF | 0.14b | 0.25 | 0.34b | 0.50 | 0.62b | 0.83 | 0.06b | 0.11 | 0.06b | 0.15 |
| Penetration rate (peak CSF/serum) (%)c | 14.4 | 16.9 | 19.6 | 15.7 | 11.4 | |||||
| GCase activity in lymphocytes, nmol/mg protein/h (% of control) | ||||||||||
| Baseline | Post‐Tx | Baseline | Post‐Tx | Baseline | Post‐Tx | Baseline | Post‐Tx | Baseline | Post‐Tx | |
| 3.2 (13.7) | 10.1 (43.0) | 5.8 (24.7) | 12.5 (53.2) | 7.1 (30.1) | 24.7 (105.0) | 4.3 (18.1) | 14.2 (60.3) | 23.6d (100.6) | 34.0 (145.0) | |
| Control (n = 68) 23.5 ± 5.3 | ||||||||||
| CSF GlcSph levels, pg/mL | Baseline | Post‐Tx | Baseline | Post‐Tx | Baseline | Post‐Tx | Baseline | Post‐Tx | Baseline | Post‐Tx |
| 18.2 | 16.1 | 26.6 | 15.0 | 49.1 | 30.4 | 635 | 533a | 146 | 118 | |
| Control (n = 37) <10.0 | ||||||||||
The effects of treatment were analyzed at the following times: patient 1, month 36; patients 2 and 3, month 12; patient 4, month 24; and patient 5, month 6.
The timing of assessment was month 12.
The CSF trough concentration of ambroxol was calculated from each patient's penetration rate.
The penetration rate of ambroxol was calculated as the peak CSF value/peak serum value × 100 (%).
Enzyme activity after bone marrow transplantation (see text).
Neurological efficacy
Myoclonus
All patients showed clinically marked improvement in myoclonus and decreased UMRS scores (Fig. 3). In patient 1, facial myoclonus began to improve at 9 mg/kg/day, and the patient exhibited a serene look. Progressive improvements in myoclonus at rest and stimulus sensitivity were observed with increasing doses of ambroxol. The decrease in stimulus‐induced myoclonus made it much easier for caregivers to care for the patient. In patients 2 and 3, myoclonus started to improve after increasing the dose of ambroxol to 9 and 12 mg/kg/day, respectively. As action myoclonus decreased, the patients could stand steadily, balance themselves, and walk again. Also, both patients were able to go up and down the stairs and transfer to a chair with some assistance. These improvements were maintained during 36 months of ambroxol therapy (Videos S1, S2). Improvements in fine motor skills were also observed. Both patients were able to eat, operate a smartphone, pull down or up pants by themselves. The increases in GMFM and FIM scores were consistent with these changes (Figs. 4, 5). In patient 4, generalized myoclonus markedly decreased at month 3, and the patient was able to smile again when she was gently stroked (Video S3). In patient 5, myoclonus of the limbs and face promptly improved, and UMRS scores at rest reached zero at month 6. The patient is now able to operate a smartphone and enjoy seeing photos. On the other hand, no improvement in symptoms of dystonia was observed.
Figure 3.
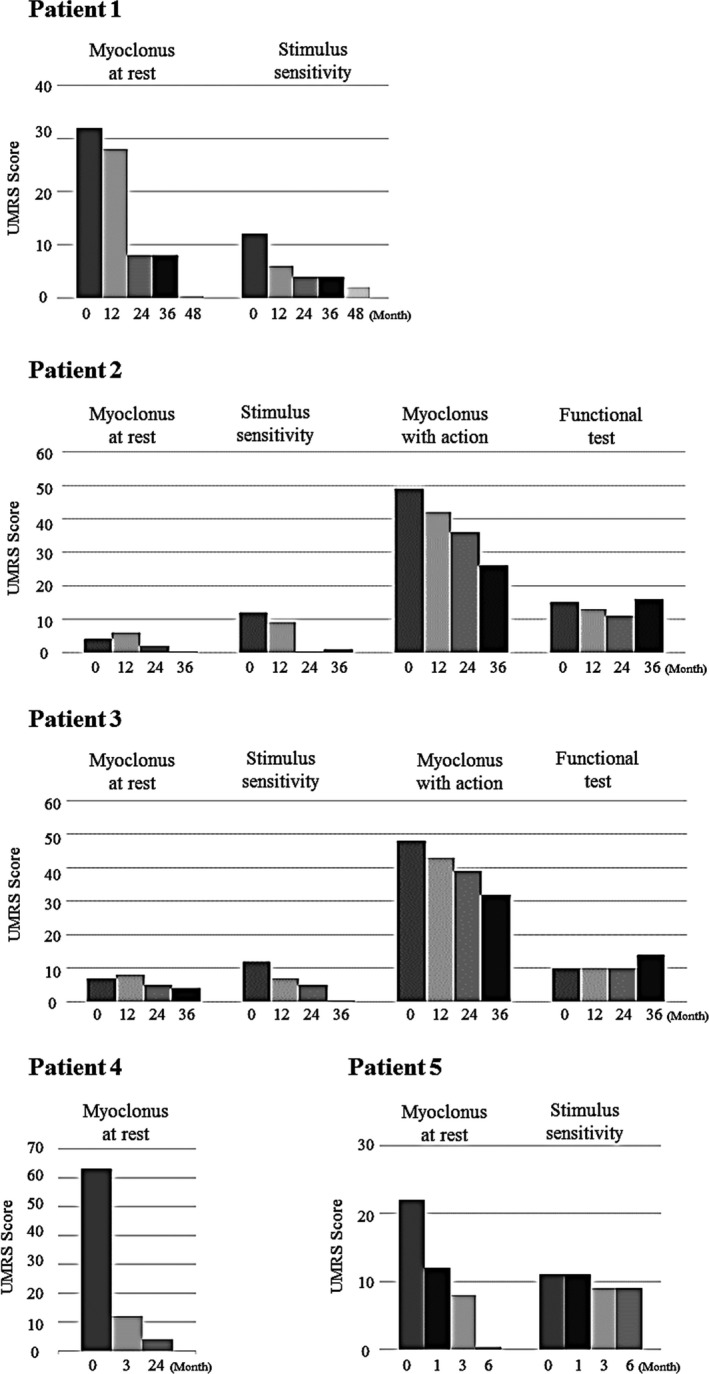
Effects of ambroxol treatment on myoclonus. The Unified Myoclonus Rating Scale (UMRS) was used to evaluate the response of myoclonus to ambroxol therapy in patients with nGD. We evaluated myoclonus at rest in all patients and myoclonus with action, functional tests, and stimulus sensitivity in testable patients. High scores on UMRS reflect a severe condition.
Figure 4.
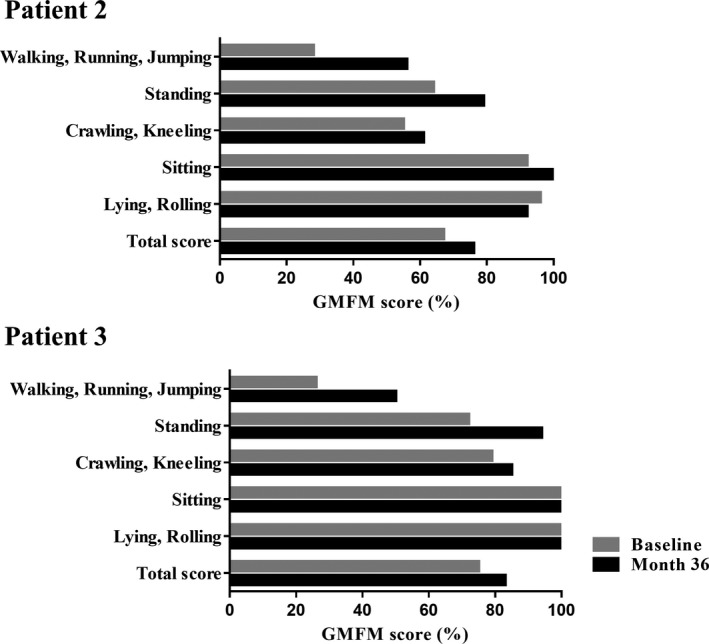
Effects of ambroxol treatment on gross motor function. The graph shows changes in the Gross Motor Function Measure (GMFM) scores after ambroxol treatment for patients 2 and 3, who could be sufficiently examined. The GMFM score of 100% means that the patient's gross motor functions are equivalent to those of a typical 5‐year‐old individual.
Figure 5.
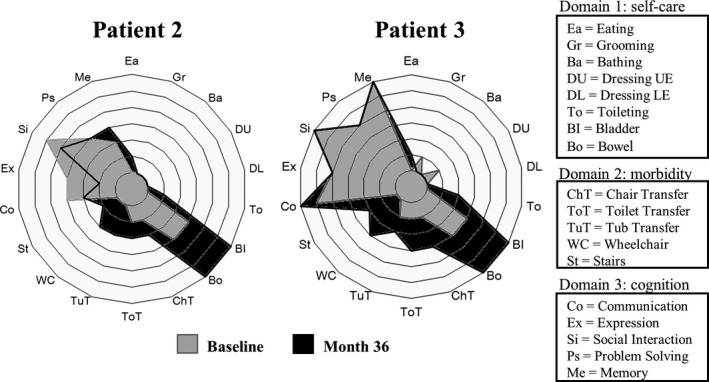
Effects of ambroxol treatment on functional status. The polar graph shows changes in the Functional Independence Measure (FIM) ratings after ambroxol treatment for each individual item. The background rings represent scores of 1–7 with an inside‐out order. All items were graded on a 1‐ to 7‐point scale (score 1: total assistance, score 7: complete independence) and the expanding rings indicate the FIM rating.
Seizures
In patient 1, the baseline MGSE frequency per 28 days was 28. These frequencies decreased to 3.7 per 28 days after stimulus‐induced myoclonus decreased following an increase in ambroxol to 15 mg/kg/day. In patient 2, the frequency of myoclonic seizures of the trunk per 28 days decreased from 28 to 5.8 at month 1 (9 mg/kg/day). Falls, which resulted from myoclonic seizures of the trunk, decreased, and she was no longer confined to a wheelchair. However, the frequency of GTCs did not change. In patient 3, the frequency of GTCs did not change, but the duration of seizures decreased from 15 to 20 min/single event at baseline to 1–2 min at month 2 (15 mg/kg/day) and remained stable during follow‐up. In patient 4, the frequency of tonic seizures did not change; however, the frequency of generalized convulsive status epilepticus decreased from three times per year (at baseline) to zero (at months 12 and 24). In patient 5, the baseline GTC seizure frequency per 28 days decreased from 12 to 3 at month 6.
Neuro‐ophthalmologic testing: PLR and horizontal saccadic latency
Figure 6 presents the graphic representation of PLR. The initial constriction rate and latency of PLR were impaired in all patients to a variable degree at baseline (Table 4). After therapy, both parameters recovered in all patients. Figure 7A presents the graphic representation of EOG at baseline, and irregularly stepped saccadic eye movements were obvious. The mean values of horizontal saccadic latency in patient 2 (0.82 ms, P < 0.0001) and patient 3 (0.44 ms, P < 0.0001) were significantly delayed compared with those in control subjects (0.19 ± 0.04 ms; Fig. 7B). At the 6‐month follow‐up, the mean latencies significantly improved in both patient 2 (0.26 ms, P < 0.001) and patient 3 (0.28 ms, P = 0.02) from baseline, and the waveform changed dramatically, particularly in patient 3.
Figure 6.
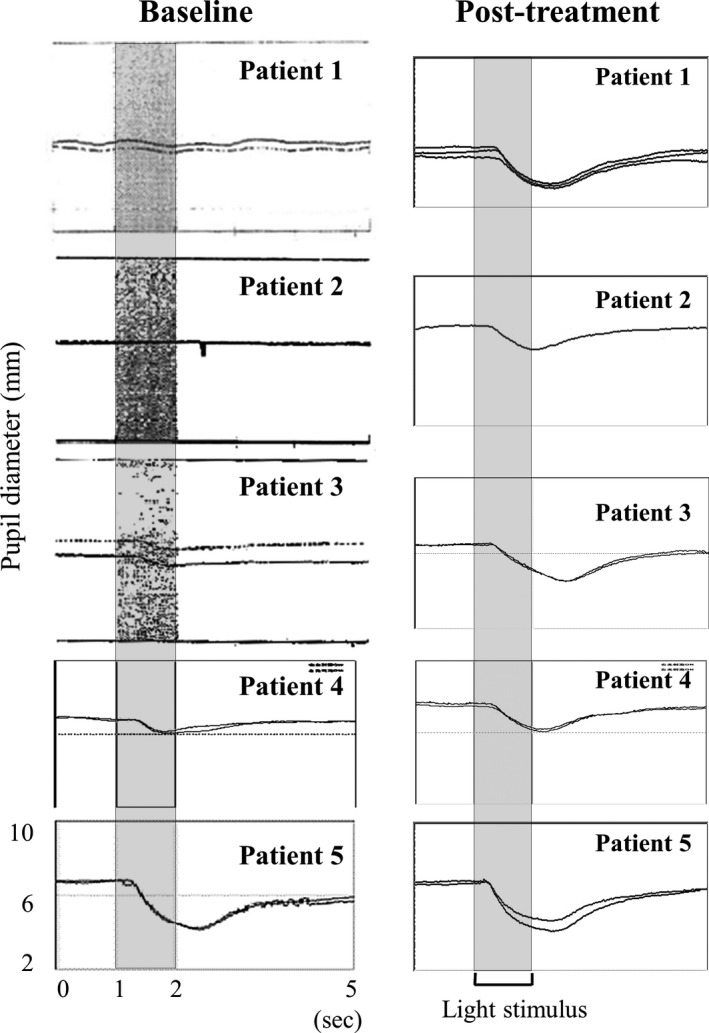
Effects of ambroxol on PLR to monochromatic light stimulation in nGD patients. Assessments were performed at the following times posttreatment: patient 1, month 48; patients 2 and 3, month 36; patient 4, month 24; and patient 5, month 6.
Table 4.
Mean pupillometry changes from baseline values
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | |
|---|---|---|---|---|---|
| Initial constriction rate, % (SD) | |||||
| Baseline | 0 (ND) | 0 (ND) | 13.0 (−5.2 SD) | 11.0 (−7.5 SD) | 38.5 (−0.5 SD) |
| Posttreatment | 21.8 (−0.8 SD) | 18.8 (−4.4 SD) | 31.5 (−1.9 SD) | 24.5 (−4.0 SD) | 36.0 (−0.9 SD) |
| Control (adult)a: 41.3 ± 5.5; Control (infant)b: 48.0 ± 5.8 | |||||
| Latency, ms (SD) | |||||
| Baseline | ND | ND | 426.7 (+8.6 SD) | 366.7 (+8.3 SD) | 308.4 (+2.3 SD) |
| Posttreatment | 463.3 (+8.6 SD) | 354.2 (+0.9 SD) | 355.6 (+4.5 SD) | 316.7 (+4.9 SD) | 275.0 (+0.6 SD) |
| Control (adult)a: 264.1 ± 19.0; Control (infant)b: 243.3 ± 14.9 | |||||
The effects of treatment were analyzed at the following times: patient 1, month 48; patients 2 and 3, month 36; patient 4, month 24; and patient 5, month 6. The results of controls represent the mean ±SD. ND, not detected.
Data were acquired from 32 healthy controls (n = 30, median age: 23 years; range: 22–37 years).
Data were acquired from 4 healthy controls (n = 4, median age, 3.8 years; range, 3–4 years).
Figure 7.
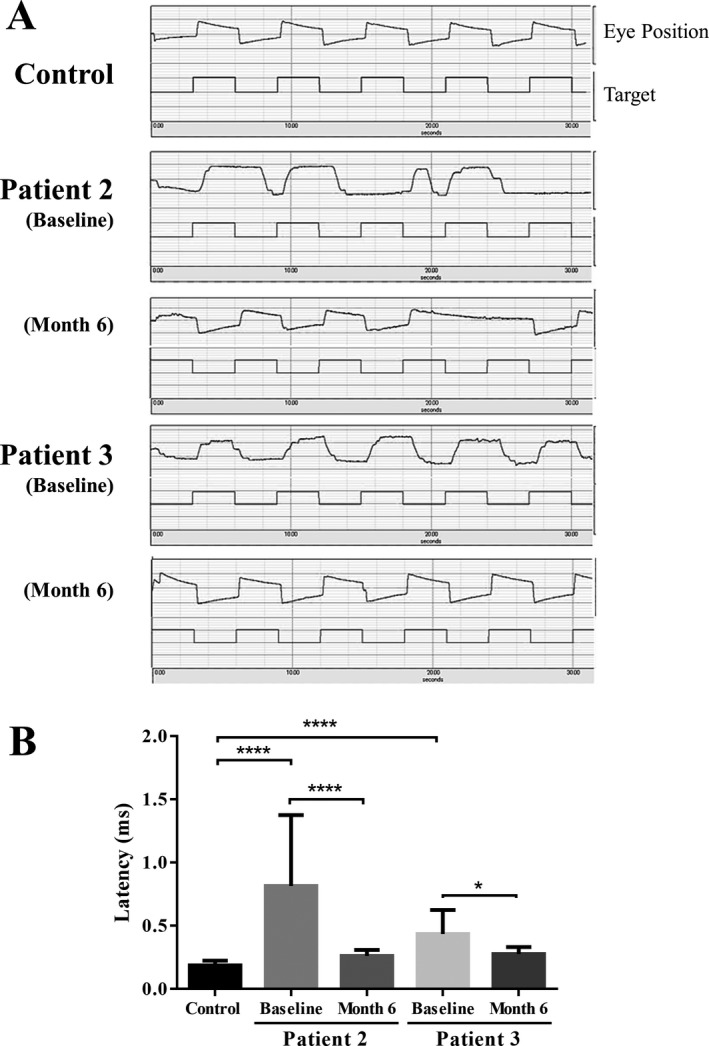
Effects of ambroxol on horizontal saccadic eye movements. Data were acquired from two testable patients (patients 2 and 3) and six healthy controls (median age = 22 years; range = 22–34 years). (A) Representation of slowed and stepped saccadic eye movements presenting with large latencies in patients at baseline. (B) Comparison of horizontal saccadic latencies in patients with nGD and normal controls.The results present the mean ± SEM. Statistically significant differences between the control and baseline were elicited, using Mann‐Whitney U‐test. Statistically significant differences between the baseline and month 6 were elicited, using Wilcoxon signed‐rank test. *P < 0.05, ****P < 0.0001.
Electrophysiological assessment: SEP and VEP
Giant SEPs were observed in patients 2 and 3 at baseline, and decreased amplitude was noted in only patient 3 (50 μV at baseline versus 21.7 μV at month 36). Giant VEPs were observed in patients 1 and 5 at baseline, and decreased amplitude was noted in only patient 1 (25 μV at baseline vs 14.9 μV at month 48).
Discussion
This is the first proof of concept study to show the safety, tolerability, and neurological efficacy of PCT in human lysosomal storage diseases. Several case studies have demonstrated the potential neurological efficacy of high‐dose ERT or ERT plus SRT (miglustat).30, 31, 32 Nevertheless, progressive neurological deterioration has been frequently reported, and there are as yet no useful therapies that can alter the intractable natural course of neurological manifestations. Our pilot study revealed that all patients showed remarkable improvement in neurological manifestations, despite exhibiting symptoms of advanced‐stage disease, and remained stable over months or years.
This study was limited by the small number of patients, resulting in an inability to conduct a randomized trial. To date, no large‐scale studies of the natural history of nGD, particularly GD3, have been performed because of the rarity of the disease compared with GD1 worldwide. Additionally, wide phenotypic variation, including the responsiveness to specific therapies, makes it difficult to evaluate the effects of treatment on neurological manifestations. Despite these limitations, this study highlights the clinical usefulness of PCT with ambroxol for neurological manifestations because the clinical courses in our patients following ambroxol therapy were generally unpredictable based on previously published works describing the clinical course of patients with nGD.
The primary objective of our pilot study was safety, and no serious side effects were identified during this study. Drug repositioning (i.e., finding new indications for existing drugs or drug candidates) has the advantage of reducing the time and cost required for bring a new drug to the market and is an attractive approach for the fulfillment of unmet medical needs for rare diseases, such as nGD. On the other hand, not much is known about the long‐term effects of high‐dose ambroxol; therefore, continued careful observation is needed to determine potential risks and benefits.
The goal of PCT is to enhance mutant enzyme activity, decrease the rate of toxic substrate accumulation, and eventually recover neuronal function and prevent neuronal loss. First, regarding the biochemical efficacy, the target dose of ambroxol satisfactorily increased mutant GCase activities in the lymphocytes. The targeted CSF ambroxol levels, which were estimated from previous studies in fibroblasts (i.e., trough levels of >0.3 μmol/L),20 were not achieved in three of the five patients; however, the clinical improvements and CSF GlcSph reductions observed in these three patients were comparable to those in patients who achieved the targeted levels. Our preliminary data indicated that the target dose of ambroxol may be sufficient for enhancing mutant GCase activity and favorably influencing GlcSph accumulation in the CNS, even with lower CSF concentrations than expected. Further studies are required to set the target level in CSF to achieve better results.
Next, regarding the neurological efficacy of ambroxol, the reduction of myoclonus and seizures contributed to the improvement of motor function, provided relief from discomfort, and consequently had a very favorable influence on the quality of life of patients and their families. The myoclonus and myoclonic seizures observed in nGD are thought to be caused by cortical neuronal hyperexcitability. However, the pathophysiology of nGD is still unknown. The role of excess GlcCer and GlcSph on neurological manifestations is also poorly understood. GlcSph, a lysoglycosphingolipid, is a direct derivative of GlcCer. The idea of investigating GlcSph was derived from the psychosine hypothesis in Krabbe disease, which assumes an active role of lysoglycosphingolipids (psychosine) in brain pathology.33 Several investigations have shown massively elevated GlcSph levels in the brains of patients with nGD compared with those in patients with GD1 and control subjects.1, 3 GlcSph is presumed to contribute to intracellular signal transduction pathways,34, 35 altered intracellular calcium homeostasis,36 and membrane lipid biosynthesis.37 The reduction in GlcSph accumulation following ambroxol therapy may help minimize the neurotoxicity and lead to the improvement of several neurological manifestations.
Finally, because of the phenotypic heterogeneity of GD, there is currently no recognized quantitative clinical endpoint for the neurological manifestations of GD. In this study, pupillometry could detect impairment of PLR with minimal patient cooperation, providing a sensitive method for detection of changes following therapy. Pupillometry may be a helpful quantitative method for analysis of the effects of novel therapies in patients with nGD, though further studies are needed to determine the underlying mechanisms of PLR dysfunction in nGD.
In conclusion, high‐dose oral ambroxol is a promising therapy for nGD. We expect that PCT will achieve more drastic clinical effects for GD patients with presymptomatic or early‐stage disease; therefore, early intervention is highly desirable not only for patients with nGD but also patients with GD1, because neurological manifestations, particularly parkinsonism and peripheral neuropathy, can also develop in GD1.38 Additionally, a pilot study reported the therapeutic benefits of ambroxol on visceral and hematologic manifestations in patients with GD1.39 Thus, further randomized controlled trials are needed to establish whether ambroxol shows protective effects against neurological complications, the efficacy as a single‐agent therapy on visceral and hematologic manifestations like SRT, and the synergistic effects of ambroxol plus ERT in treatment of refractory manifestations such as lung or bone lesions. Many PCs are currently being developed, and PCT has been applied not only to lysosomal storage diseases but neurodegenerative diseases like Parkinson's disease and other synucleinopathies, and many other protein misfolding diseases.40 We hope that this report will stimulate clinical trials of PCT for treating these intractable diseases that are not treatable at present.
Authors Contributions
KO conceived the project and acquired funding. KO and AN designed the study. AN, KS, SI, NK, RT, AH, TK, KY, YW, YN, AT, and MT provided inpatient admissions and patient care. AN performed all in vitro experiments and analyzed the data. AM conducted the GCase assays. AI and KM conducted the UMRS, GMFM, and FIM assessments. CF analyzed VEP and SEP. SK, HN, and HN analyzed CSF GlcSph levels and ambroxol concentrations. AN acquired and analyzed data and wrote the report. HS, YE, KH, EN, YS, YM, KO, and YS provided scientific input and constructive criticism of the report. All of the authors reviewed and approved the final version for publication.
Conflicts of Interest
None to report.
Supporting information
Data S1. Supplementary methods.
Video S1. Myoclonus and the effects of ambroxol in patient 2. At baseline, arrhythmic and asymmetric myoclonus of the limbs had made the patient bedridden. Myoclonic seizures of the trunk produce a sudden backward fall, and the patient could not maintain a sitting posture. She was barely able to stand and take a few steps while holding onto a rail. Three weeks after initiation of ambroxol, myoclonus of the limbs and myoclonic seizures of the trunk were markedly ameliorated, which enabled her to walk with or without minimal support. This spectacular improvement persisted during the follow‐up period of 3 years.
Video S2. Myoclonus and the effects of ambroxol in patient 3. At baseline, continuous myoclonus of the trunk and limbs disturbed ambulation. Severe myoclonus of the upper limbs caused difficulties in holding a handrail firmly and standing safely. Four weeks after the initiation of ambroxol, marked improvements of arrhythmic massive myoclonus of both the upper limbs and the trunk were noted. Her gait was steady with minimal support. This remarkable improvement persisted during the subsequent 3 years of follow‐up.
Video S3. Myoclonus and the effects of ambroxol in patient 4. At baseline, generalized myoclonus appeared continuously. Three months after the initiation of ambroxol, recurrent long‐lasting myoclonus decreased. After 2 years, myoclonus has almost disappeared, and the ability to smile was regained.
Acknowledgments
This study was supported by grants from the Ministry of Health, Labor and Welfare of Japan (H23‐25 Nanji‐Ippan‐002). We thank the families of our patients for allowing their children to participate in this clinical study, the physicians who referred their patients to this protocol, and all the physicians, nurses, and pharmacists for their dedicated patient care.
References
- 1. Orvisky E, Park JK, LaMarca ME, et al. Glucosylsphingosine accumulation in tissues from patients with Gaucher disease: correlation with phenotype and genotype. Mol Genet Metab 2002;76:262–270. [DOI] [PubMed] [Google Scholar]
- 2. Schueler UH, Kolter T, Kaneski CR, et al. Toxicity of glucosylsphingosine (glucopsychosine) to cultured neuronal cells: a model system for assessing neuronal damage in Gaucher disease type 2 and 3. Neurobiol Dis 2003;14:595–601. [DOI] [PubMed] [Google Scholar]
- 3. Nilsson O, Svennerholm L. Accumulation of glucosylceramide and glucosylsphingosine (psychosine) in cerebrum and cerebellum in infantile and juvenile Gaucher disease. J Neurochem 1982;39:709–718. [DOI] [PubMed] [Google Scholar]
- 4. Grabowski GA, Petsko GA, Kolodny EH. The Online Metabolic and Molecular Basis of Inherited Disease (OMMBID) Chapter 146, 2014. [Google Scholar]
- 5. Grabowski GA. Phenotype, diagnosis, and treatment of Gaucher's disease. Lancet 2008;372:1263–12671. [DOI] [PubMed] [Google Scholar]
- 6. Charrow J, Andersson HC, Kaplan P, et al. The Gaucher registry: demographics and disease characteristics of 1698 patients with Gaucher disease. Arch Intern Med 2000;160:2835–2843. [DOI] [PubMed] [Google Scholar]
- 7. Ida H, Iwasawa K, Kawame H, et al. Characteristics of gene mutations among 32 unrelated Japanese Gaucher disease patients: absence of the common Jewish 84GG and 1226G mutations. Human Genet 1995;95:717–720. [DOI] [PubMed] [Google Scholar]
- 8. Jeong SY, Park SJ, Kim HJ. Clinical and genetic characteristics of Korean patients with Gaucher disease. Blood Cells Mol Dis 2011;46:11–14. [DOI] [PubMed] [Google Scholar]
- 9. Kaplan P, Baris H, De Meirleir L, et al. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr 2013;172:447–458. [DOI] [PubMed] [Google Scholar]
- 10. Schiffmann R, Fitzgibbon EJ, Harris C, et al. Randomized, controlled trial of miglustat in Gaucher's disease type 3. Ann Neurol 2008;64:514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Okumiya T, Ishii S, Takenaka T, et al. Galactose stabilizes various missense mutants of alpha‐galactosidase in Fabry disease. Biochem Biophys Res Commun 1995;214:1219–1224. [DOI] [PubMed] [Google Scholar]
- 12. Fan JQ, Ishii S, Asano N, Suzuki Y. Accelerated transport and maturation of lysosomal alpha‐galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat Med 1999;5:112–115. [DOI] [PubMed] [Google Scholar]
- 13. Matsuda J, Suzuki O, Oshima A, et al. Chemical chaperone therapy for brain pathology in G(M1)‐gangliosidosis. Proc Nat Acad Sci U S A 2003;100:15912–15917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin H, Sugimoto Y, Ohsaki Y, et al. N‐octyl‐beta‐valienamine up‐regulates activity of F213I mutant beta‐glucosidase in cultured cells: a potential chemical chaperone therapy for Gaucher disease. Biochim Biophys Acta 2004;1689:219–228. [DOI] [PubMed] [Google Scholar]
- 15. Luan Z, Higaki K, Aguilar‐Moncayo M, et al. Chaperone activity of bicyclic nojirimycin analogues for Gaucher mutations in comparison with N‐(n‐nonyl)deoxynojirimycin. ChemBioChem 2009;10:2780–2792. [DOI] [PubMed] [Google Scholar]
- 16. Sawkar AR, Cheng WC, Beutler E, et al. Chemical chaperones increase the cellular activity of N370S beta‐glucosidase: a therapeutic strategy for Gaucher disease. Proc Nat Acad Sci U S A 2002;99:15428–15433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun Y, Liou B, Xu YH, et al. Ex vivo and in vivo effects of isofagomine on acid beta‐glucosidase variants and substrate levels in Gaucher disease. J Biol Chem 2012;287:4275–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Patnaik S, Zheng W, Choi JH, et al. Discovery, structure‐activity relationship, and biological evaluation of noninhibitory small molecule chaperones of glucocerebrosidase. J Med Chem 2012;55:5734–5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maegawa GH, Tropak MB, Buttner JD, et al. Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J Biol Chem 2009;284:23502–23516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Luan Z, Li L, Higaki K, et al. The chaperone activity and toxicity of ambroxol on Gaucher cells and normal mice. Brain Dev 2013;35:317–322. [DOI] [PubMed] [Google Scholar]
- 21. Lei K, Ninomiya H, Suzuki M, et al. Enzyme enhancement activity of N‐octyl‐beta‐valienamine on beta‐glucosidase mutants associated with Gaucher disease. Biochim Biophys Acta 2007;1772:587–596. [DOI] [PubMed] [Google Scholar]
- 22. Keimatsu M, Sakai K, Mihoya M, et al. Bioequivalence of NA872ET versus ambroxol hydrochloride sustainable release capsule. J New Remedies Clinics 2014;63:1964–1980. [Google Scholar]
- 23. Fukase H. Bioequivalence between NA872D and NA872 solution. J New Remedies Clinics 2004;53:388–415. [Google Scholar]
- 24. Frucht SJ, Leurgans SE, Hallett M, Fahn S. The unified myoclonus rating scale. Adv Neurol 2002;89:361–376. [PubMed] [Google Scholar]
- 25. Russel DJ, Rosenbaum PL, Wright M, et al. Gross motor function measure (GMFM‐66 and GMFM‐88) user's manual, 2nd ed London: Mac Keith Press, 2013. [Google Scholar]
- 26. Keith RA, Granger CV, Hamilton BB, Sherwin FS. The functional independence measure: a new tool for rehabilitation. Adv Clin Rehab 1987;1:6–18. [PubMed] [Google Scholar]
- 27. Narita A, Shirai K, Kubota N, et al. Abnormal pupillary light reflex with chromatic pupillometry in Gaucher disease. Ann Clin Transl Neurol 2014;1:135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miyahara A, Saito Y, Sugai K, et al. Reassessment of phenytoin for treatment of late stage progressive myoclonus epilepsy complicated with status epilepticus. Epilepsy Res 2009;84:201–209. [DOI] [PubMed] [Google Scholar]
- 29. Oosterhuis B, Storm G, Cornelissen PJ, et al. Dose‐dependent uricosuric effect of ambroxol. Eur J Clin Pharmacol 1993;44:237–241. [DOI] [PubMed] [Google Scholar]
- 30. Kraoua I, Sedel F, Caillaud C, et al. A French experience of type 3 Gaucher disease: Phenotypic diversity and neurological outcome of 10 patients. Brain Dev 2011;33:131–139. [DOI] [PubMed] [Google Scholar]
- 31. Capablo JL, Franco R, de Cabezon AS, et al. Neurologic improvement in a type 3 Gaucher disease patient treated with imiglucerase/miglustat combination. Epilepsia 2007;48:1406–1408. [DOI] [PubMed] [Google Scholar]
- 32. Cox‐Brinkman J, van Breemen MJ, van Maldegem BT, et al. Potential efficacy of enzyme replacement and substrate reduction therapy in three siblings with Gaucher disease type III. J Inherit Metab Dis 2008;31:745–752. [DOI] [PubMed] [Google Scholar]
- 33. Suzuki K. Twenty five years of the “psychosine hypothesis”: a personal perspective of its history and present status. Neurochem Res 1998;23:251–259. [DOI] [PubMed] [Google Scholar]
- 34. Giri S, Khan M, Rattan R, et al. Krabbe disease: psychosine‐mediated activation of phospholipase A2 in oligodendrocyte cell death. J Lipid Res 2006;47:1478–1492. [DOI] [PubMed] [Google Scholar]
- 35. Hannun YA, Bell RM. Lysosphingolipids inhibit protein kinase C: implications for the sphingolipidoses. Science 1987;235:670–674. [DOI] [PubMed] [Google Scholar]
- 36. Wong K, Sidransky E, Verma A, et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Gen Metab 2004;82:192–207. [DOI] [PubMed] [Google Scholar]
- 37. Sohal PS, Cornell RB. Sphingosine inhibits the activity of rat liver CTP:phosphocholine cytidylyltransferase. J Biol Chem 1990;265:11746–11750. [PubMed] [Google Scholar]
- 38. Cherin P, Rose C, de Roux‐Serratrice C, et al. The neurological manifestations of Gaucher disease type 1: the French Observatoire on Gaucher disease (FROG). J Inherit Metab Dis 2010;33:331–338. [DOI] [PubMed] [Google Scholar]
- 39. Zimran A, Altarescu G, Elstein D. Pilot study using ambroxol as a pharmacological chaperone in type 1 Gaucher disease. Blood Cells Mol Dis 2013;50:134–137. [DOI] [PubMed] [Google Scholar]
- 40. Suzuki Y. Emerging novel concept of chaperone therapies for protein misfolding diseases. Proc Jpn Acad Ser B Phys Biol Sci 2014;90:145–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplementary methods.
Video S1. Myoclonus and the effects of ambroxol in patient 2. At baseline, arrhythmic and asymmetric myoclonus of the limbs had made the patient bedridden. Myoclonic seizures of the trunk produce a sudden backward fall, and the patient could not maintain a sitting posture. She was barely able to stand and take a few steps while holding onto a rail. Three weeks after initiation of ambroxol, myoclonus of the limbs and myoclonic seizures of the trunk were markedly ameliorated, which enabled her to walk with or without minimal support. This spectacular improvement persisted during the follow‐up period of 3 years.
Video S2. Myoclonus and the effects of ambroxol in patient 3. At baseline, continuous myoclonus of the trunk and limbs disturbed ambulation. Severe myoclonus of the upper limbs caused difficulties in holding a handrail firmly and standing safely. Four weeks after the initiation of ambroxol, marked improvements of arrhythmic massive myoclonus of both the upper limbs and the trunk were noted. Her gait was steady with minimal support. This remarkable improvement persisted during the subsequent 3 years of follow‐up.
Video S3. Myoclonus and the effects of ambroxol in patient 4. At baseline, generalized myoclonus appeared continuously. Three months after the initiation of ambroxol, recurrent long‐lasting myoclonus decreased. After 2 years, myoclonus has almost disappeared, and the ability to smile was regained.