

Abstract
Progressive multifocal leukoencephalopathy (PML), a demyelinating disease of the brain, is typically diagnosed in immunocompromised persons. Here, we describe the diagnostic challenge of PML in an apparently immunocompetent patient. Thorough analyses, including cytokine release assays and whole exome sequencing, revealed a deficit in the antiviral interferon gamma production capacity of this patient and compound heterozygous mutations in BCL‐2‐associated athanogene 3. Interestingly, both factors are associated with reduced expression of John Cunningham virus T‐antigen, a protein that plays a key role in viral replication in infected cells. After validation in other patients, our findings may contribute to novel insights into the etiology and possibly treatment of PML.
Introduction
Progressive multifocal leukoencephalopathy (PML) is a destructive demyelinating disease of the central nervous system caused by the John Cunningham virus (JCV), which belongs to the family of polyoma viruses. The onset of the disease is subacute, with a broad range of clinical features. Its course is progressive and often fatal.1 Carriership of JCV is common among healthy individuals and it remains latent in the kidney, lymphoreticular, or brain tissue. Approximately 70% of adults are seropositive for JCV. Reactivation of the virus, resulting in PML, occurs typically under immunosuppressive conditions.1 The growing number of immunosuppressive and immunomodulatory therapeutics has resulted into an increased number of individuals at risk for PML.2 Although the common denominator in all these conditions is suppression of cellular immunity (either iatrogenic or endogenous), PML may also occur in patients with minimal or occult immune suppression (e.g., idiopathic CD4+ lymphocytopenia, chronic kidney, or liver disease)3 and even occasionally in apparently immunocompetent patients.4 Here, we present a case of PML in an apparently immunocompetent patient in whom a deficit of the interferon γ (IFNγ) pathway, and compound heterozygozity for mutations in BCL2‐associated athanogene 3 (BAG3) were identified.
Patient and Methods
A 49‐year‐old Caucasian male presented with rapidly progressive symptoms of aphasia, dyscalculia, hyperesthesia of the right arm, and headache. Two months after the first symptoms, his clinical condition worsened acutely with an inability to speak and to carry out activities of daily living, apraxia, confusion, and severe headache. His vital signs were normal and neurologic examination confirmed a nonfluent aphasia, dysgraphia, mild facial paresis on the right, a right‐sided hemianopsia, hemi‐hypaesthesia, and hyperreflexia without extensor plantar responses. He used no medication and had no significant medical history. The family history was unremarkable for neurologic conditions. Laboratory tests showed normal leukocyte counts, including CD3, CD4, and CD8 counts (ratio T4/T8), low infectious parameters, negative serology for human immunodeficiency virus (HIV), Lues, Borrelia, and Herpes Simplex Virus (HSV). Cerebrospinal fluid (CSF) analysis showed normal cell counts, protein and glucose levels, no oligoclonal bands or markers of neurodegeneration (except for a slightly elevated Tau [431 ng/L], normal <300 ng/L), and cytological analysis showed no signs of malignancy. Cerebral MRI showed confluating subcortical white matter T2 hyperintensities predominantly in the left hemisphere, which extended on consecutive MRIs without contrast enhancement or mass effect (Fig. 1). PCR for JCV in serum and CSF was negative.
Figure 1.
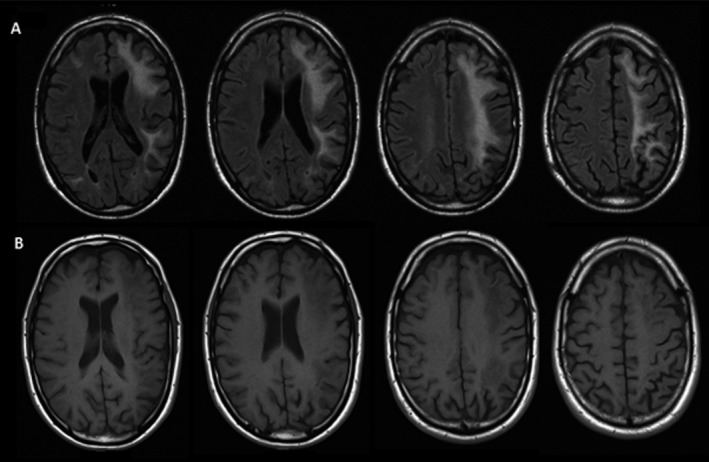
MRI images 4 months after first presentation. (A) Fluid‐attenuated inversion recovery (FLAIR) MRI: large confluating asymmetric white matter hyperintensities lesions in the frontal and parietal lobes. (B) T1‐gadolinium sequences without contract enhancement.
A stereotactic biopsy of the left frontal lobe showed perivascular lymphocytic infiltration with sporadic enlarged nuclei, which resembled astrocytes. No pathologic oligodendrocyte nuclei were found and immunohistochemical staining for SV‐40 (polyomavirus) was negative. However, a positive PCR alone is no confirmation of active virus replication. The PCR for JCV on the biopsy material, however, was positive. The normal immune status and the lack of evidence for an active JCV infection led to the primary diagnosis of tumefactive multiple sclerosis, and the patient was treated consecutively with methylprednisolone intravenously, glatiramer acetate (20 mg/mL once daily), acetaminophen (1000 mg four times a day), and plasmapheresis.5 As deterioration continued the biopsy material was re‐examined 7 months after symptom onset, in a tertiary center with extensive expertise on PML. The presence of foam cells, some bizarre astrocytes, and sporadic oligodendrocytes with ground glass appearance combined with the clinical course led to a revision of the diagnosis to PML, and the patient was started on mirtazapine (15 mg once daily). Unfortunately his condition was progressive and he died 9 months after the initial presentation due to cardiopulmonary complications of a bilateral pneumonia. Autopsy confirmed the diagnosis of PML. Macroscopy showed extensive white matter hyperintensities in both hemispheres, with focal gray glass lesions, suggestive for PML. Microscopy showed large white matter hyperintensities with prominent demyelination, and a granular tissue loss. Extensive reactive astrocytosis, and astrocytes with enlarged polymorph hyperchromatic nuclei were found, indicating a viral cytopathogenic effect. A few oligodendrocytes with enlarged nuclei, perivascular foamy macrophages, and some lymphocytic infiltration were seen.
Immunological assessment
After the diagnosis of PML an assessment of the capacity of cells isolated from the patient to respond to microbial stimuli was initiated. Peripheral blood mononuclear cells were isolated from blood collected from the patient, and stimulated with the TLR4‐ligand lipopolysaccharide (Escherichia coli LPS 10 ng/mL), the TLR3‐ligand PolyI:C (5 μg/mL), and a fungal stimulus (heat‐killed Candida albicans 105 microorganisms/mL). IFNγ production capacity was measured using an enzyme‐linked immunosorbent assay.
Genetic analysis
Whole exome sequencing was performed as described earlier.6, 7 In brief, DNA was isolated from whole blood, enriched with SureSelect v2 exome (Agilent Technologies, Santa Clara, CA) (50 Mb) and sequenced on SOLiD 4 (Life Technologies, Foster City, CA). Variants were called using high stringency settings and annotated with an in‐house pipeline containing information from dbSNP134. Variant filtering was applied as previously reported; in brief, we only selected variants affecting coding exons, microRNAs, and canonical splice sites. Subsequently, synonymous variants were filtered out, and only rare variants (frequency of <0.25% in both dbSNP134 and our in‐house database containing >2000 exomes) with high quality were reported (Table 1). All genes with rare nonsynonymous variants were systematically checked for involvement role in immunity; based on Gene Ontology terms, mouse knockout phenotypes, information from the Kyoto Encyclopedia of Genes and Genomes or direct interaction with JCV according to NCBI (the latter was performed by searching for JCV in NCBI, and selecting for genes in humans).
Table 1.
Exome sequencing statistics and the filter settings applied to exclude noncoding, nonsynonymous, common, and low‐quality variants
| Sample | PML patient |
|---|---|
| Total mapped bases (Gb) | 5.5 |
| On and near target (%) | 82.0% |
| Median fold coverage | 57.6 |
| Average fold coverage | 81.7 |
| % Covered >onefold | 94.1% |
| % Covered >10‐fold | 82.8% |
| % Covered >20‐fold | 74.5% |
| Total variants | 35,322 |
| Coding, canonical, microRNA | 14,595 |
| Nonsynonymous | 7211 |
| Rare variants <0.25% SNP, in‐house exomes | 188 |
| Variant reads >5% and >25% | 106 |
| Variants in immune‐related pathways | 21 |
| Associated with JCV (NCBI) | 2 variants, 1 gene (BAG3) |
PML, progressive multifocal leukoencephalopathy; BAG3, BCL2‐associated athanogene 3; JCV, John Cunningham virus.
Results
Immunological assessment
While cytokine production upon stimulation with LPS and C. albicans was normal compared to eight healthy individuals. IFNγ production induced by PolyI:C was severely impaired in the patient versus control stimulation (Fig. 2). We were unable to perform extensive immunophenotyping of T‐, B‐, and NK‐cell subpopulations as our patient deceased shortly after the diagnosis of PML.
Figure 2.
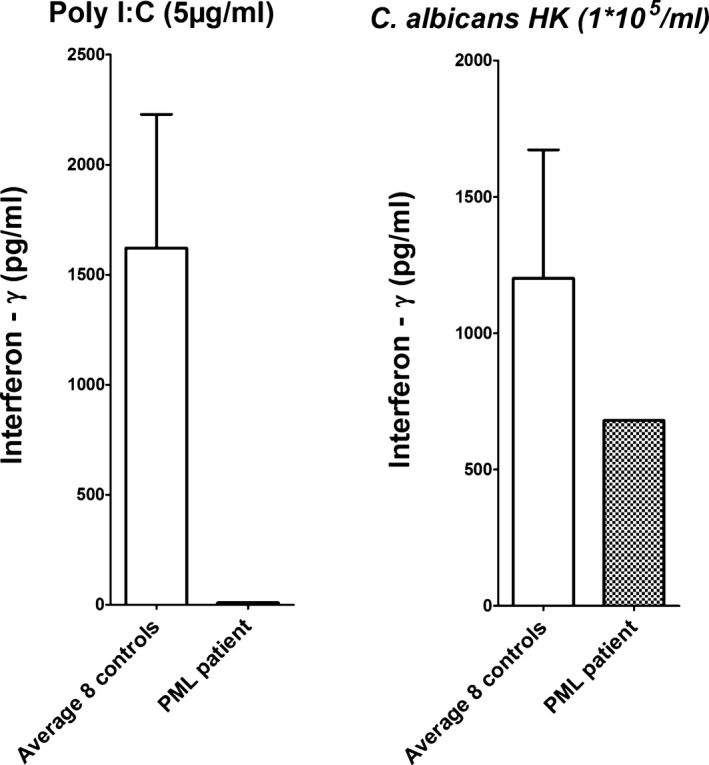
Interferon γ (IFN γ) production capacity was measured in the progressive multifocal leukoencephalopathy (PML) patient and compared with eight healthy controls upon stimulation with the TLR3 ligand PolyI:C (5 μg/mL) (left) and Candida albicans (1 × 105 microorganisms/mL) (right). The PML patient showed a clear defect in IFN γ production upon PolyI:C stimulation compared to controls, whereas stimulation with Candida does not result in significant differences.
Genetic analysis
Whole exome sequencing provided 5.5 Gb of mapped sequencing data, resulting in an average coverage of the exome of 81.7‐fold. Standard variant filtering resulted in 106 rare, nonsynonymous, and canonical splice site variants. Only 17 genetic variants remained after filtering for genes with a possible role in immunity (Table 2). A genome‐wide search in NCBI for genes associated with JCV interaction resulted in 21 genes. An overlap between these 21 genes, and the 106 rare variants resulted in one gene for which JCV interaction was described earlier8, 9; we found two very rare heterozygous variants in exon 2 of the BAG3. Cosegregation analysis within the family shows that both unaffected parents carry one of the variants in a heterozygous state, and both variants are absent in the healthy brother (Fig. 3). The variants (p. P77L; p.I94F) have been reported at population allele frequencies of 0.029% and 0.076%, respectively. In addition, rare (<1%) homozygous protein‐altering variants are only reported in 16 of >60,000 controls.10
Table 2.
Seventeen rare variants with association to immune system for the respective genes
| Selection criteria | Gene name | Gene component | mRNA change | Amino acid change | ExAC allele frequency | ExAC # of alleles/total | SIFT prediction | Polyphen prediction | PhyloP* | Grantham score |
|---|---|---|---|---|---|---|---|---|---|---|
| A(2), B | BAG3 | Exon | 230C>T | p.P77L | 0.0002883 | 35/121,382 | Tolerated | Benign | −0.157 | 98 |
| A(2), B | BAG3 | Exon | 280A>T | p.I94F | 0.000758 | 92/121,376 | Deleterious | Probably damaging | 2.098 | 21 |
| B | CDC73 | Exon | 685A>G | p.R229G | 0 | 0 | Deleterious | Probably damaging | 2.305 | 125 |
| B | TP53BP2 | Exon | 808T>C | p.M399V | 0.000239 | 29/121,318 | Tolerated | Benign | 2.459 | 21 |
| B | CPN1 | Exon | 166C>A | p.E56X | 0 | 0 | N/A | N/A | 0.354 | 1000 |
| B | KDM5A | Exon | 1885C>T | p.V629M | 0.00002485 | 3/120,732 | Deleterious | Possibly damaging | 4.234 | 21 |
| B | HYDIN | Exon | 2804T>C | p.N935S | 0 | 0 | Not scored | Probably damaging | 4.186 | 46 |
| B | CBFA2T2 | Exon | 757G>A | p.G282S | 0.000008237 | 1/121,404 | Tolerated | Benign | 1.669 | 56 |
| B | NIPBL | Exon | 3851A>G | p.N1284S | 0.00002486 | 3/120,678 | Deleterious | Benign | 2.76 | 46 |
| B | CALCR | Exon | 396T>G | p.E132D | 0 | 0 | Deleterious | Benign | 0.02 | 45 |
| B | ATP7A | Exon | 3107A>G | p.H1036R | 0 | 0 | Deleterious | Probably damaging | 4.925 | 29 |
| B, D | AHNAK | Exon | 16255C>T | p.D5419N | 0.0004119 | 50/121,374 | Deleterious | Probably damaging | 0.387 | 23 |
| B, E, F | ITGA4 | Exon | 722A>G | p.K241R | 0 | 0 | Tolerated | Benign | −3.015 | 26 |
| C | IFI16 | Exon | 494G>A | p.R165H | 0.0001236 | 15/121,400 | Tolerated | Benign | −3.32 | 29 |
| D | KYNU | Exon | 1303G>A | p.V435M | 0 | 0 | Deleterious | Probably damaging | 3.041 | 21 |
| E | ARRB1 | Canonical SA site | N/A | N/A | 0.000008239 | 1/121,376 | N/A | N/A | 5.068 | 0 |
| F | LAMB3 | Exon | 2632G>A | p.R878C | 0.00005767 | 7/121,396 | Deleterious | Probably damaging | 1.177 | 180 |
BAG3, BCL2‐associated athanogene 3; JCV, John Cunningham virus. A, NCBI human gene name interaction with JCV and number of PubMed publications (N); B, mouse knockout phenotype “immune”; C, Gene Ontology term “virus”; D, Gene Ontology term “interferon”; E, Kyoto Encyclopedia of Genes and Genomes class “immune”; F, Kyoto Encyclopedia of Genes and Genomes class “infectious.” PhyloP* relates to the amino acid conservation among 46 species. ExAC (Exome Aggregation Consortium10).
Figure 3.
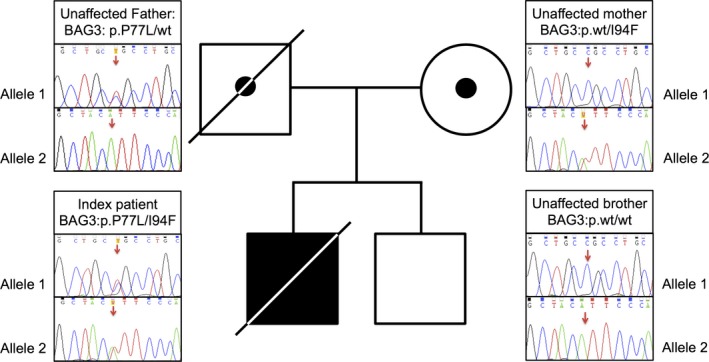
Family pedigree of our progressive multifocal leukoencephalopathy (PML) case; the unaffected parents were both carrier of one rare BCL2‐associated athanogene 3 (BAG3) variant (paternal variant p.P77L, c.230C>T; maternal variants p.I94F, c.280A>T). The compound heterozygosity for these BAG3 variants most likely affects the resistance against John Cunningham virus (JCV) in the index patient. Neither of the two variants is present in the unaffected brother.
Discussion
Although rare, similar cases of PML in apparently immunocompetent individuals have been reported.4, 11, 12 Due to its variable demographics, presenting symptoms, and prognosis, the diagnosis of PML remains a challenge, especially in apparently immunocompetent individuals. The diagnosis can be strengthened by a positive PCR for JCV in the CSF, which has a high sensitivity and specificity. However, the severity of the immunosuppression seems to determine the accuracy of the available tests, as is illustrated by the low JCV DNA copy numbers in the CSF of highly active anti‐retroviral therapy treated (HAART) HIV patients, as well as in nearly half of the multiple sclerosis patients treated with the monoclonal antibody natalizumab who were diagnosed with PML.13, 14, 15 Moreover, JCV PCR in CSF of PML cases with occult, minimal, or no detected immune suppression was often found negative.4 Also, substantial variability exists with regard to detection levels and consistency between laboratories even in testing the same sample set. If clinical suspicion remains high and JCV PCR in CSF is negative, a brain biopsy should therefore be performed. It is important to realize that JCV DNA is widespread in the brain of healthy adults and that the viral DNA load seems to increase in immunosuppresive conditions, as was observed in an autopsy study in HIV patients without PML (no clinical symptoms during life and no neuropathologic evidence of PML).16 Expression of viral proteins, which can be demonstrated by immunohistochemistry is, however, considered a pathognomonic sign of PML. In our case, viral DNA was found in the biopsy material, however, expression of viral proteins was not observed. Therefore, PML was initially not considered the most likely diagnosis in this immunocompetent patient.
Identifying factors that determine JCV reactivation in an immunocompetent patient will profoundly contribute to the understanding of pathogenesis of PML.17 Moreover, it might contribute to novel treatment options. IFNγ is an important cytokine in human antiviral cellular immune response. The observation that the IFNγ response in our patient was decreased after stimulation with PolyI:C, a synthetic TLR3 ligand used to simulate viral infections, suggests a specific deficit in the cellular antiviral immune response. Interestingly, IFNγ was recently reported to inhibit expression of JCV T‐antigen, the major viral regulatory protein.18 In addition, this study showed a significant decrease of JCV DNA copies in cells upon IFNγ treatment in vitro. The observed IFNγ deficit is most likely an important factor in the pathogenesis of PML in our patient; whole exome sequencing was performed in order to identify any genetic defects that could contribute to this deficit. Compound heterozygous BAG3 variants in our patient are worthy candidates for increased susceptibility to PML as BAG3 is implicated in autophagy and apoptosis through intracellular protein control. It was previously shown that overexpression of BAG3 results in a decrease of the JCV replication, and reduced T‐antigen expression through autophagic degradation, thereby controlling the JCV lytic cycle and its interaction with host cells.9 Although it remains speculative, the observed genetic variants in BAG3 might compromise the response against JCV T‐antigen in our patient, leading to insufficient IFNγ production, and subsequently PML.
Obviously this case report has several limitations and the results should be interpreted with caution. First, although the segregation analysis showed an autosomal recessive inheritance, the family is rather small, which makes the pathogenicity uncertain. Validating our findings in other cases is therefore crucial, however, due to the rarity of PML in apparent immunocompetent patients this remains extremely challenging. We took up this challenge and performed genetic testing for BAG3 variants in two previously reported apparent immunocompetent PML cases.11, 12 Unfortunately, no BAG3 variants were discovered in these patients. Given the number of factors suggested to be involved in the reactivation of JCV in order to develop PML and therefore increased heterogeneity between these patients, this might not be surprising.17, 19 In addition to the genetic variants in BAG3, we also provide evidence for low INFγ levels in this case. However, a possible association between the IFNγ deficit and the BAG3 variants remains to be shown.
This case report shows that specific defects in the IFNγ production upon stimulation may be present in PML patients without known immune deficits. Patients suspected of PML without immunosuppression should, in addition to regular immunologic screening, be tested for IFNγ deficiency to confirm our findings. In these conditions treatment with IFNγ might potentially be an option for PML, similar to other conditions treated with recombinant IFNγ.20
Author Contributions
N. M. K., P. A., and I. W. M. U. drafted the manuscript. N. M. K., I. W. M. U., F. L. V., and B. A. J. were involved in the clinical treatment of the patient. P. A. and A. H. performed the genetic analysis. M. G. N. and F. L. V. organized the cytokine analysis. M. G. N. and A. H. were responsible for the laboratory supervision. All authors reviewed the manuscript.
Conflict of Interest
None declared.
Acknowledgments
The authors thank Professor Halvor Naess, Neurologist in the Haukeland University Hospital, Norway, and Professor Jan O. Aasly, Neurologist in the St. Olavs Hospital, Norway, for their willingness and effort to send us DNA samples of their apparently immunocompetent PML cases. P. A. was supported by a grant from the Nijmegen Institute for Molecular Life Sciences; F. L. V. and A. H. were supported by Veni grants of The Netherlands Organisation for Scientific Research; M. G. N. was supported by a Vici grant of The Netherlands Organisation for Scientific research and an ERC Consolidator Grant (#310372).
References
- 1. Tan CS, Koralnik IJ. Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: clinical features and pathogenesis. Lancet Neurol 2010;9:425–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Clifford DB, DeLuca A, Simpson DM, et al. Natalizumab‐associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: lessons from 28 cases. Lancet Neurol 2010;9:438–446. [DOI] [PubMed] [Google Scholar]
- 3. Gheuens S, Pierone G, Peeters P, Koralnik IJ. Progressive multifocal leukoencephalopathy in individuals with minimal or occult immunosuppression. J Neurol Neurosurg Psychiatry 2010;81:247–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kastrup O, Göricke S, Kretzschmar H, et al. Progressive multifocal leukoencephalopathy of the brainstem in an immunocompetent patient – JC and BK polyoma‐virus coinfection? A case report and review of the literature. Clin Neurol Neurosurg 2013;115:2390–2392. [DOI] [PubMed] [Google Scholar]
- 5. Bastiaans DET, van Uden IWM, Ruiterkamp RA, de Jong BA. Removal of valproic acid by plasmapheresis in a patient treated for multiple sclerosis. Ther Drug Monit 2013;35:8–10. [DOI] [PubMed] [Google Scholar]
- 6. de Ligt J, Willemsen MH, van Bon BWM, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 2012;367:1921–1929. [DOI] [PubMed] [Google Scholar]
- 7. Arts P, Plantinga TS, van den Berg JM, et al. A missense mutation underlies defective SOCS4 function in a family with autoimmunity. J Intern Med 2015;278:203–210. [DOI] [PubMed] [Google Scholar]
- 8. Basile A, Darbinian N, Kaminski R, et al. Evidence for modulation of BAG3 by polyomavirus JC early protein. J Gen Virol 2009;90:1629–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sariyer IK, Merabova N, Patel PK, et al. Bag3‐induced autophagy is associated with degradation of JCV oncoprotein, T‐Ag. PLoS One 2012;7:e45000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Exome Aggregation Consortium (ExAC), Cambridge, MA. Available at: : http://exac.broadinstitute.org (accessed April 2015). [Google Scholar]
- 11. Johansen KK, Torp SH, Rydland J, Aasly JO. Progressive multifocal leukoencephalopathy in an immunocompetent patient? Case Rep Neurol 2013;5:149–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Naess H, Glad S, Storstein A, et al. Progressive multifocal leucoencephalopathy in an immunocompetent patient with favourable outcome: a case report. BMC Neurol 2010;10:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Marzocchetti A, Di Giambenedetto S, Cingolani A, et al. Reduced rate of diagnostic positive detection of JC virus DNA in cerebrospinal fluid in cases of suspected progressive multifocal leukoencephalopathy in the era of potent antiretroviral therapy. J Clin Microbiol 2005;43:4175–4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ryschkewitsch CF, Jensen PN, Monaco MC, Major EO. JC virus persistence following progressive multifocal leukoencephalopathy in multiple sclerosis patients treated with natalizumab. Ann Neurol 2010;68:384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang Y, Kirby JE, Qian Q. Effective use of JC virus PCR for diagnosis of progressive multifocal leukoencephalopathy. J Med Microbiol 2009;58:253–255. [DOI] [PubMed] [Google Scholar]
- 16. Bayliss J, Karasoulos T, McLean CA. Immunosuppression increases JC polyomavirus large T antigen DNA load in the brains of patients without progressive multifocal leukoencephalopathy. J Infect Dis 2013;207:133–136. [DOI] [PubMed] [Google Scholar]
- 17. Wollebo HS, White MK, Gordon J, et al. Persistence and pathogenesis of the neurotropic polyomavirus JC. Ann Neurol 2015;77:560–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. De‐Simone FI, Sariyer R, Otalora Y‐L, et al. IFN‐gamma inhibits JC virus replication in glial cells by suppressing T‐antigen expression. PLoS One 2015;10:e0129694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ferenczy MW, Marshall LJ, Nelson CDS, et al. Molecular biology, epidemiology, and pathogenesis of progressive multifocal leukoencephalopathy, the JC virus‐induced demyelinating disease of the human brain. Clin Microbiol Rev 2012;25:471–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marciano BE, Wesley R, De Carlo ES, et al. Long‐term interferon‐gamma therapy for patients with chronic granulomatous disease. Clin Infect Dis 2004;39:692–699. [DOI] [PubMed] [Google Scholar]