

Abstract
Objective
Inaccessibility of the inflammation compartmentalized to the central nervous system (CNS) may underlie the lack of efficacy of immunomodulatory treatments in progressive multiple sclerosis (MS). The double blind combination of Rituximab by IntraVenous and IntraThecAl injection versus placebo in patients with Low‐Inflammatory SEcondary progressive MS (RIVITALISE; NCT01212094) trial was designed to answer: (1) Whether an induction dose of intravenous and intrathecal rituximab efficiently depletes CNS B cells? and (2) If so, whether this leads to global inhibition of CNS inflammation and slowing of CNS tissue destruction?
Methods
Patients aged 18–65 years were randomly assigned to rituximab or placebo. Protocol‐stipulated interim analysis quantified the efficacy of B‐cell depletion.
Results
The efficacy on cerebrospinal fluid (CSF) biomarkers failed to reach criteria for continuation of the trial. B‐cell‐related CSF biomarkers (sCD21 and B‐cell activating factor) changed only in the active‐treatment arm. While CSF B cells were killed robustly (median −79.71%, P = 0.0176), B cells in CNS tissue were depleted inadequately (~−10–20%, P < 0.0001). Consequently, the T‐cell‐specific CSF biomarker sCD27 decreased slightly (−10.97%, P = 0.0005), while axonal damage marker, neurofilament light chain did not change. Insufficient saturation of CD20, lack of lytic complement, and paucity of cytotoxic CD56dim NK cells contribute to decreased efficacy of rituximab in the CNS.
Interpretation
Biomarker studies reliably quantified complementary pharmacodynamic effects of rituximab in the CNS, exposed causes for poor efficacy and determined that RIVITALISE trial would be underpowered to measure efficacy on clinical outcomes. Identified mechanisms for poor efficacy are applicable to all CNS‐inflammation targeting monoclonal antibodies.
Introduction
Immunomodulatory disease‐modifying treatments (DMTs) exert discernable clinical benefit only in patients in early stages of multiple sclerosis (MS), called relapsing‐remitting MS (RRMS). This lack of clinical efficacy, together with a decreased frequency of clinical relapses and contrast‐enhancing lesions (CELs) on brain MRI, has been interpreted as evidence that the vital disability drivers in progressive stages of MS are neurodegenerative, rather than immune mediated.1
Yet, this conclusion contradicts pathological observations of continued neuroinflammation in patients with progressive MS.2, 3 An alternative explanation presupposes that pathogenic immune responses in progressive MS are not accessible to current DMTs because of their compartmentalization to central nervous system (CNS) tissue. Indeed, levels of cerebrospinal fluid (CSF) T‐cell‐ and B‐cell‐specific biomarkers in both progressive MS subtypes (i.e., secondary progressive [SPMS] and primary progressive [PPMS]) are comparable to those observed in untreated RRMS.4 However, while immune responses in RRMS consist predominantly of migratory cells detected in the CSF, T, and B cells are mostly embedded in CNS tissue of progressive MS.4 As compartmentalization (and eventual establishment of tertiary lymphoid follicles in the affected tissue2) results from chronic/repeated activation of adaptive immunity in the particular compartment, it represents a continuous, rather than dichotomized (i.e., compartmentalized or not) process.4 Consistent with this explanation, functional assays also revealed higher levels of terminal differentiation of intrathecal T cells in progressive MS as compared to RRMS.5
Thus, lack of therapeutic efficacy of current DMTs in progressive MS could be explained by the combination of advanced CNS compartmentalization and terminal differentiation of pathogenic immune responses. Whether this intrathecal inflammation drives accumulation of clinical disability can be determined only after its effective silencing, which has not been convincingly achieved by any therapeutic strategy thus far, including bone marrow transplantation.6
Rituximab is a chimeric monoclonal antibody that targets CD20, which is exclusively expressed on pre‐B and mature B cells, but not on plasma cells.7 Clinical trials of intravenous rituximab have demonstrated reductions in MRI and clinical activity in RRMS patients, who, as a group, have opened blood–brain barrier (BBB) in the CNS areas with concentrated inflammation (as measured by CELs on brain MRI). However, rituximab had no efficacy on clinical outcomes in PPMS, who lack CELs,8 strongly supporting the notion that deficient penetration of the therapeutic antibody to affected CNS tissue may underlie lack of its efficacy.
Therefore, the purpose of the RIVITALISE trial was to investigate whether intrathecal and intravenous administration of rituximab can effectively deplete B cells and inhibit activation of T cells in the CNS compartment of SPMS patients. We report the prespecified interim analysis for the efficacy of B‐cell depletion and subsequent mechanistic studies, which revealed causes for differential efficacy of therapeutic antibodies in blood versus CNS compartments.
Materials and Methods
Patients and regulatory approval
RIVITALISE is a single center, randomized, double‐blind, placebo‐controlled study. Patients were prospectively enrolled at the National Institutes of Health (NIH), USA. Eligible patients were aged 18–65 years and had a diagnosis of MS according to the McDonald's criteria9; had an entry score of 3.0–7.0 on the expanded disability status scale (EDSS); diagnosed as SPMS with lack of MS relapse in the preceding 1 year and nonremitting/sustained progression of disability over 3 months; had not received any DMTs for a period of at least 1 month prior to enrollment; provided informed consent; agreed to commit to the use of an accepted method of birth control. Patients were excluded if they had a diagnosis of PPMS; had past history or signs of immunodeficiency or chronic infections; carried diagnosis of any serious medical disorder; had clinically relevant abnormal blood tests (including IgM and IgG abnormalities); had a positive pregnancy test; had positive CSF or serum quantitative PCR for JC virus. The study was approved by the NIH Institutional Review Board and all patients provided a written informed consent.
Randomization and masking
Patients were randomly assigned to receive rituximab or placebo. Randomization was done by the NIH pharmacy using a table of random numbers. Participants were divided into two strata: younger than 50 years of age (numbered 101–130) and 50 years of age and older (numbered 201–230). At the start of the study (September 2010), the randomization allocation was 1:1. In November 2012, with the protocol amendment to institute a second intrathecal dose at month (Mo) 1.5 after observing inadequate depletion of CSF B cells on the first three treated subjects, the randomization allocation was changed to 2:1. Using a block size of 3: within a block of three the highest two numbers were assigned to rituximab and the lowest number to placebo.
Procedures
Rituximab Investigational New Drug (IND) and selected dosing
Because only approximately 0.1% of intravenous rituximab reaches the CNS compartment under an intact BBB,10 depletion of B cells in CNS lymphomatous meningitis or CNS lymphoma is significantly higher after intrathecal, in comparison to intravenous administration.11 Based on literature review (Table S1), we selected 25 mg as the highest, well‐tolerated single intrathecal dose. We amended the protocol to administrate this intrathecal dose twice (6 weeks apart) after we observed inadequate depletion of CSF B cells in the first three subjects.
The selected intravenous induction dose (200 mg × 2; at Mo 0 and Mo 0.5) is lower than oncology dosing (375 mg/m2, weekly × 4 weeks) or doses previously used for MS (1000 mg × 2, 2 weeks apart). However, based on literature review12, we concluded that this dose (which represented fivefold cost saving) would saturate CD20 and should lead to lasting depletion of B cells from peripheral circulation.
We obtained IND (#107,648) from the Food Drug Administration (FDA) based on the cross‐referencing IND application of Dr. Rubenstein (UCSF, San Francisco), which included toxicology and pharmacodynamic data obtained in nonhuman primates.10
Trial design
The trial design scheme is shown in Fig. 1A. After premedication with 100 mg IV methylprednisolone, 50 mg of diphenhydramine, 650 mg of acetaminophen, and 1 mg of lorazepam, 20 cc of CSF was withdrawn by lumbar puncture (LP) followed by injection of rituximab (25 mg; 1:1 dilution in normal saline [NS]) or placebo (only NS) over 2 min, followed by 8 cc of NS flush (Mo 0). Patients were observed for 4 h in Trendelenburg position (to facilitate the flow of CSF from lumbar cistern toward hemispheres) before intravenous rituximab (200 mg) or placebo infusion was initiated. The second intravenous dose of rituximab or placebo (Mo 0.5), the second (Mo 1.5) and third (Mo 12) intrathecal rituximab or placebo doses were administered using analogous procedures.
Figure 1.
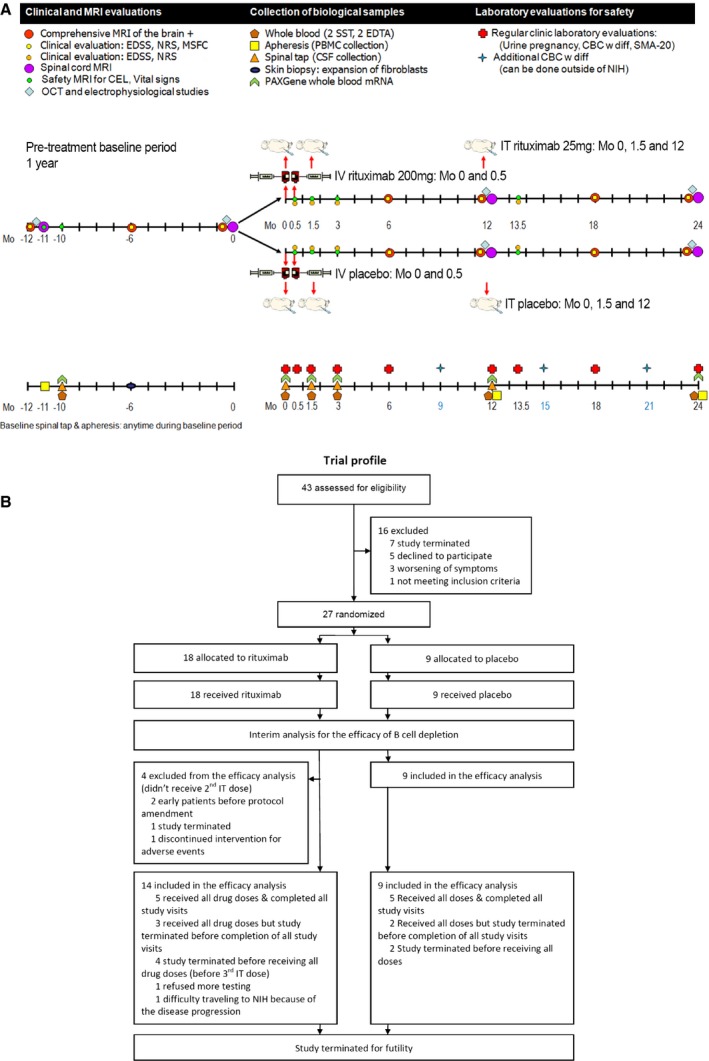
Trial design. (A) Trial design scheme; (B) CONSORT trial diagram. IT, intrathecal; NIH, National Institutes of Health.
Clinical and imaging evaluation
The clinical evaluations were done every 6 months with EDSS,13 Scripps Neurological Rating Scale (NRS14), and MS functional Composite Scale (MSFC15). Neuroimaging evaluation was performed every 6 months with routine spin‐echo and gradient‐echo T1‐weighted images were collected following intravenous administration of 0.1 mmol/kg gadopentetate dimeglumine as described.16 CELs were quantified according to the consensus of two neurologists with neuroimmunology subspecialty training (B.B. and M.K.) based on precontrast T1‐ and T2‐weighted images.
CSF collection, processing, and immunophenotyping
CSF was collected on ice and processed according to written standard operating procedures.17 A portion of CSF was sent to measure IgG index and presence of oligoclonal bands (OCBs) by isoelectric focusing. Research CSF aliquots were transferred to the Neuroimmunological Diseases Unit (NDU) laboratory, assigned alpha‐numeric codes, and centrifuged at 335 g for 10 min at 4°C within 15 min of collection. A minimum of 1 × 104 viable CSF cells were analyzed immediately by 12‐color flow cytometry to enumerate absolute numbers of 14 subsets of CSF immune cells as described.17 The CSF supernatant was aliquotted and stored in polypropylene tubes at −80°C until blinded analysis of biomarkers.
Measurement of rituximab concentration and other biomarkers
Electrochemiluminescent assays were developed and optimized to quantify the concentrations of selected biomarkers in the serum and CSF using the Meso Scale Discovery® (MSD; Meso Scale Diagnostics, Rockville, MD) system as described.4 The concentration of interleukin (IL)‐12p40 was measured by MSD V‐plex using the manufacturer's protocol. The assays for rituximab, soluble CD21 (sCD21), sCD27, sCD14, B‐cell activating factor (BAFF), and C‐X‐C motif chemokine 13 (CXCL13) were developed in the NDU laboratory. Neurofilament light protein (NFL) concentration was measured using a commercial ELISA (UmanDiagnostics, Umea, Sweden). All samples excluding IL‐12p40 and NFL were run in duplicate. Each assay contained a minimum of two additional reference samples per plate to evaluate intra‐ and inter‐assay reliability. The details of the reagents, manufacturer, detection limits, and intra‐assay coefficients of variance are depicted in Table 1.
Table 1.
Methodological details of biomarker measurements
| Molecule | Manufacturer (Antibodies/Kit) | Serum | CSF | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Dilution factor | Lower detection limita | Intra‐assay variationb | Inter‐assay variationb | Dilution factor | Lower detection limita | Intra‐assay variationb | Inter‐assay variationb | ||
| IL‐12p40 | Meso Scale Diagnostics, Rockville, MD (K15050D) | n.a. | n.a. | n.a. | n.a. | 1 | 1.6 pg/mL | 7.6% | 8.8% |
| sCD21 | R&D Systems (MAB4909, BAF4909) | 200 | 688.7 pg/mL | 3.4% | 4.5% | 2 | 12.2 pg/mL | 2.6% | 4.1% |
| sCD27 | Sanquin, Amsterdam, the Netherlands (M1960) | n.a. | n.a. | n.a. | n.a. | 20 | 0.4 U/mL | 3.6% | 4.3% |
| CXCL13 | R&D Systems (MAB801, BAF801) | 10 | 254.2 pg/mL | 2.3% | 9.7% | 1 | 25.5 pg/mL | 4.8% | 8.1% |
| BAFF | R&D Systems (DY124) | 10 | 11.6 pg/mL | 4.9% | 7.2% | 3 | 2.1 pg/mL | 4.7% | 8.6% |
| Rituximab | AbD Serotec, Oxford, UK (HCA186, MCA2260) | 5 | 83.0 pg/mL | 9.5% | 7.5% | 2 | 37.5 pg/mL | 3.6% | 7.1% |
| NFL | UmanDiagnostics, Umea, Sweden (10‐7001) | n.a. | n.a. | n.a. | n.a. | 2 | 200 pg/mL | 2.9% | 6.0% |
IL‐12p40, interleukin (IL)‐12p40; sCD21, soluble CD21; CXCL13, C‐X‐C motif chemokine 13; BAFF, B‐cell activating factor; NFL, Neurofilament light protein; n.a., not applicable.
When diluted CSF was used, detection limit is recalculated to reflect the utilized concentration factor.
Median value of concentration CV.
In vitro B‐cell surface and intracellular rituximab saturation assay
B cells were isolated by negative selection (MACS Human B cell Isolation Kit II; Miltenyi Biotec, Auburn, CA) from fresh peripheral blood mononuclear cell (PBMC) of three healthy donors. Purified B cells were then rested overnight at 37°C and 5% CO2 in X‐VIVO 15 media (Lonza) and 1 × 105 of B cells were cultured 1, 2, 4 h, or overnight without or with rituximab concentration of either 20 ng/mL (average concentration measured in CSF), 1 μg/mL (average concentration measured in serum), or 10 μg/mL. Subsequently, cells were incubated with fluorescently conjugated antibody against rituximab (MB2A4; AbD Serotec, Oxford, UK) either for surface or intracellular staining, washed, and analyzed on a LSR II flow cytometer (BD Biosciences, San Diego, CA, USA). Mean fluorescent intensity (MFI) was measured on the CD19‐gated B‐cell population.
In vitro NK cell cytotoxicity assay
NK cells and B cell subtypes were isolated by negative selection (MACS Human NK cell Isolation Kit; MACS Human naïve B cell Isolation Kit II and switched memory B cell isolation kit; Miltenyi Biotec) from fresh PBMC of three healthy donors. NK cells were either used without further purification, or stained with fluorescently conjugated antibody against CD3 (UCHT1), CD44 (G44‐26), CD56 (B159; all from BD Biosciences), and sorted into CD3‐/CD56dim or CD3‐/CD56bright NK cells. Purified cells were rested overnight and 5 × 104 of each cell type were seeded with equal number of either naïve B cells, memory B cells, or K562 cells with CD107a/b antibody (H4A3/H4B4; BD Biosciences) present during co‐culture period, in the presence of 20 ng/mL or 1 μg/mL of rituximab. Control wells included effectors (NK cells only) without targets (B cells or K562 cells) and targets without effectors. MHC‐I‐negative K562 cells served as a positive control for NK cytotoxicity. After 6 h, the cells were stained for surface expression of CD19 (SJ25‐C1; Life Technologies), dead cells (LIVE/DEAD fixable dead cell stain kit; Life Technologies), CD56 (B159; BD Biosciences) and intracellular perforin (delta G9; eBiosciences, San Diego, CA, USA) and analyzed by flow cytometer. NK cell degranulation (CD107a/b+) and perforin release (perforin−) were compared between NK cells cultured without and with different targets. Absolute numbers of live CD19 + B cells were acquired using fluorescent beads for normalization.5 Cytotoxicity was calculated as: % cytotoxicity = (live B cell numbers in the sample/live B cell numbers in B cell control) × 100.
In vitro B‐cell cytotoxicity assay
To determine whether memory B cells (dominant B cell subtype in the CSF) are more resistant to rituximab‐driven depletion than naïve B cells, and also whether B cells under rituximab treatment are predominantly killed by complement‐dependent cytotoxicity (CDC) versus antibody‐dependent cellular cytotoxicity (ADCC), 1 × 105 of purified naïve B cells and memory B cells were cultured 4 h with rituximab (1 μg/mL) or a control antibody (daclizumab 1 μg/mL; Roche) in the presence of 50% of serum (SSTTM Serum Separation Tubes; BD Vacutainer®, Franklin Lakes, NJ, USA) or 50% of pooled MS CSF samples (to assure reproducible conditions for all experiments). Subsequently, cells were incubated with fluorescently conjugated antibody against CD19 and dead cells, and analyzed by a flow cytometer. Live B cells were gated on CD19 and dead cell staining, and their relative numbers were calibrated with fluorescent beads.5
Statistical analyses
Repeated measures analysis of variance (ANOVA) was used to evaluate the change in biomarker and clinical score variables from baseline to the follow‐up visits. For both CSF and blood markers, the average of Mo ‐12 and Mo 0 was used as baseline. For clinical score, the average of Mo ‐12, Mo ‐6, and Mo 0 was used as baseline. Dunnett's method was used to adjust for multiple comparisons with the baseline. There were three follow‐up visits for CSF biomarkers: 1.5, 3, and 12 months, eight follow‐up visits for blood markers: 0.03, 0.5, 0.53, 1.5, 1.53, 3, 12, and 12.03 months, and four follow‐up visits for clinical scores: 6, 12, 18, and 24 months. For blood markers, Mo 12.03 was also compared with Mo 12 with Tukey method. The above analyses were applied to two groups separately: active‐treatment group and placebo group. Box–Cox transformation was applied to biomarker and clinical score variables. For cytotoxicity assays, paired t‐test was performed to compare two culture conditions. SAS 9.2 (SAS Institute Inc., Cary, NC, USA) was used for the statistical analyses.
Results
From September 14, 2010 to February 20, 2015, 43 patients were assessed for eligibility and 27 patients were randomly assigned to receive either rituximab (n = 18) or placebo (n = 9; Fig. 1B). Of the 18 patients assigned to a treatment group, 14 received at least two doses of intrathecal rituximab and were included in the interim analysis as per protocol. Baseline characteristics were similar for both groups (Table 2).
Table 2.
Demographic of subjects
| Placebo | Active treatment | |
|---|---|---|
| Patients (n) | 9 | 14 |
| Sex (% females) | 77.8% | 50.0% |
| Age at baseline, years | 60.1 (39.3–64.8) | 55.2 (42.0–66.0) |
| Disease duration at baseline, years | 26.0 (10.4–43.6) | 24.4 (16.5–38.5) |
| Total duration of follow‐up, months | 30.0 (18.0–36.0) | 24.0 (13.5–36.0) |
| EDSS at baseline | 6.5 (5.0–6.5) | 6.5 (2.5–7.0) |
| IgG index at baseline | 0.71 (0.46–1.8) | 0.86 (0.40–2.36) |
| OCB positivity at baseline | 88.9% (8/9) | 100% (14/14) |
| CEL positivity (Mo −12/Mo 0) | 25.0%/11.1% | 14.3%/0% |
Data are median (range), unless otherwise stated.
EDSS, expanded disability status scale; OCB, oligoclonal bands; CEL, contrast‐enhancing lesion in MRI; Mo, month.
Selected dosing leads to measurable CSF concentrations of free rituximab, sustained for several months
Free (i.e., non‐cell bound) rituximab levels were measured by sensitive electrochemiluminescence ELISA. In the serum, we observed a significant increase in rituximab levels 1 day after each intravenous dose (Mo 0.03 and Mo 0.53) in the active‐treatment patients only (Fig. 2A). Consequently, serum rituximab levels were significantly elevated at months 0.5, 1.5, and 3, but had returned to baseline at Mo 12. Measured serum levels of free rituximab in this study were lower (average 1036 ng/mL, median 822 ng/mL) in comparison to reported levels of rituximab under oncology dosing (i.e., 375 mg/m2 IV × 4; Cmax 486 μg/mL).18 Lower concentrations can be explained by ≥fivefold lower intravenous dose in this protocol and by the fact that postinfusion concentrations were measured >12 h after completion of the infusion (which is expected to significantly diminish Cmax based on reported serum rituximab half‐life of 76.3 h18). Rituximab serum concentrations also significantly increased after intrathecal dose administrated at Mo 12 (Fig. 2A), consistent with neonatal Fc‐receptor (n‐FcR)‐mediated efflux of CSF immunoglobulins to the blood.11, 19
Figure 2.
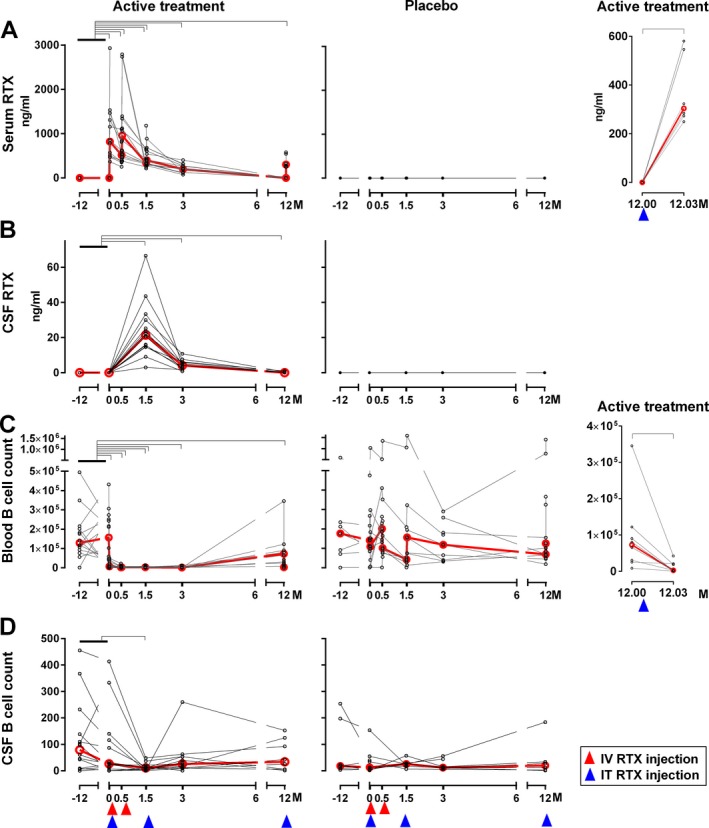
Intrathecal dosing leads to measurable cerebrospinal fluid (CSF) concentrations of rituximab sustained for several months, but depletion of CSF B cells is incomplete and transient. Free rituximab (RTX) levels in the serum (A) and CSF (B) were measured in coded samples (active‐treatment cohort: n = 14, placebo cohort: n = 9) using electrochemiluminescence assay. Absolute number of CD19‐positive B cells in blood (C) and CSF (D) were counted by flow cytometry. Red lines show median values of biomarkers at each follow‐up visit. Red and blue arrows show the timing of intravenous (IV) or intrathecal (IT) injection of rituximab or placebo, respectively. Black brackets represent statistical significance (P < 0.05) based on adjusted P‐value (Dunnett's method). Concentrations of rituximab and counts of B cells in blood and CSF for the active‐treatment cohort (left panels) and placebo (middle panels) were compared to baseline (average of visit Months ‐12 and 0) and each follow‐up visit (serum: Months 0.5, 0.53, 1.5, 1.53, 3, 12, and 12.03; CSF: Months 1.5, 3, and 12). Additionally, concentration of rituximab in serum and absolute numbers of the blood B cell number (right panels) for the active‐treatment cohort was compared between visit Months 12 and 12.03 (Tukey's method).
In the CSF, we observed a significant increase in rituximab levels in the active‐treatment cohort only, at months 1.5, 3, and 12 (Fig. 2B). CSF rituximab levels (average 24 ng/mL, median 21 ng/mL) were approximately 50‐fold lower in comparison to serum levels, which still represent a 20‐fold increase in bioavailability of rituximab in the intrathecal compartment in comparison to intravenous administration only (i.e., 2% of serum values measured in current study vs. 0.1% of serum values achievable without intrathecal dosing10).
While depletion of peripheral B cells is complete and lasting, depletion of CSF B cells is incomplete and transient
Consistent with the measurable levels of cell‐free rituximab in both compartments, we observed significant depletion of blood and CSF B cells from patients on active therapy only (Fig. 2C, D). While B‐cell depletion from the blood was almost complete (median −98.79% at Mo 3; P < 0.0001) and sustained for at least 12 months (at which point blood levels started to re‐populate with naïve B cells), CSF B‐cell levels decreased significantly only at Mo 1.5 (median −79.71%; P = 0.0176).
Interestingly, the efflux of intrathecally administered rituximab to systemic circulation at Mo 12.03 (i.e., one day after intrathecal injection) lead to a measurable decrease in the absolute numbers of blood B cells (Fig. 2C), demonstrating that the resultant concentration of rituximab in the serum (median 304 ng/mL) has a robust B‐cell‐depleting efficacy in the peripheral circulation.20
Significantly greater and lasting increase of serum BAFF in comparison to mild/transient increase in CSF BAFF levels is consistent with partial depletion of CNS B cells
As BAFF is consumed by B cells and plasma cells, we observed significant increase (median +141.86%; P < 0.0001) at Mo 3 in serum BAFF levels in the active‐treatment group only (Fig. 3A) consistent with published literature.21 Although CSF levels of BAFF also increased (by median of +8.39% at Mo 3, P = 0.0164), it did not reach the protocol‐stipulated predetermined threshold of at least 50% increase, required for study continuation.
Figure 3.

Soluble B‐cell‐related biomarkers corroborate robust and lasting depletion of blood B cells, but poor depletion of central nervous system B cells by rituximab (RTX) treatment. Concentration of serum and cerebrospinal fluid (CSF) B‐cell activating factor (BAFF) (A), C‐X‐C motif chemokine 13 (CXCL13) (B), and soluble CD21 (sCD21) (C) were measured in coded samples (active‐treatment cohort: n = 14, placebo cohort: n = 9; placebo data not shown) using electrochemiluminescence assay. Red lines show median values of biomarkers at each follow‐up visit. Red and blue arrows show the timing of intravenous (IV) or intrathecal (IT) injection of RTX. Black brackets represent statistical significance (P < 0.05) based on adjusted P‐value (Dunnett's method). Concentrations of BAFF, CXCL13, and sCD21 were compared between baseline (average of visit Months ‐12 and 0) and each follow‐up visit (serum: Months 0.5, 0.53, 1.5, 1.53, 3, 12, and 12.03; CSF: Months 1.5, 3, and 12).
CSF levels of CXCL13 are not affected by intrathecal rituximab therapy
In the serum, we observed a significant decrease in the levels of CXCL13 at Mo 1.5 (median −31.51%, P = 0.0039) and Mo 3 (median −58.43%, P = 0.0014), but the CSF levels of this chemokine remained unchanged in the rituximab‐treated patients, even in those with the highest CSF CXCL13 levels (Fig. 3B). This second protocol prespecified criterion for continuation of study was therefore not fulfilled either.
Measured decreases in B‐cell‐specific marker, sCD21, corroborates significantly higher efficacy of B‐cell‐depletion in peripheral as compared to CNS
CD21 is a B‐cell‐specific shed surface marker, which is expressed on 100% of naïve B cells, but its surface expression decreases during the B‐cell differentiation process.4 Therefore, while it is not a sensitive diagnostic marker (i.e., normal CSF sCD21 levels do not rule out an increase in intrathecal B cells), its cell specificity for B cells makes it an excellent pharmacodynamic marker of B‐cell‐depleting therapies. We observed a significant decrease in serum sCD21 levels (median of −56.02% at Mo 0.5; P < 0.0001, median of −32.69% at Mo 3; P < 0.0001; Fig. 3C) and a lower, but still highly significant, decrease in CSF sCD21 levels (maximum decrease with median of −28.02% at Mo 3, P < 0.0001). Serum levels of sCD21 returned to pretreatment baseline values at Mo 12, consistent with observed repopulation of the blood by naïve B cells. CSF levels of sCD21 also returned to pretreatment baseline at Mo 12. The ratio of sCD21 per B cell in the CSF, which is a measure of B cells compartmentalized to the CNS tissue,4 increased to median of 424.12% at Mo 1.5 (P = 0.0045; data not shown). This result demonstrates that intrathecal rituximab preferentially eliminated the mobile (CSF) pool of B cells, while CNS tissue‐embedded B cells were not affected.
B cells under rituximab therapy are killed by CDC, and to a lesser degree by ADCC. Low CSF levels of rituximab and lytic complement and low proportion of cytotoxic CD56dim NK cells collectively underlie poor B‐cell depletion in the CNS
The afore‐mentioned experiments demonstrated that despite the lasting measurable concentrations of free rituximab in the CSF, intrathecal B cells were only partially and transiently depleted. We considered and experimentally tested several possible explanations of this phenomenon: (1) Because of proportional predominance of immunoregulatory CD56bright NK cells in the CSF as compared to blood17, we asked whether CD56dim and CD56bright NK cells differ in their ability to mediate ADCC of B cells under rituximab therapy; (2) Because of predominance of memory B cells in the CSF,22 we asked whether memory B cells are more resistant to rituximab‐driven depletion than naïve B cells; and finally, (3) Because of the controversy whether B cells under rituximab treatment are predominantly killed by CDC versus ADCC,23 we compared these two cytotoxic modalities side by side (Fig. 4).
Figure 4.
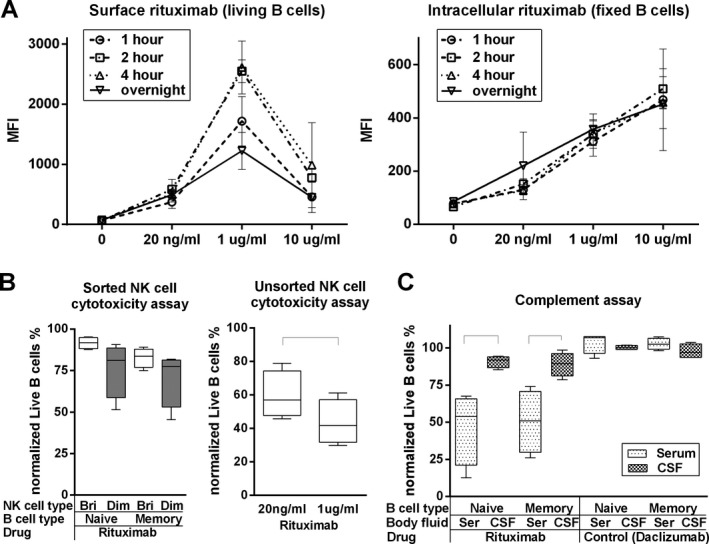
The low levels of complement components in the cerebrospinal fluid (CSF) underlies poor B‐cell depletion in the intrathecal compartment. (A) In vitro B‐cell surface and intracellular rituximab saturation assay. Purified B cells were cultured in the absence or presence of different concentrations of rituximab (10 μg/mL, 1 μg/mL [average serum concentration in current study], 20 ng/mL [average CSF concentration in current study], using time‐assay (1, 2, 4 h or overnight). Surface or intracellular rituximab mean fluorescence intensity (MFI) of living or fixed B cells was measured by flow cytometry. Error bars represent standard deviation (SD). (B left) In vitro NK cell cytotoxicity assay. Sorted CD56dim or CD56bright NK cells and negatively purified naïve and memory B cells were obtained. Each NK cell type was seeded with equal number of either Naïve B cells or Memory B cells for 6 h, in the presence of 1 μg/mL of rituximab. Cytotoxicity of NK cells to B cells was calculated as: % cytotoxicity = (relative B cell numbers in the sample/relative B cell numbers in B cell control) × 100. (B right) In vitro rituximab dose‐dependent NK cell cytotoxicity assay. Negatively selected NK cells were cultured for 6 h with equal number of negatively selected B cells, in the presence of either 1 μg/mL or 20 ng/mL of rituximab. Cytotoxicity of NK cells to B cells was determined by the % cytotoxicity formula. Black brackets represent statistical significant (P < 0.05) difference between the two conditions based on the paired t‐test. (C) In vitro B cell cytotoxicity assay. Negatively purified naïve and memory B cells was cultured 4 h with rituximab (1 μg/mL) or a control antibody (daclizumab: 1 μg/mL) in the presence of 50% serum or pooled multiple sclerosis CSF samples (to assure identical conditions for all donors). Cytotoxicity of NK cells to B cells was determined by the % cytotoxicity formula. Black brackets represent statistical significant (P < 0.05) difference between the two conditions based on the paired t‐test.
We observed that average measured CSF concentrations of rituximab (20 ng/mL) did not saturate CD20 on B cells, while the average measured concentration in the blood (1 μg/mL) did (Fig. 4A left panel). Our data also demonstrated that intravenous dose selected for this trial (i.e., 200 mg × 2) may be closer to the optimal dose for non‐oncologic indications than standardly utilized 1000 mg × 2 dose because the 10 μg/mL concentration (which is still below peak concentrations achievable by oncology dosing18) paradoxically decreased the measurable rituximab concentration on the surface of B cells, due to higher internalization of CD20/rituximab complexes (Fig. 4A right panel).
CD56dim NK cells killed rituximab‐coated B cells more efficiently in comparison to CD56bright NK cells (Fig. 4B; left panel). This observation is consistent with higher levels of perforin (data not shown) and Fc‐receptors24 on CD56dim as compared to CD56bright NK cells. However, the level of ADCC was overall mild and did not reach statistical significance. Because FACS‐sorted CD56bright & CD56dim NK cells did not efficiently kill even MHC‐I‐deficient K562 controls (data not shown), we investigated whether sorting inhibited NK cell function; indeed, we observed robust killing of B cells by unsorted (only purified by negative selection) NK cells (Fig. 4B; right panel), which was significantly higher (P = 0.0001) for blood (1 μg/mL) in comparison to CSF (20 ng/mL) rituximab concentrations.
Nevertheless, the most efficient killing of rituximab‐coated B cells was observed by CDC in the serum, but not CSF (Fig. 4C). Killing was comparable for naïve and memory B cells (P = 0.0453 and 0.0223, respectively). In contrast, when B cells were pretreated with control antibody (daclizumab; which is a nondepleting humanized antibody against CD25)24, B cell killing was not observed (Fig. 4C).
Combination of intravenous and intrathecal rituximab leads to significant, but quantitatively modest inhibition of MS‐related intrathecal T‐cell‐mediated inflammation, which does not affect CSF biomarker of axonal damage
Because an early termination of clinical trials is often associated with the ambiguity of a potential final outcome of fully executed study, we investigated the effect of rituximab on three additional CSF biomarkers that have been previously validated as biomarkers of MS‐related inflammation (i.e., sCD27, a biomarker of activated T cells4; IL‐12p40 mainly produced by activated cells of myeloid lineage and by activated B cells4) or axonal damage (i.e., neurofilament light chain; NF‐L25; data not shown). In rituximab‐treated patients, we observed significant inhibition of IL‐12p40 (median −42.42%, P = 0.0110 at Mo 3) as well as sCD27 (−10.97%, P = 0.0005 at Mo 3; −29.28%, P = 0.0024 at Mo 12), in the active therapy group only. On the other hand, CSF levels of NF‐L remained unchanged.
Clinical scores worsened during the observation period for both placebo and treated groups
Early termination makes the acquired clinical and imaging data insufficient (i.e., underpowered) for reliable analyses. Therefore, we present descriptive statistics (Fig. 5) and conclude that clinical scales were comparable between placebo and rituximab groups, while CEL counts on brain MRI remained low with trend toward their inhibition on active treatment.
Figure 5.

Clinical scores showed tendency for worsening during the observation period for both treated and placebo groups. (A) Clinical scores of expanded disability status scale (EDSS), Scripps neurological rating scale (NRS), and multiple sclerosis functional composite (MSFC) for patients in active treatment and placebo cohorts. (B) Average cumulative contrast‐enhancing lesion (CEL) counts on brain MRI for patients in active treatment and placebo cohorts.
Discussion
Current immunomodulatory treatments have limited efficacy in progressive MS, especially when patients no longer experience relapses and CELs,8, 26 although CNS inflammation remains quantitatively identical to inflammation in RRMS.4 This apparent discrepancy has two possible explanations, not mutually exclusive: 1. Compartmentalized CNS inflammation in progressive MS4 is not amenable to inhibition by current DMTs; and/or 2. The CNS inflammation, while present, no longer drives clinical disability. Though we discussed evidence supporting the first alternative2, 4, 5 in the introduction, the second alternative can be proven only by achieving a substantial inhibition of the intrathecal inflammation (ideally its complete normalization) and observing no efficacy on clinical outcomes in adequately powered studies.
Consequently, the rationale behind the RIVITALISE trial rested in the ability of intravenous rituximab to significantly decrease CSF B‐cell and T‐cell counts27 and inhibit MS relapses28 in patients with RRMS who have opened BBB, but having no effect on CSF B‐cell counts29 and disability8 in PPMS patients, who have intact BBB. Therefore, we hypothesized that combination of intravenous and intrathecal administration of rituximab will lead to an effective depletion of B cells from both systemic and CNS compartments, leading to inhibition of all MS‐related inflammation. Only if this hypothesis was correct, we could answer the most important question: whether successful inhibition of intrathecal inflammation in progressive MS decreases CNS tissue destruction and accumulation of disability. It is with this understanding that we incorporated an interim analysis for the efficacy of B‐cell depletion in the RIVITALISE trial and related trial stopping criteria for futility.
The main limitation of this approach is uncertainty whether restricted numbers of patients in the interim analysis can provide unequivocal evidence for intrathecal efficacy. While we acknowledge this drawback, we believe that our selection of multiple, biologically complementary biomarkers of B‐cell lineage and our measurements of congruent results for all of these biomarkers provide confidence to our conclusion that rituximab has significantly lower efficacy in the CNS, as compared to blood.
Use of complementary biomarkers that are either specifically released by B cells (sCD21), or enriched in meningeal lymphoid follicles (CXCL13)2 where it is predominantly produced by myeloid cells and activated B cells,4 or that are consumed by B cells and plasma cells (BAFF), provided unequivocal evidence for almost complete and lasting depletion of B cells from the peripheral blood, but only partial and transient depletion of B cells from the intrathecal compartment. The treatment‐induced rise in sCD21/CSF B‐cells ratio indicates that B cells were insufficiently depleted from CNS tissue,4 which is supported by the limited diffusion of rituximab in the brain parenchyma.30
Comparing the efficacy of CDC versus ADCC on the same mixture of effector/target cells identified CDC as dominant form of B‐cell killing by rituximab. These results are consistent with published observations that polymorphism in the complement component C1qA correlates with the efficacy of rituximab in B‐cell malignancies31 and that addition of fresh frozen plasma augments therapeutic efficacy of rituximab by providing additional complement.32 The lack of components capable to assemble the lytic terminal complement complex in the CNS, together with a lower ADCC underlies the poor CNS efficacy of rituximab in the subjects with intact BBB (Fig. 6). Proportional predominance of regulatory CD56bright NK cells over cytotoxic CD56dim NK cells in the CSF17 is probably the most important factor for diminished ADCC in the intrathecal compartment, as CD56bright NK cells either lack, or express significantly lower levels of Fc receptors in comparison to CD56dim NK cells.24 However, because the NK cells integrate inhibitory (i.e., MHC‐I/killer inhibitory receptors) and activating (e.g., Fc receptor‐ and cytokine‐mediated) signals when deciding to kill, the lack of CD20 saturation, as well as lower rituximab‐induced “cytokine storm” likely further inhibits ADCC in the CSF (Fig. 6).
Figure 6.

Schematic representation of differences between blood and intrathecal compartment that underlie poor efficacy of rituximab in depleting CNS B cells. CSF, cerebrospinal fluid; CNS, central nervous system; NK cell, natural killer cell.
The observed insufficient depletion of B cells from the CNS in RIVITALISE trial is supported by in vivo observations from neurological diseases such as anti‐NMDA encephalitis, where complement‐binding (i.e., IgG1) anti‐NMDA antibodies present in large quantities in the CSF, and CNS tissue do not cause cytotoxicity, but rather lead to internalization of NMDA molecules from the neuronal surface.33 In contrast, opening of the BBB (seen in RRMS patients and CNS B‐cell malignancies) that produces focally high concentrations of the complement may be a prerequisite for efficient depletion of B cells under rituximab therapy, but also to cause antibody‐mediated CNS damage under pathological conditions such as neuromyelitis optica.
This conclusion has far‐reaching consequences: as long as efficacy of therapeutic antibodies depends on CDC or ADCC, administration of a drug to the CSF will not lead to a substantial efficacy under situations with closed BBB. Therefore, while administration of antibodies to the CSF is feasible and relatively well tolerated (and potentially can be used as long‐term therapy via CSF pumps), antibody‐mediated efficacy needs to rely on different mechanism(s) to exert a potent therapeutic effect in CNS diseases with intact BBB. We expect that supplementing complement in addition to therapeutic antibodies may not be a viable option due to possible side effects to vital CNS tissue. Our results also highlight important role for n‐FcR mediated active efflux of antibodies from the intrathecal compartment to blood and suggest that engineered modifications of antibody structure that minimize its binding to n‐FcR can greatly increase achievable concentrations and half‐life of intrathecally administered antibodies in the CSF/CNS, while limiting (often undesired) systemic exposure.
In summary, implementation of biomarker studies to RIVITALISE trial led to its early termination based on the assessment that lower‐than‐expected depletion of intrathecal B cells and resultant limited inhibition of MS‐related inflammation is insufficient to translate to potential clinical efficacy in small Phase II trial. This study provides proof that incorporating biomarker assays that measure pharmacodynamic effects on the crucial pathogenic pathways in the CNS tissue makes drug development more efficient, by providing essential information for the funders to decide whether to proceed to expensive Phase III trial(s) and how to power these trials properly in order to assure positive results.
Author contributions
BB designed and supervised the study. IC, JO, JC, and BB participated in the enrollment of patients. MK, YL, IC, AB, JO, JC, DM, and PK acquired data. MK analyzed data. TK did the statistical analysis. MK and BB did literature search, created figures, and interpreted data. BB acquired funding. MK and BB wrote the manuscript and all authors reviewed the final version.
Conflicts of Interest
Dr. Bielekova is a co‐inventor on NIH patent related to daclizumab therapy and as such has received patent royalty payments.
Supporting information
Table S1. Review of published literature describing previous experience with intrathecal rituximab dosing that led to the selection of dosing regimen for current study
Acknowledgments
The study was funded by the Intramural Research program of the National Institute of Neurological Disorders and Stroke (NINDS) of the National Institutes of Health (NIH). We thank Dr. Rubenstein (UCSF, San Francisco, USA) for the ability to cross‐reference his IND for intrathecal rituximab administration. We thank Dr. Judy Starling from NIH pharmacy for supervising randomization and drug preparation, our DSMB members Mitchell T. Wallin, MD (DSMB chair; the MS Center of Excellence, East, VA, Baltimore, MD), Walter Royal, MD (the MS Center of Excellence, East, VA, Baltimore, MD) and Adrian Wiestner, MD, PhD (NHLBI, NIH) for trial monitoring, Alison Wichman, MD, research nurse Jenifer Dwyer, regulatory nurse Rosalind Hayden and patient schedulers Anne Mayfield and Kewounie Pumphrey for help with patient care and patient scheduling, and Elena Romm for processing of CSF samples. Finally, we thank the patients and their caregivers without whom this study would not be possible.
Reference
- 1. Stys PK, Zamponi GW, van Minnen J, Geurts JJ. Will the real multiple sclerosis please stand up? Nat Rev Neurosci 2012;13:507–514. [DOI] [PubMed] [Google Scholar]
- 2. Magliozzi R, Howell O, Vora A, et al. Meningeal B‐cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007;130(Pt 4):1089–1104. [DOI] [PubMed] [Google Scholar]
- 3. Frischer JM, Bramow S, Dal‐Bianco A, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009;132(Pt 5):1175–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Komori M, Blake A, Greenwood M, et al. CSF markers reveal intrathecal inflammation in progressive multiple sclerosis. Ann Neurol 2015;78:3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wuest SC, Mexhitaj I, Chai NR, et al. A complex role of herpes viruses in the disease process of multiple sclerosis. PLoS ONE 2014;9:e105434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Metz I, Lucchinetti CF, Openshaw H, et al. Autologous haematopoietic stem cell transplantation fails to stop demyelination and neurodegeneration in multiple sclerosis. Brain 2007;130(Pt 5):1254–1262. [DOI] [PubMed] [Google Scholar]
- 7. Reff ME, Carner K, Chambers KS, et al. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood 1994;83:435–445. [PubMed] [Google Scholar]
- 8. Hawker K, O'Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double‐blind placebo‐controlled multicenter trial. Ann Neurol 2009;66:460–471. [DOI] [PubMed] [Google Scholar]
- 9. Polman CH, Reingold SC, Edan G, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann Neurol 2005;58:840–846. [DOI] [PubMed] [Google Scholar]
- 10. Rubenstein JL, Combs D, Rosenberg J, et al. Rituximab therapy for CNS lymphomas: targeting the leptomeningeal compartment. Blood 2003;101:466–468. [DOI] [PubMed] [Google Scholar]
- 11. Rubenstein JL, Fridlyand J, Abrey L, et al. Phase I study of intraventricular administration of rituximab in patients with recurrent CNS and intraocular lymphoma. J Clin Oncol 2007;25:1350–1356. [DOI] [PubMed] [Google Scholar]
- 12. Maloney DG, Liles TM, Czerwinski DK, et al. Phase I clinical trial using escalating single‐dose infusion of chimeric anti‐CD20 monoclonal antibody (IDEC‐C2B8) in patients with recurrent B‐cell lymphoma. Blood 1994;84:2457–2466. [PubMed] [Google Scholar]
- 13. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 1983;33:1444–1452. [DOI] [PubMed] [Google Scholar]
- 14. Sharrack B, Hughes RA. Clinical scales for multiple sclerosis. J Neurol Sci 1996;135:1–9. [DOI] [PubMed] [Google Scholar]
- 15. Cutter GR, Baier ML, Rudick RA, et al. Development of a multiple sclerosis functional composite as a clinical trial outcome measure. Brain 1999;122(Pt 5):871–882. [DOI] [PubMed] [Google Scholar]
- 16. Bielekova B, Kadom N, Fisher E, et al. MRI as a marker for disease heterogeneity in multiple sclerosis. Neurology 2005;65:1071–1076. [DOI] [PubMed] [Google Scholar]
- 17. Han S, Lin YC, Wu T, et al. Comprehensive immunophenotyping of cerebrospinal fluid cells in patients with neuroimmunological diseases. J Immunol 2014;192:2551–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maloney DG, Grillo‐Lopez AJ, Bodkin DJ, et al. IDEC‐C2B8: results of a phase I multiple‐dose trial in patients with relapsed non‐Hodgkin's lymphoma. J Clin Oncol 1997;15:3266–3274. [DOI] [PubMed] [Google Scholar]
- 19. Schlachetzki F, Zhu C, Pardridge WM. Expression of the neonatal Fc receptor (FcRn) at the blood‐brain barrier. J Neurochem 2002;81:203–206. [DOI] [PubMed] [Google Scholar]
- 20. Svenningsson A, Bergman J, Dring A, et al. Rapid depletion of B lymphocytes by ultra‐low‐dose rituximab delivered intrathecally. Neurol Neuroimmunol Neuroinflamm 2015;2:e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pranzatelli MR, Tate ED, Travelstead AL, Verhulst SJ. Chemokine/cytokine profiling after rituximab: reciprocal expression of BCA‐1/CXCL13 and BAFF in childhood OMS. Cytokine 2011;53:384–389. [DOI] [PubMed] [Google Scholar]
- 22. Cepok S, Rosche B, Grummel V, et al. Short‐lived plasma blasts are the main B cell effector subset during the course of multiple sclerosis. Brain 2005;128(Pt 7):1667–1676. [DOI] [PubMed] [Google Scholar]
- 23. Edwards JC, Cambridge G. B‐cell targeting in rheumatoid arthritis and other autoimmune diseases. Nat Rev Immunol 2006;6:394–403. [DOI] [PubMed] [Google Scholar]
- 24. Bielekova B, Catalfamo M, Reichert‐Scrivner S, et al. Regulatory CD56bright natural killer cells mediate immunomodulatory effects of IL‐2R‐alpha‐targeted therapy (daclizumab) in multiple sclerosis. Proc Natl Acad Sci 2006;103:5941–5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Teunissen CE, Khalil M. Neurofilaments as biomarkers in multiple sclerosis. Mult Scler 2012;18:552–556. [DOI] [PubMed] [Google Scholar]
- 26. Kappos L, Weinshenker B, Pozzilli C, et al. Interferon beta‐1b in secondary progressive MS: a combined analysis of the two trials. Neurology 2004;63:1779–1787. [DOI] [PubMed] [Google Scholar]
- 27. Cross AH, Stark JL, Lauber J, et al. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol 2006;180:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hauser SL, Waubant E, Arnold DL, et al. B‐cell depletion with rituximab in relapsing‐remitting multiple sclerosis. N Engl J Med 2008;358:676–688. [DOI] [PubMed] [Google Scholar]
- 29. Monson NL, Cravens PD, Frohman EM, et al. Effect of rituximab on the peripheral blood and cerebrospinal fluid B cells in patients with primary progressive multiple sclerosis. Arch Neurol 2005;62:258–264. [DOI] [PubMed] [Google Scholar]
- 30. Wolak DJ, Pizzo ME, Thorne RG. Probing the extracellular diffusion of antibodies in brain using in vivo integrative optical imaging and ex vivo fluorescence imaging. J Control Release 2015;197:78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Racila E, Link BK, Weng WK, et al. A polymorphism in the complement component C1qA correlates with prolonged response following rituximab therapy of follicular lymphoma. Clin Cancer Res 2008;14:6697–6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Klepfish A, Rachmilewitz EA, Kotsianidis I, et al. Adding fresh frozen plasma to rituximab for the treatment of patients with refractory advanced CLL. QJM 2008;101:737–740. [DOI] [PubMed] [Google Scholar]
- 33. Linnoila JJ, Rosenfeld MR, Dalmau J. Neuronal surface antibody‐mediated autoimmune encephalitis. Semin Neurol 2014;34:458–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Review of published literature describing previous experience with intrathecal rituximab dosing that led to the selection of dosing regimen for current study