

Abstract
During the past six years, researchers have made major progress identifying common inherited genetic variation that increases risk for primary adult glioma. This paper summarizes knowledge about rare familial cancer syndromes that include adult glioma and reviews the available literature on the more recently discovered common inherited variation. Ten independent inherited variants in eight chromosomal regions have been convincingly associated with increased risk for adult glioma. Most of these variants increase relative risk of primary adult glioma by 20% to 40%, but the TP53 variant rs78378222 confers a two-fold relative risk (ie, 200%), and rs557505857 on chromosome 8 confers a six-fold relative risk of IDH-mutated astrocytomas and oligodendroglial tumors (ie, 600%). Even with a six-fold relative risk, the overall risk of developing adult glioma is too low for screening for the high-risk variant on chromosome 8. Future studies will help clarify which inherited adult glioma risk variants are associated with subtypes defined by histology and/or acquired tumor mutations. This review also provides an information sheet for primary adult glioma patients and their families.
Keywords: epidemiology, genetics, primary adult glioma, risk, variants
People are born with substantial inherited variation in their genomes (DNA) and epigenomes (methylation and regulatory molecules). Such variation in the DNA nucleotide sequence(s) are called inherited or germline variants. Over a lifespan, cells acquire additional variation from mutations, methylation, and structural or other molecular errors that are not repaired during cell division; these are called somatic alterations or mutations. Cancers may arise when genomic and epigenomic variation and acquired mutations in a cell cause it to divide and multiply without the usual controls. Variation leading to cancers can be inherited and/or acquired. In the field of cancer genetics, the roles of both inherited variation and acquired mutation are studied.
Three sets of ground-breaking discoveries involving inherited and acquired genomic variation have transformed concepts about gliomagenesis in adults: (i) common inherited variants in eight chromosomal regions increase risk of glioma,1–6 (ii) many gliomas have acquired mutations in IDH; these IDH mutations are both prognostic and predictive,7–10 and (iii) many gliomas have acquired mutations in the TERT gene promoter that predict age at diagnosis and outcome when considered in combination with IDH mutation and 1p/19q co-deletion.10–13 In this paper, we review current knowledge about inherited risk for primary adult glioma, specifically, glioblastoma, and grade II/III astrocytoma, oligodendroglioma, and oligoastrocytoma. We also discuss how inherited risk varies by histology and molecular subtypes characterized by acquired mutations. An information sheet is appended to help patients and their families understand these important concepts.
Prior to 2009, a glioma patient's question as to what caused their tumor typically went unanswered. The only established and strong risk factors were high-dose radiation and some very rare familial cancer syndromes (Table 1). However, since 2009, we have learned that 10 inherited variants near 8 genes influence the risk of developing glioma; we call these glioma risk variants or risk loci. While most of these risk variants are common and confer a modest increased risk of glioma, two are uncommon and confer a substantial increased risk. Table 2 provides information about the inherited variants in these 8 regions and they are discussed below.
Table 1.
Rare hereditary cancer syndromes that increase risk of glioma
| Gene | Disorder/Syndrome | Features | Associated Gliomas |
|---|---|---|---|
| NF1 | Neurofibromatosis 1 | Neurofibromas, schwannomas, café-au-lait macules | Astrocytoma, optic nerve glioma |
| NF2 | Neurofibromatosis 2 | Acoustic neuromas, meningiomas, neurofibromas, eye lesions | Ependymoma |
| TSC1, TSC2 | Tuberous sclerosis | Development of multisystem nonmalignant tumors |
Giant cell astrocytoma |
| MSH2, MLH1, MSH6, PMS2 | Lynch syndrome | Predisposition to gastrointestinal, endometrial, and other cancers | Glioblastoma, other gliomas |
| TP53 | Li-Fraumeni syndrome | Predisposition to numerous cancers, especially breast, brain, and soft-tissue sarcoma | Glioblastoma, other gliomas |
| p16/CDKN2A | Melanoma-neural system tumor syndrome | Predisposition to malignant melanoma and malignant brain tumors | Glioma |
| IDH1/IDH2 | Ollier disease/Maffucci syndrome | Development of intraosseous benign cartilaginous tumors, cancer predisposition | Glioma |
| POT1a | Associated with longer telomeres in familial melanoma | Oligodendroglioma |
This table was adapted from reference #14.
aSee reference #15.
Abbreviations: NF1/NF2, neurofibromin 1/2; TSC1/TSC2, tuberous sclerosis 1/2; MLH1, mutL homolog 1; MSH2/MSH6, mutS homolog 2/6; PMS2, postmeiotic segregation increased 2; TP53, tumor protein P53; CDKN2A, cyclin-dependent kinase inhibitor 2A; IDH, isocitrate dehydrogenase; POT1, protection of telomeres 1.
Table 2.
Odds ratios of glioma for confirmed genetic inherited variants
| Risk Variant Location |
Odds Ratioa | |
|---|---|---|
| Chromosome | Nearby Gene | |
| All glioma typesb | ||
| 3 | TERC | 1.2 |
| 5 | TERT | 1.3 |
| 7 | EGFR | 1.2 |
| 8 | CCDC26c | 1.4 |
| 9 | CDKN2B/ANRIL | 1.3 |
| 11 | PHLDB1 | 1.2 |
| 17 | TP53 | 2.4 |
| 20 | RTEL1 | 1.2 |
| Oligodendroglial and IDH-mutated astrocytic tumors | ||
| 8 | CCDC26d | 6.3 |
aOdds ratios are for number of risk alleles. The source of the odds ratios are the following references: TERC [reference #10]; TERT, EGFR, CCDC26 (for all gliomas), CDKN2B/ANRIL, PHLDB1, RTEL1 [reference #6]; TP53 [reference #3]; CCDC26 for oligodendroglial and IDH-mutated astrocytic tumors [reference #19].
bIncludes WHO grades II-IV astrocytoma, oligodendroglioma, oligoastrocytoma.
cvariant is rs4295627.
dvariant is rs55705857.
Abbreviations: TERC, telomerase RNA component; TERT, telomerase reverse transcriptase; EGFR, epidermal growth factor receptor; CCDC26, coiled-coil domain-containing protein 26; CDKN2B, cyclin-dependent kinase inhibitor 2B/ANRIL, antisense noncoding RNA in the INK4 locus; PHLDB1, pleckstrin homology-like domain, family B, member 1; TP53, tumor protein P53; RTEL1, regulator of telomere elongation helicase 1.
That several rare hereditary cancer syndromes greatly increase risk of glioma has been known for many years. These familial syndromes include neurofibromatosis, tuberous sclerosis, Lynch syndrome, Li-Fraumeni syndrome, melanoma-neural system tumor syndrome, and Ollier disease (Table 1).14 Patients diagnosed with a glioma are often asked questions to determine if their disease could be related to one of these syndromes; if warranted, the patient and family may be referred for genetic testing and counseling. The GLIOGENE study of familial glioma also recently identified mutations in POT1 as being the likely cause of oligodendroglioma in some multiply affected families.15 In familial cancer syndromes, the inherited mutations may be quite rare in the population or even completely unique to the affected family. Family members with the mutation are at high risk of developing a cancer (that is, the mutation has high penetrance).
Relative Risks of the New Inherited Adult Glioma Risk Variants
Since 2009, 8 common inherited variants near the genes TERC, TERT, EGFR, CDKN2B, PHLDB1 and RTEL1 have been associated with relatively small increased risk of glioma. These variants have odds ratios, which approximate relative risks, between 1.2 and 1.43,4,6,10,16 (Table 2). This indicates that inheriting one of these variants increases a person's risk of glioma by 20% to 40% compared with a person who did not inherit that variant. These are referred to as low penetrance variants since many people with the variant do not develop a glioma. The estimated lifetime risk of developing glioma is about 4 to 5/100017; a 30% increase in risk translates to a lifetime risk of about 6/1000.
Two less common variants discovered since 2009, one near TP53 and the other near CCDC26, have higher relative risks. People with one risk allele in the TP53 variant (allele C in rs78378222) have a 2.4-fold relative risk of developing a glioma.3 The inherited risk allele on chromosome 8, near CCDC26 (allele G in rs55705857), confers approximately a 6-fold relative risk of developing gliomas with either an oligodendroglial component or an IDH mutation.18,19 With about a 1/1000 lifetime risk for an oligodendroglial or IDH-mutated glioma in the general population, a six-fold relative risk for a person with the CCDC26 risk allele translates to an ∼6/1000 lifetime risk of developing this type of a glioma. In 2013, Enciso-Mora et al18 reported even higher relative risks associated with this variant. However, their estimates relied on imputed rather than directly genotyped data. Caution is needed in applying relative risks to estimate lifetime risks, and the numbers above should be considered rough approximations. However, since the risk of glioma is low, the risk obtained by multiplying the relative risk by the general population risk is a reasonable approximation.20
It is very important to understand that inherited genetic risk variants, whether common or rare, with low or high penetrance, are neither necessary nor sufficient for glioma formation – rather, they contribute to risk. Even for people with familial syndromes, additional acquired mutations are required for tumorigenesis. Moreover, not everyone who inherits the same mutation necessarily develops cancer or the same type of cancer.
Screening for 8q24 Variant is not Recommended
Patients may wonder why the general public are not screened for the chromosome 8 glioma risk variant, rs55705857, even though it confers a six-fold relative risk for some types of glioma. First, as noted above, the risk of gliomas remains only about 6/1000 even with this magnitude of increased relative risk. By comparison, some women are screened for BRCA1 mutations, which also confer about a six-fold relative risk for breast cancer. Because breast cancer is a much more common cancer, the lifetime risk for women who have inherited harmful variants in BRCA1 is about 65/100.21,22 (Fig. 1) Even so, BRCA1 testing is still only recommended for women who have an indication of inherited risk such as being diagnosed at age 45 or younger or having a family history. This is because even with the high lifetime risk, general population screening would identify too many false positives.
Fig. 1.
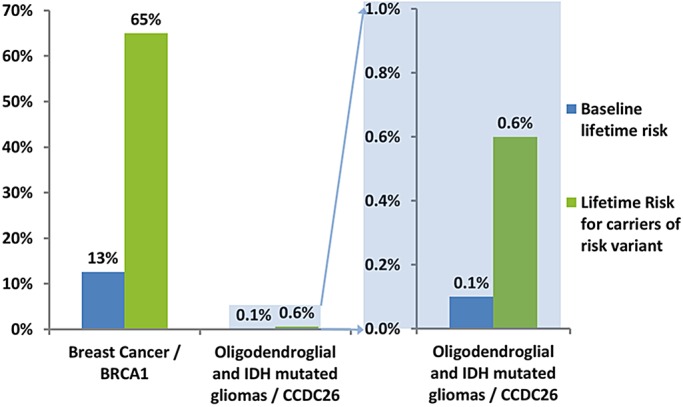
Comparison of lifetime risk for breast cancer among women with and without a risk variant in BRCA122 and lifetime risk for oligodendroglial/IDH mutated gliomas among people with and without the risk variant in rs55705857.19
To further demonstrate why the general population are not screened for the glioma risk variant rs55705857, consider how many people among 1,000,000 would have both the risk variant and glioma versus how many would have the risk variant but not have glioma. Among UCSF AGS (University of California, San Francisco Adult Glioma Study) participants with oligodendroglial or IDH-mutated gliomas, the genotype frequencies for rs55705857 are 65% AA, 32% AG, and 3% GG; in controls the frequencies are 93% AA, 7.0% AG, and <0.05% GG. Using CBTRUS (Central Brain Tumor Registry of the United States) data,23 we estimate that the annual incidence rate for these types of glioma is about 1.5 persons/100,000. So among 1,000,000 people, 5 of 15 people diagnosed with this type of glioma would have the risk allele, but 69,999 of 999,985 without the disease would also have the risk allele. Thus, the vast majority (>99%) of people with the rs55705857 risk allele will not have the disease. Consequently, screening would yield many, many more false positives than true positives.
For glioma screening based on inherited risk variants to be feasible, a subgroup of people at a much higher risk of glioma than the general population would need to be identified for targeted screening. A possible example might be patients with an indeterminate lesion coincidentally identified on MRI. Such lesions are commonly encountered in practice, do not have a specific clinical correlate, and are radiographically nonspecific, but do suggest that an infiltrating glioma should be considered. If such a patient had some combination of relevant risk alleles suggesting an increased risk for glioma well above that of the general population, there could be a compelling argument to consider biopsy or resection of the lesion if it is safe and there is acceptable neurologic morbidity based on location. More detailed studies would be required to determine if screening is warranted in this context.
A second reason for not screening for glioma risk variants is that disease screening is typically performed when an acceptable primary or secondary prevention is available, as is the case for breast, ovarian, and colorectal cancers. For example, a prophylactic mastectomy may be recommended for women who have the BRCA1 risk variant. For glioma, however, even if we could accurately identify who has an increased risk, there is currently no way to prevent the glioma from developing. Thus, knowing one's heritable glioma risk would not lead to any specific recommendations to decrease that risk, but could lead to unwarranted anxiety among those identified as being at increased risk.
Function of Inherited Glioma Risk Variants
The TP53 glioma risk variant (rs78378222) has a frequency of 1% to 2% in individuals without glioma. This variant is also associated with increased risk of basal cell carcinoma, prostate cancer, colorectal adenomas, and neuroblastoma.3,24 rs78378222 occurs in the polyadenylation signal of TP53 and the risk-associated variant disrupts the signal sequence, impairing proper termination and polyadenylation of the TP53 transcript. This variant is thought to promote oncogenesis by impairing the production of the TP53 protein, which when normal, controls the cell cycle. Interestingly this variant is distinct from the inherited mutations identified in families with Li-Fraumeni syndrome. TP53 was among the first molecules discovered to have a strong influence in human cancer risk.25
The function of the inherited variant near CCDC26 (rs55705857) is unknown. However, it is in a region of DNA that is highly conserved evolutionarily and the association remains statistically significant after other variants in the region are taken into account.19 Therefore, it is thought to be the functional variant and experiments are underway to understand the mechanism behind its association with glioma risk. It is important to note that this region is distinct from other areas of chromosome 8q24 that are associated with other types of cancers.
RTEL1, TERT, and TERC are involved in telomere maintenance. The glioma risk variants near TERT and TERC are also associated with increased leukocyte telomere length, suggesting that inherited propensity for longer telomeres may increase glioma risk.2 It is possible that additional functions of the glioma risk variants in these regions may eventually be discovered.
The practice of naming risk variants for nearby genes may be misleading. All of the glioma risk variants and most of those discovered for other cancers and many other diseases through genome-wide association studies are not in protein coding regions of genes.26 As more is learned about the regulatory functions of the noncoding regions of the genome, we will learn more about what disease risk variants actually do. We may find that these variants affect the expression of entirely different genes than the region for which they are currently named. In a well-characterized example of this, an inherited variant in the intron of FTO that had been reproducibly associated with obesity was recently found to be functionally related to expression of a completely different gene, IRX3, and not to expression of FTO.27 Another example is the potential relationship between chromosome 8q24 risk regions and the expression of the quite distant MYC gene and how these increase risk for colon and other cancers.28,29
The functions of the two risk regions near EGFR on chromosome 7 are unknown. However, EGFR is known to be amplified and overexpressed in many gliomas, so it seems plausible that the risk loci may facilitate EGFR overexpression.
The glioma risk region on chromosome 9 is near CDKN2B (cyclin-dependent kinase inhibitor 2B), but the association peak covers the entire region, which also encodes the long noncoding RNA called ANRIL; thus, it is possible that this variant operates through changes in ANRIL expression, but this has not been definitively established. It is becoming increasingly clear that much gene regulation occurs through variation in expression and splicing of long noncoding RNAs, of which ANRIL is one. Long noncoding RNA is a subset of RNA that is transcribed from DNA, but not translated to form proteins. Variation in DNA may affect the expression and splicing of noncoding RNAs.
Associations with Histologic and Molecular Groups
Figure 2 shows the distribution of infiltrating glioma by histology from CBTRUS,23 highlighting which risk variants are statistically significantly associated with different types and grades of infiltrating glioma. This figure is informed by results reported in the literature5,9,16,18,19,30–33 and by data from the UCSF AGS (Supplementary Table S1). Caution is needed in interpreting these results because there is less statistical power with the smaller samples sizes of less common histologic types and grades. Caution is also needed because the histologic types reported by the registry do not completely correspond to WHO grade. This is especially true for the category “diffuse astrocytoma,” which may include astrocytomas of various grades, and therefore does not fully correspond to grade II astrocytoma.
Fig. 2.

Histologic types of glioma (A)21 and associated inherited risk variants (B).5,9,16,18,19,30–33
We recently showed that 3 acquired molecular alterations (IDH mutation, 1p19q co-deletion, and TERT promoter mutation) in tumor cells define five groups of glioma that have distinctive characteristics with respect to association with risk variants, other risk factors, and survival experience. These five groups account for over 95% of grade II-IV gliomas.10 Figure 3 shows the estimated proportions of the molecular groups expected among people with primary grade II-IV gliomas in the general population (see Supplementary Table S2 for calculations). It also shows which inherited risk variants are associated with which molecular groups. As above, caution is needed in interpreting results for the groups with smaller sample sizes. The Cancer Genome Atlas (TCGA) has also found that lower-grade gliomas could be stratified into three distinct molecular subtypes that are associated with differences in overall survival: IDH wild type, IDH mutant with 1p/19q co-deletion, and IDH mutant without 1p/19q co-deletion.34 Grade IV gliomas with IDH mutation are sometimes referred to as secondary glioblastomas, a subset of glioblastoma originally described as preceded by diagnosis of a lower grade glioma. As this paper primarily focuses on inherited risks of first diagnosis of incident adult glioma, risk of secondary glioblastomas or recurrent tumors are not discussed. Much exciting work lies ahead to find other risk variants that may be associated with different subtypes as larger numbers of cases are categorized according to these acquired alterations.
Fig. 3.

Five molecular groups of glioma based on presence or absence of TERT promoter mutation, IDH mutation, and 1p19q co-deletion.10 (A) Estimated distribution of five molecular groups among ∼15 000 adult gliomas diagnosed in the United States each year (details in Supplementary Table S2). (B) Inherited glioma risk variants are associated with different molecular groups.
Summary
Although screening for inherited risk for glioma in the general population is not warranted, the identification of genes that increase risk for gliomas brings us closer to understanding the mechanisms underlying glioma development. Some of the glioma risk variants appear to increase risk of all or most types of glioma, while others increase risk of certain histologic or molecular groups of glioma. As larger groups of patients are studied, it is likely that we will discover more inherited risk variants. We will also learn more about which inherited variants increase risk for which groups. For example, a recent study of families with 2 or more patients with gliomas showed that inherited variation in another telomere-related gene, POT1, may be involved in risk of oligodendroglioma.15 In addition, the large Glioma International Case-Control Consortium is currently working to genotype 4000 people with glioma and 4000 people without glioma with the goal of discovering new risk variants.
Gliomagenesis is a very complex process. Fig. 4 summarizes our current understanding of the interplay of inherited risk variants and acquired alterations in gliomagenesis. The inherited risk variants are those a person is born with and has in all the cells in his/her body; we conceive of these as possible roots that enhance the possibilities of glioma developing. The early acquired mutations occur in all the tumor cells and can be considered trunk mutations; these likely first occurred in the tumor's precursor cells. The branch mutations occur later in gliomagenesis and are responsible for the particular characteristics of the resulting glioma. Every step towards a better understanding of this process brings us closer to improved glioma treatment or prevention.
Fig. 4.

Hypothesized pathways of adult glioma development, including inherited risk variants and acquired mutations and chromosomal changes. Two established risk variants in TP53 and EGFR were not included in this figure because it is not yet clear which branches of gliomagenesis they influence. Artwork was produced by Xavier Studio.
An information sheet included as a supplement to this review may be helpful in explaining inherited risk of primary adult glioma to patients and their families.
Supplementary Material
Glossary of Abbreviations
ANRIL, antisense non-coding RNA in the INK4 locus; CBTRUS, Central Brain Tumor Registry of the United States; CCDC26, coiled-coil domain-containing protein 26; CDKN2A, cyclin-dependent kinase inhibitor 2A; CDKN2B, cyclin-dependent kinase inhibitor 2B; EGFR, epidermal growth factor receptor; IDH, Isocitrate Dehydrogenase; MLH1, mutL homolog 1; MSH2/MSH6, mutS homolog 2/6; NF1/NF2, neurofibromin ½; PHLDB1, pleckstrin homology-like domain, family B, member 1; PMS2, postmeiotic segregation increased 2; POT1, protection of telomeres 1; RTEL1, regulator of telomere elongation helicase 1; TERC, telomerase RNA component; TERT- telomerase reverse transcriptase; TP53, tumor protein P53; TSC1/TSC2, tuberous sclerosis ½; UCSF AGS, University of California, San Francisco, Adult Glioma Study; WHO, World Health Organization.
Funding
The National Institutes of Health [(R01CA52689, P50CA097257, R01CA126831, R01CA139020, R25CA112355 and R01CA163687 funded work at the University of California, San Francisco), (P50CA108961, P30 CA15083, RC1NS068222Z funded work at the Mayo Clinic)]; Other funders include: the National Brain Tumor Foundation; Sence Foundation; Stanley D. Lewis and Virginia S. Lewis Endowed Chair in Brain Tumor Research; Robert Magnin Newman Endowed Chair in Neuro-oncology; family and friends of John Berardi, Helen Glaser, Elvera Olsen, Raymond E. Cooper, and William Martinusen. Bernie and Edith Waterman Foundation; Ting Tsung and Wei Fong Chao Family Foundation. National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health [NuUL1 RR024131 funded the UCSF-CTSI]; California Department of Public Health [The collection of cancer incidence data used in this study was part of the statewide cancer reporting program mandated by California Health and Safety Code Section 103885]; National Cancer Institute's Surveillance, Epidemiology and End Results Program [contract HHSN261201000140C awarded to the Cancer Prevention Institute of California, contract HHSN261201000035C awarded to the University of Southern California, contract HHSN261201000034C awarded to the Public Health Institute]; Centers for Disease Control and Prevention's National Program of Cancer Registries [agreement # U58DP003862-01 awarded to the California Department of Public Health]; Centers for Disease Control and Prevention [Agreement 5U58DP00381-04], the Sontag Foundation (www.sontagfoundation.org), Genentech (www.gene.com), the Pediatric Brain Tumor Foundation (www.curethekids.org), Novocure, Inc. (www.novocure.com), the Musella Foundation (www.virtualtrials.com), Voices Against Brain Cancer (www.voicesagainstbraincancer.org), Elekta (www.elekta.com), as well as private and in kind donations contributed to the maintenance of the CBTRUS database.
This publication's contents are solely the responsibility of the authors and do not necessarily represent the official views of any of their funders.
Acknowledgments
The authors acknowledge study participants, clinicians and research staff at participating medical centers, Katherine Cornelius, Mayo Clinic Comprehensive Cancer Center Biospecimens and Processing and Genotyping Shared Resources, and UCSF Diller Cancer Center Genomics Core. We would like to thank investigators of the UCSF and Mayo Clinic Adult Glioma Studies for their invaluable contributions to this research: Mitchel Berger, Paige Bracci, Susan Chang, Paul A. Decker, Helen Hansen, Thomas Kollmeyer, Lucie McCoy, Alexander Pico, Michael Prados, Hugues Sicotte, Ivan Smirnov and Shichun Zheng. We dedicate this work to the late Marissa Suzuki and Csaba Polony for their many years interviewing participants in the UCSF Adult Glioma Study.
Conflict of interest statement. None declared.
References
- 1.Wrensch M, Jenkins RB, Chang JS et al. Variants in the CDKN2B and RTEL1 regions are associated with high-grade glioma susceptibility. Nat Genet. 2009;41(8):905–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walsh KM, Codd V, Smirnov IV et al. Variants near TERT and TERC influencing telomere length are associated with high-grade glioma risk. Nat Genet. 2014;46(7):731–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stacey SN, Sulem P, Jonasdottir A et al. A germline variant in the TP53 polyadenylation signal confers cancer susceptibility. Nat Genet. 2011;43(11):1098–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shete S, Hosking FJ, Robertson LB et al. Genome-wide association study identifies five susceptibility loci for glioma. Nat Genet. 2009;41(8):899–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rajaraman P, Melin BS, Wang Z et al. Genome-wide association study of glioma and meta-analysis. Hum Genet. 2012;131(12):1877–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanson M, Hosking FJ, Shete S et al. Chromosome 7p11.2 (EGFR) variation influences glioma risk. Hum Mol Genet. 2011;20(14):2897–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan H, Parsons DW, Jin G et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christensen BC, Smith AA, Zheng S et al. DNA methylation, isocitrate dehydrogenase mutation, and survival in glioma. J Natl Cancer Inst. 2011;103(2):143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rice T, Zheng S, Decker PA et al. Inherited variant on chromosome 11q23 increases susceptibility to IDH-mutated but not IDH-normal gliomas regardless of grade or histology. Neuro Oncol. 2013;15(5):535–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eckel-Passow JE, Lachance DH, Molinaro AM et al. Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med. 2015;372(26):2499–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Labussiere M, Di Stefano AL, Gleize V et al. TERT promoter mutations in gliomas, genetic associations and clinico-pathological correlations. Br J Cancer. 2014;111(10):2024–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Labussiere M, Boisselier B, Mokhtari K et al. Combined analysis of TERT, EGFR, and IDH status defines distinct prognostic glioblastoma classes. Neurology. 2014;83(13):1200–1206. [DOI] [PubMed] [Google Scholar]
- 13.Killela PJ, Reitman ZJ, Jiao Y et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA. 2013;110(15):6021–6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ostrom QT, Bauchet L, Davis FG et al. The epidemiology of glioma in adults: a “state of the science” review. Neuro Oncol. 2014;16(7):896–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bainbridge MN, Armstrong GN, Gramatges MM et al. Germline mutations in shelterin complex genes are associated with familial glioma. J Natl Cancer Inst. 2015;107(1):384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walsh KM, Anderson E, Hansen HM et al. Analysis of 60 reported glioma risk SNPs replicates published GWAS findings but fails to replicate associations from published candidate-gene studies. Genet Epidemiol. 2013;37(2):222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Howlader NNA, Krapcho M, Garshell J, Miller D, Altekruse SF, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA (eds). SEER Cancer Statistics Review, 1975–2011. posted to the SEER web site, April 2014; based on November 2013 SEER data submission. Available at: http://seer.cancer.gov/csr/1975_2011/.
- 18.Enciso-Mora V, Hosking FJ, Kinnersley B et al. Deciphering the 8q24.21 association for glioma. Hum Mol Genet. 2013;22(11):2293–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jenkins RB, Xiao Y, Sicotte H et al. A low-frequency variant at 8q24.21 is strongly associated with risk of oligodendroglial tumors and astrocytomas with IDH1 or IDH2 mutation. Nat Genet. 2012;44(10):1122–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dupont WD, Plummer WD Jr.. Understanding the relationship between relative and absolute risk. Cancer. 1996;77(11):2193–2199. [DOI] [PubMed] [Google Scholar]
- 21.Gail MH, Brinton LA, Byar DP et al. Projecting individualized probabilities of developing breast cancer for white females who are being examined annually. J Natl Cancer Inst. 1989;81(24):1879–1886. [DOI] [PubMed] [Google Scholar]
- 22.Antoniou A, Pharoah PD, Narod S et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72(5):1117–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ostrom QT, Gittleman H, Liao P et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol. 2014;16(Suppl 4):iv1–i63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diskin SJ, Capasso M, Diamond M et al. Rare variants in TP53 and susceptibility to neuroblastoma. J Natl Cancer Inst. 2014;106(4):dju047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malkin D, Li FP, Strong LC et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250(4985):1233–1238. [DOI] [PubMed] [Google Scholar]
- 26.Welter D, MacArthur J, Morales J et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42(Database issue):D1001–D1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smemo S, Tena JJ, Kim KH et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014;507(7492):371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grisanzio C, Freedman ML. Chromosome 8q24-associated cancers and MYC. Genes Cancer. 2010;1(6):555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huppi K, Pitt JJ, Wahlberg BM, Caplen NJ. The 8q24 gene desert: an oasis of non-coding transcriptional activity. Front Genet. 2012;3:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Di Stefano AL, Enciso-Mora V, Marie Y et al. Association between glioma susceptibility loci and tumour pathology defines specific molecular etiologies. Neuro Oncol. 2013;15(5):542–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Egan KM, Thompson RC, Nabors LB et al. Cancer susceptibility variants and the risk of adult glioma in a US case-control study. J Neurooncol. 2011;104(2):535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Enciso-Mora V, Hosking FJ, Di Stefano AL et al. Low penetrance susceptibility to glioma is caused by the TP53 variant rs78378222. Br J Cancer. 2013;108(10):2178–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jenkins RB, Wrensch MR, Johnson D et al. Distinct germ line polymorphisms underlie glioma morphologic heterogeneity. Cancer Genet. 2011;204(1):13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brat DJ, Verhaak RG, Aldape KD et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015;372(26):2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]