

Abstract
The endothelial-mesenchymal transition (EndMT) is known to be involved in the transformation of vascular endothelial cells to mesenchymal cells. EndMT has been confirmed that occur in various pathologic conditions. Transforming growth factor β1 (TGF-β1) is a potent stimulator of the vascular endothelial to mesenchymal transition (EMT). Aspirin-triggered resolvin D1 (ATRvD1) has been known to be involved in the resolution of inflammation, but whether it has effects on TGF-β1-induced EndMT is not yet clear. Therefore, we investigated the effects of AT-RvD1 on the EndMT of human umbilical vein vascular endothelial cells line (HUVECs). Treatment with TGF-β1 reduced the expression of Nrf2 and enhanced the level of F-actin, which is associated with paracellular permeability. The expression of endothelial marker VE-cadherin in HUVEC cells was reduced, and the expression of mesenchymal marker vimentin was enhanced. AT-RvD1 restored the expression of Nrf2 and vimentin and enhanced the expression of VE-cadherin. AT-RvD1 did also affect the migration of HUVEC cells. Inhibitory κB kinase 16 (IKK 16), which is known to inhibit the NF-κB pathway, had an ability to increase the expression of Nrf2 and was associated with the inhibition effect of AT-RvD1 on TGF-β1-induced EndMT, but it had no effect on TGF-β1-induced EndMT alone. Smad7, which is a key regulator of TGF-β/Smads signaling by negative feedback loops, was significantly increased with the treatment of AT-RvD1. These results suggest the possibility that AT-RvD1 suppresses the TGF-β1-induced EndMT through increasing the expression of Smad7 and is closely related to oxidative stress.
Keywords: TGF-β1-induced endothelial-mesenchymal transition, Aspirin-triggered resolvin D1, Human umbilical vein vascular endothelial cells line (HUVECs), Inhibitor of IκB kinase, Smad7
INTRODUCTION
The epithelial-mesenchymal transition(EMT) is an important cellular mechanism in embryonic development, tissue repair, and disease. A similar process termed the endothelialmesenchymal transition (EndMT) involves the transformation of vascular endothelial cells to mesenchymal cells (Thiery et al., 2003; Garside et al., 2013; Gonzalez et al., 2014).These developmental programming mechanisms often result in the development of various diseases. At cellular and molecular level EndMT is regulated by similar factors and signaling pathways under both physiological and pathological conditions (Krizbai et al., 2015). EndMT also occurs in various pathologic conditions, including cerebral cavernous malformation, cardiac and kidney fibrosis, vein stenosis, and cancer progression, in addition, it is also involved in the formation of stem-like cells that differentiate into osteoprogenitor cells (Medici et al., 2010; Yao et al., 2013; Xiao et al., 2015). EndMT coincides with genome-wide reprogramming that allows endothelial cells to exist in a variety of phenotypic states, which contributes to disease progression. In tumors, EndMT is an important source of cancer-associated fibroblasts (CAFs), which have been demonstrated that contribute to the tumor growth and metastasis (Allen et al., 2011). Previous studies (Zeisberg et al., 2007) have shown that the EndMT process, which is regarded as an important mechanism for CAF recruitment to the tumor stroma, can be mediated by TGF-β produced in the tumor. Yet, the mechanism of EndMT in tumor is still unclear, but is to be expected to involved similar pathways as in fibrosis (Van Meeteren et al., 2012).
The EndMT process of endothelial cells, which is often stimulated with TGF-β or Notch ligands, and has also been induced by Wnt signaling pathways (Zeisberg et al., 2007; Aisagbonhi et al., 2011). The Snail family, which has been found to involve in the TGF-β induced EndMT. Moreover, the activation of Smad, MEK, P13K and p38 MAPK are also involved in this process. The TGF-β homodimer transduces its signal by bringing together two types of serine/threonine kinase receptors-two type I receptors and two type II receptors (Yan et al., 2009). Following ligand binding, the type II receptor phosphorylates the types I receptor, which in turn phosphorylates the receptor-regulated Smads (R-Smads). Activated R-Smads associate with Smad4, a common-partner Smad (Co-Smad), and translocate into the nucleus (Inoue, Y et al 2008). TGF-β induce ALK5 (TβRI) signaling leads to Smad2 and Smad3 phosphorylation resulting in inhibition of angiogenesis by inhibiting endothelial cells proliferation and migration. In contrast, TGF-β induce ALK1 (TβRII) signaling activates Smad1 and Smad5 leading to cells proliferation and migration (Goumans et al., 2003). Smad7, a negative feedback regulator of Smad signaling, is shown to form a stable complex with type I receptors, therefore leading to inhibition of R-Smad phosphorylation and the hetero-complex formation between R-Smads and Co-Smad (Inoue et al., 2008). Studies have shown (Ryoo et al., 2014) that TGF-β1-stimulate renal epithelial transition to fibroblastic cells through the Smurf1-Smad7 signaling, which is regarded as a molecular signaling linking the Nrf2-GSH pathway in human renal tubular epithelial cells.
Tight junction (TJ) is important components of paracellular pathways, and their destruction enhances vascular permeability (Zhang et al., 2013). The F-actin cytoskeleton plays a major role in TJ barrier function and the regulation of paracellular pathways in different physiological and pathological states (Edens et al., 2000). Study has been reported (Churchman et al., 2009) that TGF-β1 activated kinases (TAK) and MAPK signaling pathways contribute to Nrf2/ARE activation, and the activation of MAPK extracellular signal-regulated kinase (ERK) is associated with the disruption of TJ proteins (Kevil et al., 2000). Thus, we will observe the expression of F-actin to analyze the effect of TGF-β1 on the paracellular permeability.
The Nuclear factor-like 2 (Nrf2), which play a crucial role in oxidative stress and inflammation processes, has a protective effect on cells against reactive oxygen species and other oxidative damage (Zhang et al., 2013). The nuclear factor κB (NF-κB) pathway is know to regulate the inflammation response and is associated with vascular permeability (Zhang et al., 2013). Inhibitory κB kinase (IKK) plays a key role in the regulation of NF-κB activation. Our previous research (Shu et al., 2014) have shown that IKK 16 could inhibite the NF-κB pathway and up-regulate Nrf2-regulated heme oxygenase 1. Reactive oxygen species (ROS), which has been proposed as potent contributors to TGF-β1-mediated pathology (Kashihara et al., 2010), can be regulated by the Nrf2 antioxidant signaling pathway. Therefore, we will combine the NF-κB pathway inhibitor IKK 16 to investigate the relationship between TGF-β1 induced EndMT and Nrf2 antioxidant signaling pathway.
In our report, we described the relationship between antioxidant signaling pathways and TGF-β1-Smad7 signaling pathway in TGF-β1-induced EndMT and demonstrated the functionality of pro-resolving lipid mediator AT-RvD1, which is known to influence the resolution of inflammation and repair of damaged tissues (Cox et al., 2015; Harrison et al., 2015), could inhibit the TGF-β1-induced EMT by increasing the expression of Smad7. The inhibitory effect of AT-RvD1 was associated with Nrf2 expression.
MATERIALS AND METHODS
Reagents
Recombinant human TGF-β1 was purchased from Peprotech (Rocky Hill, NJ, USA). Aspirin-triggered resovin D1 was purchased from Cayman chemicals (AnnArbor, MI, USA). Antibody against VE-cadherin, Nrf2, β-actin and Smad7 were obtained from Santa Cruz Biotchnology Inc (Santa Cruz, CA, USA). The NF-κB inhibitor IKK 16 and IκBα antibody was purchased form Selleck (Selleck chemicals, Houston, USA) and Cell signal Technology (Cell signal Technology, Boston, USA). Fetal bovine serum (FBS) and Dulbecco’s modified Eagle’s medium (DMEM) were obtained from gibco (Carlsbad, CA, USA).
Cell culture
Human umbilical vein vascular endothelial cells line (HUVECs) was obtained from ATCC. This cells were cultured in DMEM supplemented with 10% fetal bovine serum, streptomycin, and penicillin (HyClone). The cells was grown at 37°C in a humidified 5% CO2 atmosphere.
Migration assay using transwell
Cell migration was determined using 24-well plates with transwell chamber inserts (Chemicon International, Atlanta, GA, USA), which had membranes coated with matrigel. In the upper chambers, 3×104 A549 cells were seeded in fetal bovine serum-free culture media, and media with 10% serum were added to the lower chambers. After a 24 hr incubation, the cells on the upper surface of the membrane were scraped off, and the cells that had migrated onto the lower surface of the membrane were immersed in 500 μl media with 0.5 mg/ml MTT, the results was measured with an MTT cell proliferation assay kit (Beyotime, Shanghai, China) at 490 nm absorbance using a plate reader.
Measurement of ROS
Cellular ROS levels were determined using a Reactive Oxygen Species Assay kit (Beyotime, Shanghai, China). Cells in 96-well plates were treated with TGF-β1 or AT-RvD1 for 48 hr. Then, the cells were incubated with 200 μl DMEM and a DCFH-DA fluorescent probe for 30 min at 37°C. The fluorescence intensity was measured with a microplate reader at 488 nm excitation and 525 nm emission wavelengths. Moreover, fluorescence microscopy was used to take pictures.
Measurement of F-actin
Cellular F-actin levels were determined using an Actin-Trakcer Green (Beyotime, Shanghai, China). Cells in 96-well plates were treated with TGF-β1 or AT-RvD1 for 48 hr. Then, the cells were washed twice with PBS and fixed in 4% paraformaldehyde solution in PBS for 10 min at room temperature, permeabilized twice with 0.1% Triton X-100 in PBS for 5 min. F-actin filaments staining was performed with phalloidin-FITC diluted in 3% BSA in PBS for 60 min followed by staining with Actin-Trakcer Green. The pictures were photographed by fluorescent microscopy.
Total RNA extraction and RT-PCR analysis
Total RNA was extracted from cells using TRIzol reagent (Invitrogen, Carlsbad, USA) according to the manufacturer’s instructions. The reverse transcription of 1 μg RNA was carried out using qRT SuperMix (Vazyme, Nanjing, China). A real-time-polymerase chain reaction (RT-PCR) analysis was carried out using the ABI Stepone Plus system with the AceQ SYBR Green Master Mix (Vazyme, Nanjing, China). The primers were synthesized by Bioneer, and the primer sequences for the human genes were: VE-cadherin, 5′-CCAGAATTTGCCCAGCCTA-3′ and 5′-TTACTGGCACCACGTCCTTGTC-3′; Vimentin, 5′-TGCCGTTGAAGCTGCTAACTA-3′ and 5′-CCAG-AGGGAGTGAATCCAGATTA-3′; Nrf2, 5′-TTCCCGGTCACAT-CGAGAG-3′ and 5′-TCCTGTTGCATACCGTCTAAATC-3′; Smad7, 5′-GGACAGCTCAATTCGGACAAC-3′ and 5′-GTACACCCACACACCATCCAC-3′; β-actin, 5′-AAGACCTGTACGCCAACACAGT-3′ and 5′-AGAAGCATTTGCGGTGGACGAT-3′. Amplified expressions of gene transcriptions were normalized to β-actin expression. Cycle threshold (ΔΔCt) values were calculated for each experimental group, which indicated the number of available template cDNA in each reaction. The gene expression levels were compared using values of 2−ΔΔCt.
Total protein extraction and western blot analysis
The total protein extracted from cells was analyzed using a radio immunoprecipitation assay (RIPA) and PMSF (Beyotime, Shanghai, China). Protein concentrations were measured using the BCA method. The protein samples were separated by electrophoresis on 6–10% SDS-polyacrylamide gels and transferred to polyvinylidene fluoride (PVDF) membranes (Invitrogen, Carlsbad, USA). The membranes were blocked with 5% skim milk for 2 hr and then incubated with the appropriate antibodies against Nrf2, Smad7 and β-actin et al in 4% BSA at 4°C overnight. The membranes were further incubated for 2 hr with a peroxidase-conjugated secondary antibody (Beyotime, Shanghai, China) at room temperature, and the results were detected using a ChemiDoc system (Bio-Rad) and analyzed by Image J software.
Statistical analysis
All data were expressed as the means ± s.e.m. and were analyzed with One-way analysis of variance (ANOVA) followed by Newman-Keuls Multiple Comparison Test (GraphPad Prism, USA). Data were recorded from at least three independent experiments. Statistical significance was defined at *p<0.05.
RESULTS
TGF-β1-induced EndMT increased F-actin levels and reduced the expression of Nrf2 in HUVEC cells
TGF-β1-induced EndMT in HUVEC cells enhanced the expression of the mRNA of mesenchymal markers, such as vimentin, and reduced the expression of the endothelial marker VE-cadherin (Fig. 1B). These findings were consistent with previous studies (Kaneda et al., 2011; Choi et al., 2015). The ROS levels were not changed but the F-actin levels were significantly increased in HUVEC cells by incubation with TGF-β1 (Fig. 1A). The migration of HUVEC cells were enhanced by TGF-β1 (Fig. 2). Nrf2 play a critical role in anti-oxidative stress process, and TGF-β1 could reduce its expression in renal epithelial cells (Kobayashi et al., 2005; Ryoo et al., 2014). The effect of TGF-β1 in HUVEC cells was also confirmed by western blot analyses (Fig. 1C). In addition, the expression of Smad7, which plays a crucial role in the regulators of TGF-β1/Smad signaling by negative feedback loops (Yan et al., 2009), was increased by TGF-β1 incubation (Fig. 1B). These results suggested that TGF-β1-induced EndMT increased the F-actin levels and reduced the expression of Nrf2 in HUVEC cells
Fig. 1.

TGF-β1-induced EndMT increased F-actin levels and AT-RvD1 inhibited this process in HUVEC cells. (A) Cells were pretreated with AT-RvD1 for 30 min. The HUVEC cells in 96-well plates were then treated with different concentrations of TGF-β1 (20 ng/ml) for 48 hr. We used a reactive oxygen species assay kit and actin-trakcer green, a microplate reader and fluorescence microscope as described in Materials and Methods. (B) Real time PCR of EndMT markers in HUVEC cells was treated with TGF-β1. (C) Western blot analysis on the expression of Nrf2 and VE-cadherin. β-actin was used as a control. The values were normalized to each control and the means ± SE from three independent experiments were presented. *p<0.05 compared with control groups.
Fig. 2.
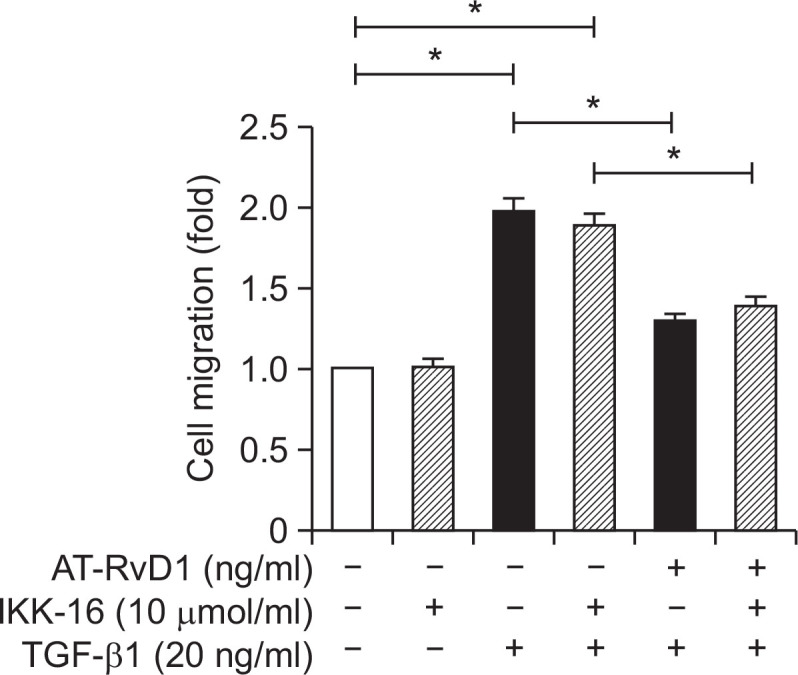
The effects of AT-RvD1 on TGF-β1-induced cell migration. Cells were pretreated with AT-RvD1 for 30 min. The HUVEC cells in24-well plates with transwell chamber inserts were then treated with different concentrations of TGF-β1 (20 ng/ml) for 48 hr. The result was measured with an MTT cell proliferation assay kit at 490 nm absorbance using a plate reader. The results showed that ATRvD1 inhibited TGF-β1-induced cell migration.
AT-RvD1 inhibited TGF-β1-induced EndMT and F-actin levels by increasing the expression of Nrf2 and Smad7
To elucidate the mechanism involved in the effects of aspirin-triggered resolvin D1 on the TGF-β1-induced process, different concentrations of AT-RvD1 were used to interfere with cells 30 min before the TGF-β1 was added. AT-RvD1 treatment blocked the expression of the mesenchymal marker vimentin and restored the expression of VE-cadherin. The effects of AT-RvD1 on the TGF-β1-treated VE-cadherin and vimentin of HUVEC cells were confirmed by RT-PCR (Fig. 3B). Moreover, AT-RvD1 could reduce the F-actin levels of TGF-β1-treated HUVEC cells but had no effect on the levels of ROS (Fig. 1A). The reduced expression of Nrf2 in TGF-β1-treated HUVEC cells was restored by AT-RvD1 (Fig. 3B, 3C). AT-RvD1 enhanced the expression of Nrf2 would be one of a reason for the higher expression of Smad7. Furthermore, ATRvD1 clearly reduced the concentration-dependent TGF-β1-induced migration properties of the TGF-β1-treated HUVEC cells (Fig. 2). Our previous research (Shu et al., 2014) had shown that IKK 16 could inhibite the NF-κB pathway and up-regulated Nrf2-regulated heme oxygenase 1. So we would investigate the effect of IKK16 on TGF-β1-induced EndMT. Our studies had shown that the IKK16 enhanced the expression of Nrf2 but it had no obviously effect on the TGF-β1-induced EndMT. We next sought to explore the relationship between IKK16 and Nrf2 and determined whether the Nrf2 expression was associated with NF-κB pathway. The inhibitor of nuclear factor kappa B (IκB) α protein is the main regulator of NF-κB pathway, we found that IKK 16 had the ability to enhance the expression of IκBα and Nrf2 (Fig. 4) but it had no effect on Smad7 expression (Fig. 3A, 3B). In addition, the AT-RvD1 could also enhance the expression of IκBα (Fig. 4).
Fig. 3.

AT-RvD1 inhibited TGF-β1-induced EndMT by increasing the expression of Nrf2 and Smad7 and the effect of IKK16 on this process. (A) Cells were pretreated with IKK 16 (10 μmol/ml) for 1 hr. The HUVEC cells were then treated with TGF-β1 as mentioned. The effects of the Nrf2 and Smad7 gene expressions by IKK 16 in the TGF-β1-induced EndMT process had been shown. (B) Cells were treated with an IKK 16 (10 μmol/ml) for 1 hr. The cells were then cultured with TGF-β1 (20 ng/ml) and AT-RvD1 (20 ng/ml). After incubation, the gene expressions of Nrf2 and Smad7 were determined by RT-PCR. The effects of the Nrf2 and Smad7 gene expressions by AT-RvD1 in the TGF-β1-induced EndMT process had been altered. (C) The protein expression of Nrf2 and Smad7 were changed by AT-RvD1 and IKK 16.
Fig. 4.

The effect of IKK 16 on the TGF-β1-induced EndMT process in HUVEC cells. Cells were treated with an IKK 16 (10 μmol/ml) for 1 hr. The cells were then cultured with TGF-β1 (20 ng/ml) and AT-RvD1 (20 ng/ml). After incubation, the protein expression of Nrf2 and IκBα were determined by Western blot. Our studies had shown that the IKK16 enhanced the expression of Nrf2 and was associated with the inhibitory effect of AT-RvD1 on TGF-β1-induced EndMT.
DISCUSSION
Endothelial-to-Mesenchymal Transition (EndMT) is a form of the more widely known and studied Epithelial-to-Mesenchymal Transition (EMT). Like EMT, EndMT can also be induced by TGF-β1 (Van Meeteren et al., 2012). Our results had shown that TGF-β1-induced EndMT in HUVEC cells enhanced the expression of the mRNA of mesenchymal markers, such as vimentin, and reduced the expression of the endothelial marker VE-cadherin. The ROS levels were not changed but the F-actin levels were significantly increased in HUVEC cells. The ROS levels were associated with oxidative stress and the Keap1-Nrf2 antioxidant signaling pathways. Treatment with TGF-β1 enhance the levels of ROS and suppress Nrf2 activity in human renal tubular epithelial HK2 (Ryoo et al., 2014). The EndMT process leads to the loss of endothelium and subsequently causes the badly perfuse tissue and tissue damage (Benton et al., 2009; Van Meeteren et al., 2012). Previous studies (Lee et al., 2013) have shown that RvD1 and RvD2 both suppress TGF-β1-induced EMT in lung cancer cells, and RvD1 inhibit the TGF-β1-induced EMT via the ALX/FPR2 and GPR32 receptors. Specialized pro-resolving lipid mediators, such as lipoxin and resolving have the ability to prevent excessive neutrophil trafficking to inflammation sites and thereby promote damaged tissue recovery (Serhan et al., 2002; Spite et al., 2009; Wu et al., 2010). Aspirin-Triggered Resolvin D1 is a potent pro-resolution metabolite of DHA which can curb the inflammatory effects in various acute injuries (Cox et al., 2015; Harrison et al., 2015), yet the effect of AT-RvD1 on TGF-β1 induced EMT and EndMT has not been investigated. Therefore, we investigated the effects of the available aspirin-triggered resolvin D1 on the TGF-β1-induced EndMT of HUVEC cells.
AT-RvD1 suppressed the TGF-β1-induced expression of vimentin and restored the endothelial marker VE-cadherin in HUVEC cells. Moreover, AT-RvD1 also inhibited the TGF-β1-induced migration of HUVEC cells. Previous studies (Lee et al., 2013) have shown that RvD1 inhibite the TGF-β1-induced EMT by reducing the expression of ZEB1. LXA4, another mediator of resolution, has also been reported to inhibit the EMT process by CTGF induction (Wu et al., 2010). In HUVEC cells, RvD1 can inhibit the NF-κB pathway to protect against impairment of endothelial barrier function induced by LPS through up-regulating the expression of TJ protein, which is associated with vascular permeability (Zhang et al., 2013). So we used the NF-κB pathway inhibitor IKK 16 to clarify the possible mechanism. Research findings had shown that it had no significantly effect on TGF-β1-induced EndMT, but the expression of Nrf2 was enhanced. These results had shown that the NF-κB signaling pathway might not considered to be one of the main mechanisms and the effect of AT-RvD1 in suppressing the TGF-β1-induced EndMT of HUVEC cells by other ways.
Reactive oxygen species (ROS) have been reported to promote the TGF-β1-induced EMT genetic changes (Kashihara et al., 2010). However, high concentrations of ROS can also damage cellular components and compromise cell viability (Anastasiou et al., 2011). The F-actin cytoskeleton detemines cell shape and participates in the regulation of TJ proteins and is associated with paracellular permeability (Zhang et al., 2013). Our results had shown that the levels of ROS could not be changed by AT-RvD1, but the morphological changes were reversible. The cultured HUVEC cells are polygonal with the cobblestone morphology (Jaffe et al., 1973; Emmanuel et al., 2013). TGF-β1 induced elongation and streamlining of endothelium could be reversed by AT-RvD1.These were possible associated with the expression of F-actin. We also examined the expression of Nrf2, which is one of the key proteins involved in antioxidant signaling pathways. The expression of Nrf2 could be suppressed by TGF-β1 but AT-RvD1 partially raised it expression. Studies has been reported (Churchman et al., 2009) that TGF-β1 activated kinases (TAK) and MAPK signaling pathways contribute to Nrf2/ARE activation and subsequent induction of protective antioxidant defence genes such as HO-1 in aortic smooth muscle cells, but with the increasing expression of Smad7, the effect of TGF-β1 is weakened and inhibite TGF-β1-mediated induction of HO-1 expression. In addition, treatment with TGF-β1 enhance the levels of ROS and suppress Nrf2 activity in human renal tubular epithelial HK2 (Ryoo et al., 2014), and the protein level of Smad7 is elevated by the activation of Nrf2. These finding suggested that there was a relationship between Nrf2 anti-oxidant signaling pathways and the TGF-β1/Smad pathway on the trans-differentiation process. Smad7, as an important negative regulator factors, will plays a key role in the process.
Inhibitory Smads, including Smad6 and Smad7, are key regulators of TGF-β/bone morphogenetic protein (BMP) signaling by negative feedback loops (Yan et al., 2009). Smad7 is a general antagonist of TGF-β family, while Smad6 is specific for BMP signaling. Smad7 can antagonize TGF-β family signaling mainly through some kinds of mechanisms (Yan et al., 2009). Smad7 can form a stable complex with type I receptors, and then lead to inhibition of R-Smad phosphorylation and the hetero-complex formation between R-Smads and Co-Smad. On the other hand, the HECT type of E3 ubiquitin ligases Smurf1 and Smurf2 can be recruited, Smad7 binds to Smurfs in the nucleus and translocates into the cytoplasm in response to TGF-β and recruits the ubiquitin ligases to the activated type I receptors, leading to the degradation of the receptor through the proteasomal pathway (Ng et al., 2005; Yan et al., 2009). In our studies, we found that the expression of Smad7 was enhanced on TGF-β1-induced EndMT, this might indicate that HUVEC cells promoted the expression of Smad7 to play an inhibitory effect on TGF-β1-induced EndMT. The purpose was to maintain cells survival but could not prevent the trans-differentiation of HUVEC cells.
Our previous research (Shu et al., 2014) have shown that the inhibition of NF-κB signaling pathway is associated with up-regulated Nrf2-regulated heme oxygenase 1. In our current study, IKK 16 also increased the expression of Nrf2 but it had no effect on TGF-β1-induced EndMT. Our studies had also shown that AT-RvD1 enhanced the expression of Smad7 and reduced the gene expression of Smurf1. A recent study (Ryoo et al., 2014) demonstrate that pure activation of Nrf2 can modulate the TGF-β1/Smad signaling, the protein level of Smad7 is elevated and diminish the expression of Smurf1, indicating that Smurf1-Smad7 may be a molecular signaling linking the Nrf2 pathway to TGF-β1-induced EMT changes. Smad7 has also been reported to enhance the transcription of IκB, which is a key inhibitor of NF-κB signaling pathway (Ng et al., 2005). It is also disrupt the TRAF-TAK1-TAB2/3 complex, thus inhibiting NF-κB signaling (Yan et al., 2009). So we suggested that the inhibitory effect of AT-RvD1 on the expression of IκBα was possible associated with Nrf2. To explored the relationship between Smad7 and Nrf2 and determined whether the Nrf2 expression was associated with the IκBα expression, we observed the influence on the Nrf2 and IκBα expression by IKK 16. Our results suggested that IKK-16 had the ability to enhance the expression of IκBα and Nrf2. This results indicated a hypothesis that the high expression of Smad7 up-regulated the expression of Nrf2 possibly through inhibiting the NF-κB signaling pathway and formed a positive feedback loops by the Nrf2 activation.
There are a few limitations to our experiments. We illustrate that there is a relationship between Nrf2 antioxidant signaling pathways and TGF-β1/Smad pathway; however, the complex dynamics are very complicated and require further investigation. In addition, the effect of AT-RvD1 has not been confirmed for in vivo animal models. We will focus our efforts on exploring the other mechanisms responsible for the expression of Smad7 as affected by AT-RvD1 and also carry out animal experiments.
Our results suggest the possibility that AT-RvD1 suppresses the TGF-β1-induced EndMT through increasing the expression of Smad7 and is closely related to oxidative stress.
Acknowledgments
The anthors wish to thank Yusheng Shu for linguistic advice, and Xinxin Li, Ling Cao, Xiaolong Yuan, Wenhui Li, Qianqian Cao for technical assistance.
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
- Allen M, Louise Jones J. Jekyll and Hyde: the role of the microenvironment on the progression of cancer. J Pathol. 2011;223:162–176. doi: 10.1002/path.2803. [DOI] [PubMed] [Google Scholar]
- Aisagbonhi O, Rai M, Ryzhov S, Atria N, Feoktistov I, Hatzopoulos AK. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis Model Mech. 2011;4:469–483. doi: 10.1242/dmm.006510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, Bellinger G, Sasaki AT, Locasale JW, Auld DS, Thomas CJ, Heiden MGV, Cantley LC. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334:1278–1283. doi: 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton RL, Maddie MA, Dincman TA, Hagg T, Whittemore SR. Transcriptional activation of endothelial cells by TGFβ coincides with acute microvascular plasticity following focal spinal cord ischaemia/reperfusion injury. ASN Neuro. 2009;1:181–184. doi: 10.1042/AN20090008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchman AT, Anwar AA, Li FY, Sato H, Ishii T, Mann GE, Siow R. Transforming growth factor-β1 elicits Nrf2-mediated antioxidant responses in aortic smooth muscle cells. J Cell Mol Med. 2009;13:2282–2292. doi: 10.1111/j.1582-4934.2009.00874.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox R, Jr, Phillips O, Fukumoto J, Fukumoto I, Tamarapu Parthasarathy P, Arias S, Cho Y, Lockey RF, Kolliputi N. Aspirin-Triggered Resolvin D1 Treatment Enhances Resolution of Hyperoxic Acute Lung Injury. Am J Respir Cell Mol Biol. 2015;53:422–435. doi: 10.1165/rcmb.2014-0339OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HY, Lee HG, Kim BS, Ahn SH, Jung A, Lee M, Lee JE, Kim HJ, Ha SK, Park HC. Mesenchymal stem cell-derived microparticles ameliorate peritubular capillary rarefaction via inhibition of endothelial-mesenchymal transition and decrease tubulointerstitial fibrosis in unilateral ureteral obstruction. Stem Cell Res Ther. 2015;6:18. doi: 10.1186/s13287-015-0012-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edens HA, Parkos CA. Modulation of epithelial and endothelial paracellular permeability by leukocytes. Adv Drug Deliv Rev. 2000;41:315–328. doi: 10.1016/S0169-409X(00)00049-1. [DOI] [PubMed] [Google Scholar]
- Emmanuel C, Huynh M, Matthews J, Kelly E, Zoellner H. TNF-α and TGF-β synergistically stimulate elongation of human endothelial cells without transdifferentiation to smooth muscle cell phenotype. Cytokine. 2013;61:38–40. doi: 10.1016/j.cyto.2012.09.017. [DOI] [PubMed] [Google Scholar]
- Garside VC, Chang AC, Karsan A, Hoodless PA. Co-ordinating Notch, BMP, and TGF-β signaling during heart valve development. Cell Mol Life Sci. 2013;70:2899–2917. doi: 10.1007/s00018-012-1197-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal. 2014;7:re8. doi: 10.1126/scisignal.2005189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, Karlsson S, ten Dijke P. Activin receptor-like kinase (ALK) 1 is an antagonistic mediator of lateral TGFβ/ALK5 signaling. Mol. Cell. 2003;12:817–828. doi: 10.1016/S1097-2765(03)00386-1. [DOI] [PubMed] [Google Scholar]
- Harrison JL, Rowe RK, Ellis TW, Yee NS, O’Hara BF, Adelson PD, Lifshitz J. Resolvins AT-D1 and E1 differentially impact functional outcome, post-traumatic sleep, and microglial activation following diffuse brain injury in the mouse. Brain Behav Immun. 2015;47:131–140. doi: 10.1016/j.bbi.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y, Imamura T. Regulation of TGF-β family signaling by E3 ubiquitin ligases. Cancer Sci. 2008;99:2107–2112. doi: 10.1111/j.1349-7006.2008.00925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe EA, Nachman RL, Becker CG, Minick CR. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J Clin Invest. 1973;52:2745–2756. doi: 10.1172/JCI107470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizbai IA, Gasparics Á, Nagyőszi P, Fazakas C, Molnár J, Wilhelm I, Bencs R, Rosivall L, Sebe A. Endothelial-Mesenchymal Transition of Brain Endothelial Cells: Possible Role during Metastatic Extravasation. PLoS One. 2015;10:e0119655. doi: 10.1371/journal.pone.0119655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kevil CG, Oshima T, Alexander B, Coe LL, Alexander JS. H2O2-mediated permeability: role of MAPK and occludin. Am J Physiol Cell Physiol. 2000;279:C21–C30. doi: 10.1152/ajpcell.2000.279.1.C21. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal. 2005;7:385–394. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- Kashihara N, Haruna Y, Kondeti VK, Kanwar YS. Oxidative stress in diabetic nephropathy. Curr Med Chem. 2010;17:4256–4269. doi: 10.2174/092986710793348581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda H, Arao T, Matsumoto K, De Velasco MA, Tamura D, Aomatsu K, Kudo K, Sakai K, Nagai T, Fujita Y, Tanaka K, Yanagihara K, Yamada Y, Okamato I, Nakagawa K, Nishio K. Activin A inhibits vascular endothelial cell growth and suppresses tumour angiogenesis in gastric cancer. Br. J. Cancer. 2011;105:1210–1217. doi: 10.1038/bjc.2011.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Park MK, Lee EJ, Lee CH. Resolvin D1 inhibits TGF-β1-induced epithelial mesenchymal transition of A549 lung cancer cells via lipoxin A4 receptor/formyl peptide receptor 2 and GPR32. Int J Biochem Cell Biol. 2013;45:2801–2807. doi: 10.1016/j.biocel.2013.09.018. [DOI] [PubMed] [Google Scholar]
- Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med. 2010;16:1400–1406. doi: 10.1038/nm.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng YY, Hou CC, Wang W, Huang XR, Lan HY. Blockade of NFκB activation and renal inflammation by ultrasound-mediated gene transfer of Smad7 in rat remnant kidney. Kidney Int Suppl. 2005;67:S83–S91. doi: 10.1111/j.1523-1755.2005.09421.x. [DOI] [PubMed] [Google Scholar]
- Ryoo IG, Ha H, Kwak MK. Inhibitory role of the KEAP1-NRF2 pathway in TGFβ1-stimulated renal epithelial transition to fibroblastic cells: a modulatory effect on SMAD signaling. PLoS One. 2014;9:e93265. doi: 10.1371/journal.pone.0093265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu YS, Tao W, Miao QB, Zhu YB, Yang YF. Improvement of ventilation-induced lung injury in a rodent model by inhibition of inhibitory κB kinase. J Trauma Acute Care Surg. 2014;76:1417–1424. doi: 10.1097/TA.0000000000000229. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, Moussignac RL. Resolvins a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med. 2002;196:1025–1037. doi: 10.1084/jem.20020760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spite M, Norling LV, Summers L, Yang R, Cooper D, Petasis NA, Flower RJ, Perretti M, Serhan CN. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature. 2009;461:1287–1291. doi: 10.1038/nature08541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–746. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Van Meeteren LA, Ten Dijke P. Regulation of endothelial cell plasticity by TGF-β. Cell Tissue Res. 2012;347:177–186. doi: 10.1007/s00441-011-1222-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SH, Zhang YM, Tao HX, Dong L. Lipoxin A4 inhibits transition of epithelial to mesenchymal cells in proximal tubules. Am J Nephrol. 2010;32:122–136. doi: 10.1159/000315121. [DOI] [PubMed] [Google Scholar]
- Xiao L, Kim DJ, Davis CL, McCann JV, Dunleavey JM, Vanderlinden AK, Xu N, Pattenden SG, Frye SV, Xu X, Onaitis M, Monaghan-Benson E, Burridge K, Dudley AC. Tumor endothelial cells with distinct patterns of TGFβ-driven endothelial-to-mesenchymal transition. Cancer Res. 2015;75:1244–1254. doi: 10.1158/0008-5472.CAN-14-1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Jumabay M, Ly A, Radparvar M, Cubberly MR, Boström KI. A role for the endothelium in vascular calcification. Circ Res. 2013;113:495–504. doi: 10.1161/CIRCRESAHA.113.301792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Liu Z, Chen Y. Regulation of TGF-β signaling by Smad7. Acta Biochim Biophys Sin (Shanghai) 2009;41:263–272. doi: 10.1093/abbs/gmp018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007;67:10123–10128. doi: 10.1158/0008-5472.CAN-07-3127. [DOI] [PubMed] [Google Scholar]
- Zhang X, Wang T, Gui P, Yao C, Sun W, Wang L, Wang H, Xie W, Yao S, Lin Y, Wu Q. Resolvin D1 reverts lipopolysaccharide-induced TJ proteins disruption and the increase of cellular permeability by regulating IκBα signaling in human vascular endothelial cells. Oxid Med Cell Longev. 2013;1857;2013:15. doi: 10.1155/2013/185715. [DOI] [PMC free article] [PubMed] [Google Scholar]